


Abstract
1-Aminobenzotriazole (ABT) has been extensively used as a nonspecific inhibitor of cytochromes P450 (P450s) in animals for mechanistic studies, and antipyrine (AP) has been used as a probe for hepatic oxidative metabolic capacity determination in vivo. The method of use of ABT has been variable from lab to lab due largely to unknown pharmacokinetics of ABT itself and incomplete information on various P450s inhibited. The oral pharmacokinetic profiles of ABT were generated in rats, dogs, and monkeys in the dose range of 5 to 200 mg/kg. The results showed that after single oral doses of 50 mg/kg in rats, and 20 mg/kg in dogs and monkeys, the plasma concentrations were high and were sustained for over 24 h. In vitro, inhibition of various expressed P450s upon 30-min preincubation with ABT (0–500 μM) showed that CYP1A2, 2B6, 2C9, 2C19, 2D6, and 3A4 were inhibited in a dose-dependent manner. The intravenous pharmacokinetics of AP also was affected in a dose-dependent manner in all species, treated 2 h earlier with ABT. Thus, the plasma clearance of AP was inhibited by 88% in rats pretreated with 50 mg/kg ABT and 96% in dogs and 83% in monkeys pretreated with 20 mg/kg ABT. Based on these data in rats, dogs, and monkeys, and the established safety profile of ABT in rats dosed up to 100 mg/kg, a pretreatment at 2 h with a single oral dose of ABT at 100 mg/kg in rats (providing 93% inhibition) and 20 mg/kg in dogs and monkeys effectively inhibited the clearance of the probe compound.
ABT1has been extensively used as a nonspecific inhibitor of cytochromes P450 in animals for mechanistic studies (Mico et al., 1988; Huijzer et al., 1989; Mugford and Tarloff, 1995; Su et al., 1998; Constan et al., 1999; Marczylo and Ioannides, 1999). The method of use of ABT has been quite variable from lab to lab, for example, single versus multiple dosing of animals prior to the test compound, p.o. versus i.v. route of administration, different pretreatment times, and different dose levels. Moreover, the pharmacokinetics of ABT in different species have not been reported. Also there is incomplete information in the literature on the inhibition of P450s by ABT (Knickle and Bend, 1992;Su et al., 1998; Di Re et al., 1999). To provide a guideline for the pretreatment regimen of ABT, 1) single dose oral pharmacokinetic studies were conducted in rats, dogs, and monkeys at several dose levels; 2) in vitro inhibition of various recombinant P450s by ABT was determined; and 3) the effect of selected doses of ABT on the intravenous pharmacokinetics of antipyrine, a probe for measuring efficiency of hepatic oxidative metabolism, was determined. The results from these studies are described in this report.
Materials and Methods
ABT and antipyrine (AP) were obtained from Sigma-Aldrich (St. Louis, MO). Single oral dose pharmacokinetics of ABT, in 0.5% aqueous methylcellulose, were studied in fasted male Sprague-Dawley rats at 10, 50, and 200 mg/kg (n ≥ 3) and in Beagle dogs and Cynomolus monkeys at 5, 20, and 100 mg/kg (n = 2 each). All animals were obtained from Charles River Laboratories (Wilmington, MA). The plasma samples were collected over a period of 72 h, and the concentrations of ABT as well as of antipyrine (from experiments described below) were determined simultaneously by precipitation of protein from 0.1 ml of plasma by 0.2 ml of acetonitrile, followed by liquid chromatography/tandem mass spectrometry on a SCIEX API 4000 (PerkinElmerSciex Instruments, Boston, MA) using a Monolithic RP-18 column (4.6 × 100 mm, 2 μ). The mobile phase involved a mixture of acetonitrile and water containing 0.1% formic acid flowing at 2.5 ml/min.
In vitro inhibition of rP450 activities was measured in a 96-well plate format involving 30-min preincubation of individual human P450 supersomes (40 nM) with ABT (0–500 μM) in the presence of 1 mM NADPH and 0.05 M potassium phosphate buffer, at 37°C. The incubation mixtures were then diluted 10 times with NADPH and marker substrates at 100 μM: 7-benzyloxy-4-trifluoromethylcoumarin (CYP1A2, 3A4), 4-(Aminomethyl)-7-methoxycoumarin (CYP2D6), 7-methoxy-4-trifluoromethylcoumarin (CYP2C9), or 7-ethoxy-4-trifluoromethylcoumarin (CYP2B6 and 2C19). The metabolism was determined by fluorometric assays as described by GENTEST (www.gentest.com). The substrates, rP450s and fluorescent metabolite standards, were obtained from BD Gentest Corporation (Woburn, MA). The metabolism was determined by fluorometric assays as described by GENTEST.
Two hours prior to administration of AP (20 mg/kg, i.v.), ABT was administered orally to rats at 50 and 100 mg/kg (n = 5 each), and dogs and monkeys at 20 (n = 2) and 100 mg/kg (n = 1), for the interaction study. The control groups involved dosing of animals (n = 2) with 20 mg/kg AP alone and with a low dose of ABT alone (n = 1). Plasma samples were collected up to 24 h and processed and analyzed as above by the liquid chromatography/tandem mass spectrometry method for simultaneous quantitation of both the analytes.
Results and Discussion
ABT is a nonspecific and effective inhibitor of P450s. It is a metabolism-based inactivator of cytochromes P450 by the mechanism ofN-alkylation of heme moiety (Ortiz de Montellano and Mathews, 1981; Su et al., 1998; Wong et al., 1999). Other enzymes, like NADPH cytochrome c reductase, glutathioneS-transferase, and glucuronyl transferase are not affected by this inhibitor (Mugford et al., 1992), and hence ABT is a fairly selective P450 inhibitor. [14C]ABT is known to be well absorbed (≥70%) in rats and radioactivity distributed widely throughout the body, with liver, kidney, and adrenals showing the highest levels (Town et al., 1993). Thus, the inhibition of P450s is expected in many organs. ABT has been demonstrated to be safe in rats on multiple dosing and also at acute high doses (Mico et al., 1988;Meschter et al., 1994), making it an attractive agent for differentiating between metabolite- and parent compound-based toxicities of test materials in safety assessment studies. ABT has been implicated in the inhibition of CYP1A, 2A, 2B, 2C, 2E, 3A, 4A, and 4B in various organs of different species (Knickle and Bend, 1992; Mathews et al., 1996; Su et al., 1998; Di Re et al., 1999; Jackson et al., 2000) but without any reports on CYP2D inhibition.
AP is a nonspecific substrate of cytochromes P450 (Engel at al., 1996;Matzke et al., 2000). It is water soluble, has low hepatic extraction ratio of 0.01 (Soda and Levy, 1975) and low plasma protein binding of 13% (Rane et al., 1977), properties ideal for investigating intrinsic hepatic metabolic clearance of the compound, free from dependence on blood flow and protein binding. AP is almost completely metabolized by hepatic cytochrome P450 enzymes, with very little excreted unchanged in human urine (Brodie and Axelrod, 1950). Approximately 99% of the radioactivity was recovered in urine following a14C-labeled drug administration in humans (Uchino et al., 1983). Alterations in the disposition of AP by different drugs or disease state have been used as an indicator of hepatic metabolic dysfunction (Gurley et al., 1997; Takeda et al., 1999; Jorquera et al., 2001).
The plasma concentration-time profiles of ABT following single oral doses in fasted male Sprague-Dawley rats at 10, 50, and 200 mg/kg (n ≥ 3), and beagle dogs and cynomolgus monkeys at 5, 20, and 100 mg/kg (n = 2 each), are shown in Fig.1. In all species, the AUC0–72h increased in a greater than dose proportional manner, as expected for this inhibitor (Table1). At the mid and high doses in all animals, plasma concentrations showed plateaus of high concentrations for over 24 h (Fig. 1), an indication of extensive and prolonged inhibition of metabolism. Thus, a single oral dose administration—instead of multiple doses—of ABT could be effective when the test compound is given well within the plateau range.

Plasma concentration-time profiles of ABT after single oral doses in rats, dogs, and monkeys.
Mean/average pharmacokinetic parameters after single oral doses of 1-aminobenzotriazole in rats, dogs, and monkeys
The results of the in vitro inhibition of rP450 activities following a 30-min preincubation of individual human CYP1A2, 2B6, 2C9, 2C19, 2D6, and 3A4 supersomes with ABT (0–500 μM) showed that all P450s were inhibited dose dependently by ABT pretreatment. ABT has previously been shown to inhibit CYP2E1 as well (Mathews et al., 1996; Jackson et al., 2000). The CYP3A4 was affected the most while CYP1A2 the least (Fig.2). Thus, CYP2D6 was also effectively inhibited by ABT, information that is complementary to the existing inhibition profile of ABT in the literature. Pretreatment for longer times would give greater inhibition. Thus, the above conditions could easily be adopted for in vitro P450 inhibition assays.

In vitro inhibition of marker activities for human rP450 isoforms following 30-min preincubation with ABT.
The extent of metabolism was measured by fluorescence of metabolite generated.
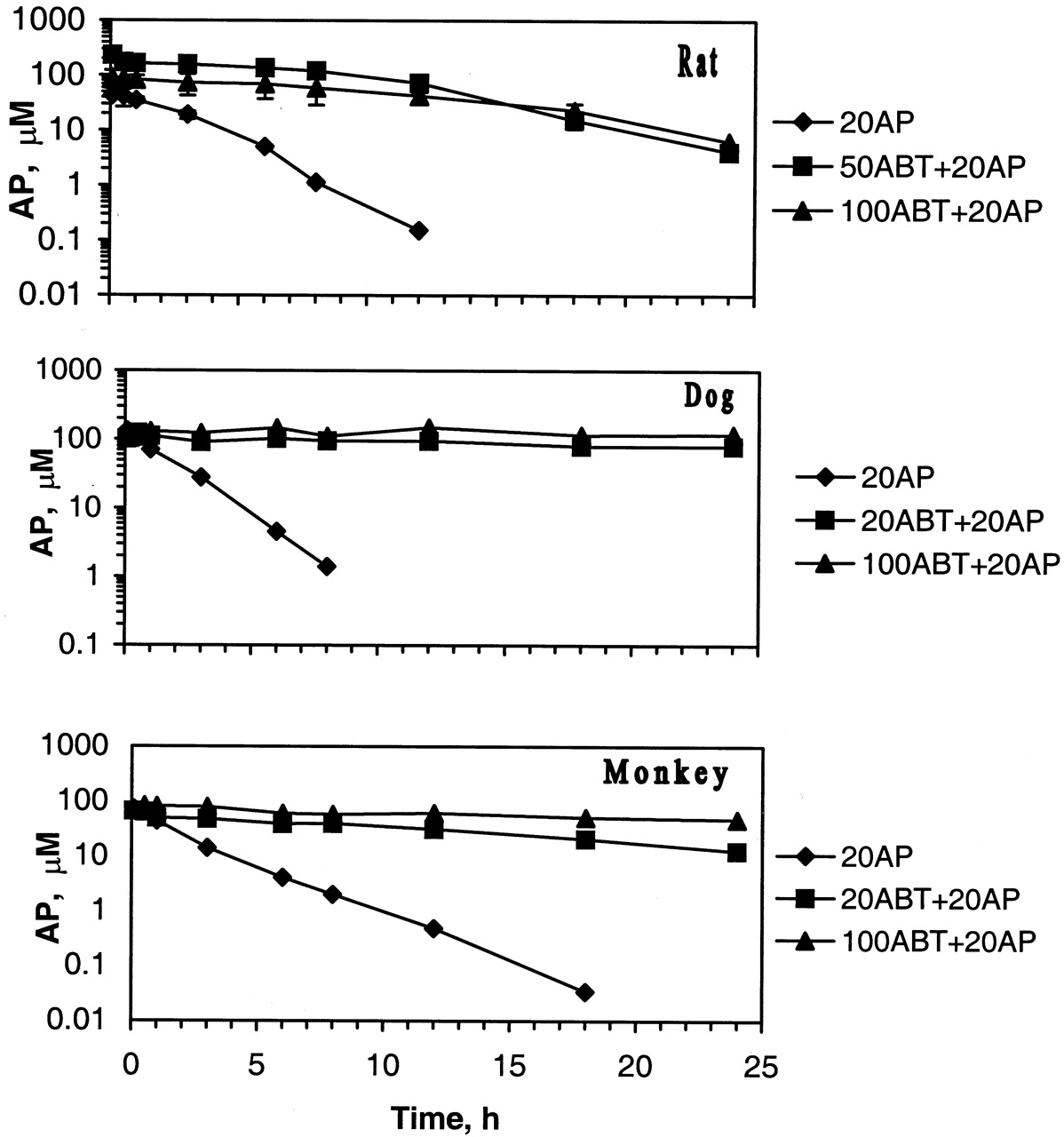
Based on the observed, high plasma concentrations in animals and the potential for inhibition of P450s to a significant extent after 30-min ABT pretreatment, ABT doses of 50 and 100 mg/kg for rats, and 20 and 100 mg/kg for dogs and monkeys, were selected for coadministration with AP. The control studies involved dosing of animals with 20 mg/kg AP alone and with a low dose of ABT alone. The pretreatment of animals 2 h prior to the intravenous AP administration was considered to provide substantial inhibitory effect on the P450s, based on reports of profound loss of cytochrome P450 contents of liver and kidneys in rats 2 h post the ABT dose (Mugford et al., 1992). The plasma concentration-time profiles of AP are shown in Fig.3. The ABT concentrations in these experiments were comparable to those seen or approximated from the earlier discrete studies (Fig. 1). Coadministration with AP did not alter the plasma concentration-time profiles of ABT in these species. However, ABT caused dose-dependent changes in the pharmacokinetics of AP in all species. Thus, the metabolism, and hence the plasma clearance of AP was inhibited by approximately 88% in rats pretreated with 50 mg/kg ABT, and 96% in dogs and 83% in monkeys each pretreated with 20 mg/kg ABT (Table 2). The corresponding values of the AUC0–24h were approximately 8-fold higher for rats, 9-fold for dogs, and 5-fold for monkeys. In rats, ABT has been shown to be safe in acute high and chronic (100 mg/kg) dose studies. Thus, in rats single oral doses of ABT at 100 mg/kg could be employed to achieve even better, 93% inhibition of the plasma clearance of AP. Similar chronic studies in dogs and monkeys have not been reported. However, ABT has been successfully used in our labs for many single dose mechanistic studies without any overt toxicity. Thus, 2 h prior treatment with a single oral dose of ABT at 100 mg/kg for rats, and 20 mg/kg for dogs and monkeys, would provide safe and notable changes in pharmacokinetics for low metabolic clearance compounds to ascertain toxicities due to metabolites. For compounds with high hepatic extraction ratio, which are cleared largely through oxidative metabolism, doses lower than the above could be employed. Also at the recommended dose levels, single oral dose, instead of multiple dose, pretreatment of animals with ABT (2 h prior to test compound administration) could provide measurable inhibition of oxidative metabolism for mechanistic or bioactivation studies.

Plasma concentration-time profiles of antipyrine (20 mg/kg i.v.) in rats, dogs, and monkeys pretreated (2 h) orally with ABT.
Mean/average pharmacokinetic parameters of antipyrine (20 mg/kg i.v.) with and without pretreatment of 1-aminobenzotriazole (p.o.) in rats, dogs and monkeys
Acknowledgments
We thank Drs. Check Quon and Gerald Miwa for their helpful discussions.
Footnotes
-
↵2 Current address: Millennium Pharmaceuticals, Inc., 75 Sidney Street, Cambridge, MA 02139.
- Abbreviations used are::
- ABT
- 1-aminobenzotriazole
- P450
- cytochromes P450
- rP450
- recombinant cytochrome P450
- AP
- antipyrine
- AUC
- area under the curve
- Received March 13, 2002.
- Accepted June 11, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}