


Abstract
The effect of microsomal protein concentration on the inhibitory potency of a series of CYP3A inhibitors was assessed in vitro using diazepam 3-hydroxylation (yielding temazepam) as an index of CYP3A activity. With diazepam concentrations fixed at 100 μM, inhibition of temazepam formation by fixed concentrations of ritonavir, ketoconazole, itraconazole, OH-itraconazole, norfluoxetine, and fluvoxamine decreased substantially as active protein concentrations increased from 0.0625 to 3.0 mg/ml. However protein concentration had only a small effect on the inhibitory activity of fluconazole. Equilibrium dialysis indicated extensive microsomal binding of all inhibitors except fluconazole; binding increased with higher protein concentrations. Based on the CYP3A content of liver microsomes, decrements in inhibitory potency of stronger inhibitors (ketoconazole and ritonavir) could be explained by specific binding, whereas nonspecific binding is anticipated to account for the effect on weaker inhibitors (norfluoxetine and fluvoxamine). Thus, microsomal binding (specific, nonspecific, or a combination of both) may have a major effect on estimation of inhibitory potency of P450 inhibitors and may contribute to variations among laboratories. The effect can be minimized by use of the lowest possible microsomal protein concentration for in vitro studies of metabolic inhibition.
Microsomal binding of drug substrates and/or metabolic inhibitors is increasingly recognized as a potential source of artifact arising in the course of in vitro studies of drug metabolism. Nonspecific binding of substrate to microsomal protein can influence availability of the substrate to the metabolizing enzyme or enzymes in vitro, and thereby yield biased estimates of enzyme kinetic parameters. These, in turn, may produce inaccurate predictions when in vitro data are used to estimate in vivo pharmacokinetics (Obach, 1996,1997; Obach et al., 1997; McLure et al., 2000; Venkatakrishnan et al., 2000c, 2001; Kalvass et al., 2001). Inhibitor binding to microsomal systems may likewise influence estimation of potency of metabolic inhibitors (Gibbs et al., 1999a). The influence of binding has been termed “inhibitor depletion” when the inhibitor interacts with the active site (Gibbs et al., 1999a). However, inhibitor depletion could also refer to nonspecific inhibitor binding to microsomal preparations, as well as to actual microsomal consumption of the inhibitor itself through biotransformation.
The present study evaluated the influence of microsomal protein concentration on the inhibitory capacity of known CYP3A inhibitors. In vitro 3-hydroxylation of diazepam to temazepam was used as an index reaction to profile CYP3A activity. Two selective serotonin reuptake inhibitors (fluvoxamine and norfluoxetine), three antifungal azole agents (itraconazole, ketoconazole, and fluconazole), a metabolite of itraconazole (OH-itraconazole), and the human immunodeficiency virus protease inhibitor ritonavir were used as representative CYP3A inhibitors. These agents have different intrinsic inhibitory capacities and different binding affinities.
Materials and Methods
Incubation Procedures and Index Reaction Characteristics.
Liver samples from individual human donors with no known liver disease were provided by the International Institute for the Advancement of Medicine (Exton, PA), the Liver Tissue Procurement and Distribution System (University of Minnesota, Minneapolis, MN), or the National Disease Research Interchange (Philadelphia, PA). All samples were of the CYP2D6 and CYP2C19 normal metabolizer phenotype based on prior in vitro phenotyping studies. Microsomes were prepared by ultracentrifugation; microsomal pellets were suspended in 0.1 M potassium phosphate buffer containing 20% glycerol and stored at −80°C until use. Chemical reagents and drug entities were purchased from commercial sources or kindly provided by their pharmaceutical manufacturers (von Moltke et al., 1993, 1994a,b, 1996a,b;Venkatakrishnan et al., 2000a,b,c, 2001a,b).
Diazepam 3-hydroxylation to form temazepam was used as the index reaction for CYP3A activity (Andersson et al., 1994; Ono et al., 1996;Jung et al., 1997; Venkatakrishnan et al., 2000). Methanolic solutions of diazepam (100 μM) were added to 2-ml microcentrifuge tubes, and the solvent was evaporated to dryness under conditions of mild vacuum at 40°C. Incubation mixtures containing an isocitrate/isocitric dehydrogenase regenerating system in 50 mM phosphate buffer, 5 mM Mg2+, and 0.5 mM NADP+ were then added to the tubes and equilibrated in a 37°C water bath for 5 min.
Metabolic Inhibitors.
Incubations were performed with diazepam alone (no inhibitor), and with coaddition with fixed concentrations of the following established CYP3A inhibitors: ketoconazole, 0.05 μM; itraconazole, 1.0 μM; OH-itraconazole, 0.25 μM; fluconazole, 10 μM; ritonavir, 0.05 μM; norfluoxetine, 25 μM; and fluvoxamine, 50 μM (von Moltke et al., 1994b, 1995, 1996a,b, 1998a,b; Venkatakrishnan et al., 2000b).
Influence of Active Protein Concentration.
Reactions were initiated by addition of varying amounts of microsomal protein (0, 0.0625, 0.125, 0.25, 0.5, 0.75, 1, 2, and 3 mg/ml) with final volume of 250 μl, and incubated for 10 min. Previous studies had demonstrated time-dependent linearity of temazepam formation up to 20 min. After the incubation period, 100 μl of 2% butylated hydroxytoluene in acetonitrile was added to stop the reactions. A benzodiazepine analog (U31485, 100 ng) (Greenblatt et al., 1981) was added as internal standard. Samples were then extracted by addition of 1.2 ml of toluene/isoamyl alcohol (98.5:1.5), vortexing vigorously for approximately 60 s, and centrifuging at 3000 rpm for 10 min. The organic phase was then transferred into 2-ml autosampling vials for gas chromatographic (GC1) analysis. All reactions were performed in duplicate, and all studies were performed using microsomal preparations from four different human livers.
Influence of Inactive Protein Concentration.
Diazepam (100 μM) with or without ketoconazole (0.05 μM), OH-itraconazole (0.25 μM), or fluconazole (10 μM) was added into 2-ml microcentrifuge tubes and dried under mild vacuum at 40°C. Buffer containing isocitrate/isocitric dehydrogenase system (as described above) was added and the tubes were incubated at 37°C for 5 min. Reactions were initiated by addition of varying amounts of heat-inactivated microsomal protein (0, 0.25, 0.75, 1.75, and 2.75 mg/ml), with a constant amount of active microsomal protein (0.25 mg/ml). The final volume was 250 μl. After 10 min of incubation, 100 μl of 2% butylated hydroxytoluene in acetonitrile was added to stop the reactions. U31485 was added as internal standard, and samples were extracted with toluene/isoamyl alcohol for GC analysis as described above.
Analytical Conditions for Gas Chromatography.
The GC (model 6890A; Hewlett Packard, Palo Alto, CA) was equipped with an electron-capture detector, automatic sampler, and data processor integrator The column was 6 feet in length, 2 mm in internal diameter, and packed with 3% SP-2250 on 80/100 Supelcoport (Supelco, Bellefonte, PA). The chromatographic conditions were oven temperature, 275°C; injection port temperature, 310°C; and detector temperature, 310°C. The carrier gas was argon/methane (95:5) at a flow rate of 30 ml/min. The retention times for diazepam (substrate), desmethyldiazepam (N-demethylated metabolite), temazepam (3-hydroxylated metabolite), and U31485 (internal standard) were 2.6, 3.7, 5.4, and 8.5 min, respectively.
Studies of Inhibitor Consumption.
Ketoconazole (5 μM), with or without diazepam (100 μM), was added into 2-ml microcentrifuge tubes and dried under mild vacuum at 40°C. Reactions were initiated by addition of microsomal protein, and stopped by addition of 100 μl of acetonitrile. Terfenadine (15 μg) was added as internal standard. The samples were centrifuged at 14,000 rpm for 10 min. Twenty-five microliters of the supernatant was injected for HPLC analysis of remaining ketoconazole.
Equilibrium Dialysis Studies.
Ketoconazole or fluconazole (initial added concentration, 100 μM) was added to 2-ml microcentrifuge tubes and dried under mild vacuum conditions at 40°C. Varying concentrations of microsomal protein (0, 0.2, 0.5, 1.0, and 3.0 mg/ml) were added into tubes along with buffer A (50 mM phosphate buffer and 5 mM MgCl2 without cofactors) to a final volume of 1.0 ml. Then 400 μl of mixtures from each tube was injected into membrane bags (Spectra/Por Biotech Regenerated Cellulose membrane tubing, molecular weight cutoff 15 kDa; Spectrum, Inc., Los Angeles, CA). The membrane bags were immersed in 6 ml of buffer A in 15-ml centrifuge tubes and incubated at 37°C for 6 h (Venkatakrishnan et al., 2000c). Dialysates, along with calibration standards, were mixed with microsomal protein at a final concentration of 1.5 mg/ml in 0.4 ml.
Norfluoxetine and fluvoxamine, at concentrations of 25 and 50 μM, respectively, were mixed with human liver microsomes (0.25–3.0 mg/ml) in a volume of 0.15 ml, and dialyzed versus 0.15 ml of phosphate buffer (100 mM, pH 7.4) in a 96-well equilibrium dialysis apparatus at 37°C for 6 h. Hydroxyitraconazole and ritonavir, at concentrations of 1.0 μM in human liver microsomes (0.25–3.0 mg/ml), were dialyzed under the same conditions using a Spectra-Por dialysis apparatus (Spectrum, Los Angeles, CA) for 5.5 h at 37°C, using #4 membranes prepared as per instructions from the manufacturer. After the dialysis period, the samples were removed from the apparati and stored at −20°C until analysis. Drugs in the microsomal matrix were incubated at 37oC for the dialysis period to ensure stability of the analytes through the dialysis process.
For analysis of ketoconazole, terfenadine (25 μg) was added as internal standard. Acetonitrile (100 μl) was added to precipitate microsomal protein. Mixtures were centrifuged for 10 min. Twenty-five microliters of the supernatant was injected for HPLC analysis. For analysis of fluconazole, the dialysate and calibration standards with phenacetin (0.5 μg) as internal standard were alkalinized with 150 μl of 1 N NaOH and extracted twice with 2 ml of ethyl acetate. The organic extract was evaporated to dryness and reconstituted with 250 μl of mobile phase. Fifty microliters was injected for HPLC analysis.
For fluvoxamine, norfluoxetine, ritonavir, and hydroxyitraconazole, dialyzed microsomal samples were diluted with buffer, and buffer samples were diluted with microsomes to ensure an identical matrix for each sample, at a given microsomal protein concentration. The volume of retentate or dialysate used in analysis was varied according to the microsomal protein concentration. This was necessary to ensure that the detector responses in the liquid chromatography/mass spectrometry assay were in the linear range for all samples. For fluvoxamine and norfluoxetine samples, microsomal proteins were precipitated using two volumes of acetonitrile containing the internal standard (150 pmol/ml fluvoxamine for norfluoxetine samples, and 250 pmol/ml norfluoxetine for fluvoxamine samples) followed by centrifugation, and supernatants were injected on the column for liquid chromatography/mass spectrometry analysis. For ritonavir and hydroxyitraconazole samples, ketoconazole (10 ng in 20 μl of methanol) was added as an internal standard followed by extraction with 3 ml of methyl t-butyl ether. The extracts were evaporated (N2, 35°C) and reconstituted in 0.025 ml of HPLC mobile phase.
Due to poor recovery from the dialysis system, equilibrium dialysis studies of itraconazole could not be performed.
HPLC Conditions: Analysis of Ketoconazole and Fluconazole.
The analytical column was 30 cm in length containing C18 μBondapak (10-μm particle size). The mobile phase for ketoconazole was 50 mM NH4H2PO4/CH3CN/CH3OH (55:40:5) at a flow rate of 1.5 ml/min. The ultraviolet absorbance detector was set at 206 nm. The retention times for ketoconazole and terfenadine were approximately 13 and 19.5 min, respectively. For fluconazole analysis, the mobile phase was methanol/10 mM sodium acetate (40:60) at a flow rate of 1.0 ml/min, and UV detection was at 261 nm. The retention times for fluconazole and phenacetin were approximately 8 and 12.5 min, respectively.
HPLC-Mass Spectrometry Conditions: Analysis of Hydroxyitraconazole, Fluoxetine, Fluvoxamine, and Ritonavir.
Samples were injected onto a Phenomenex Luna C18column (2.0 × 50 mm, 5 μm) equilibrated in 20 mM acetic acid (pH adjusted to 4 with NH4OH) containing 23% CH3CN at a flow rate of 0.5 ml/min. The system consisted of a CTC PAL autosampler (CTC Analytics, Carrboro, NC), model 1100 quaternary gradient pump and solvent degasser (Agilent, Palo Alto, CA), and an API100 mass spectrometer (PE Sciex, Thornhill, ON, Cananda) fitted with a TurboIonspray interface. The initial mobile phase conditions were maintained for 1 min followed by a linear gradient to 77% CH3CN at 6 min and held for 3 min. The flow was split approximately 50:50 into the mass spectrometer. The mass spectrometer was operated in the positive ion mode with an orifice voltage of 20 V and a source temperature of 400°C. The monitored ions and their respective retention times (Rt) were m/z 296 (norfluoxetine, Rt = 4.0 min);m/z 319 (fluvoxamine, Rt = 3.7 min); m/z 531 (ketoconazole,Rt = 4.4 min); m/z 721 (hydroxyitraconazole, Rt = 6.1 min); and m/z 721 (ritonavir, Rt= 6.1 min).
Data Analysis.
At each microsomal protein concentration, rates of formation of temazepam from diazepam with inhibitor present were expressed as a fraction (Rv) of the control velocity without inhibitor, as follows:
Iu can be calculated from the added (free plus bound) inhibitor concentration (Ia) and the inhibitor free fraction (FF) in the microsomal mixture, as follows:
Results
CYP3A Inhibition Studies: Effect of Active Protein Concentration.
Temazepam formation in the absence of inhibitors increased as microsomal protein concentration increased to 3 mg/ml. This was accompanied by some degree of substrate consumption, based on GC analysis of diazepam remaining in the reaction mixture (Fig.1). The inhibitory capacity of the various CYP3A inhibitors declined as microsomal protein concentration increased (Table 1). At low concentrations of microsomal protein (0.0625 mg/ml), inhibitors reduced temazepam formation rate to 23 to 49% of control. At the highest concentration of microsomal protein (3 mg/ml), these inhibitors reduced metabolite formation to no less than 80% of control velocity. The only exception was 10 μM fluconazole, for which inhibitory capacity was less dependent on protein concentration.

Mean (±S.E.) remaining fraction of initial diazepam concentration in relation to active microsomal protein concentrations for control (inhibitor-free) incubations.
Effect of active microsomal protein concentration on inhibitory effect of CYP3A inhibitors, based on diazepam 3-hydroxylation as an index reaction
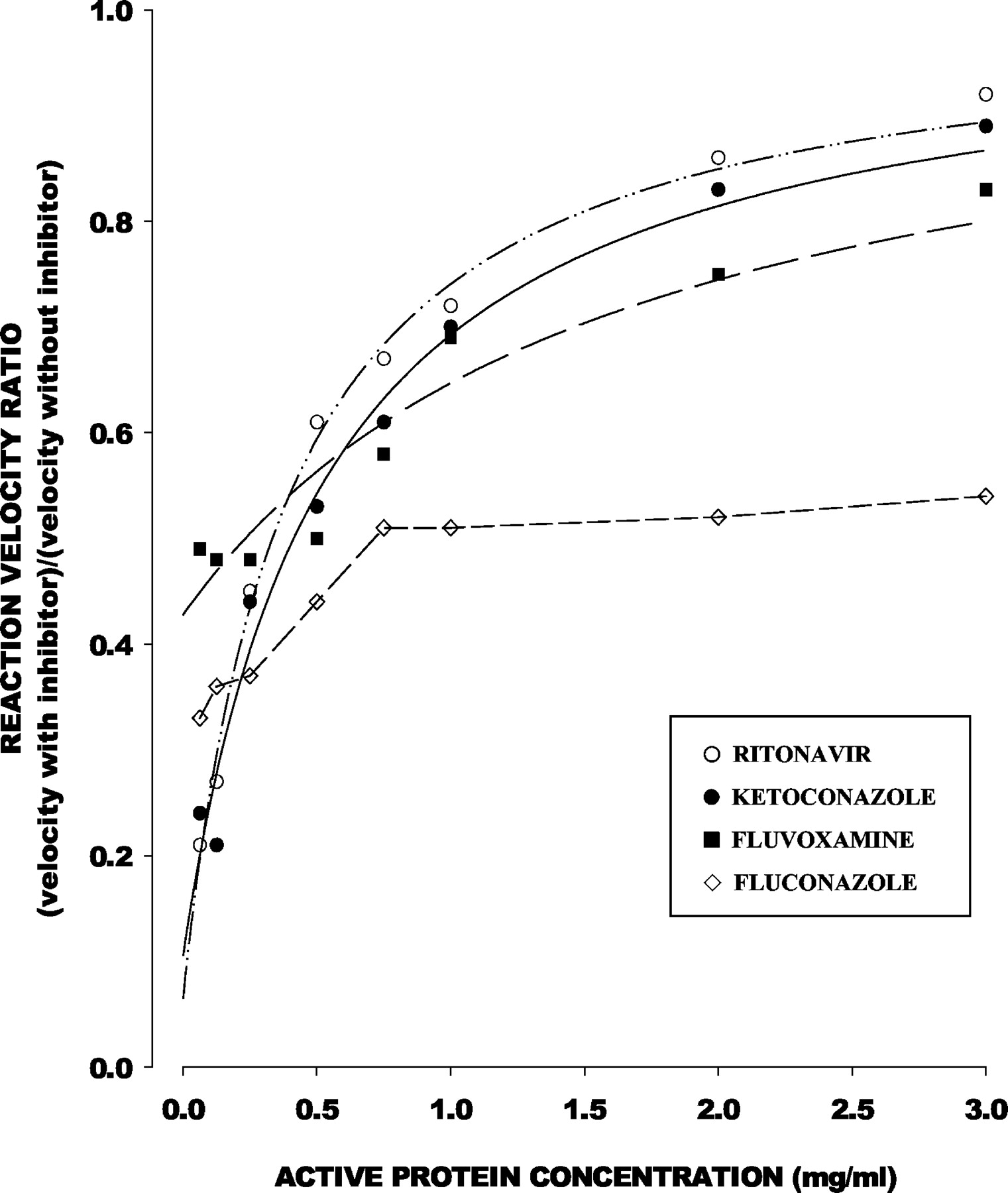
The relation of Rv to total microsomal protein concentration was consistent with eq. 5 for ritonavir (r2 = 0.995), ketoconazole (r2 = 0.979), fluvoxamine (r2 = 0.928), norfluoxetine (r2 = 0.968), itraconazole (r2 = 0.959), and OH-itraconazole (r2 = 0.982), but not for fluconazole (r2 = 0.74) (Fig.2). Estimated unbound IC50 values are shown in Table 1.

Fractional decrement in reaction velocity relative to inhibitor-free control (y-axis) in relation to active microsomal protein concentration (x-axis) for representative CYP3A inhibitors.
Data points are mean values from Table 1. For ritonavir, ketoconazole, and fluvoxamine, data points are consistent with eq. 5; lines represent functions of best fit, with unbound IC50 values shown in Table 1. Data points for fluconazole were not consistent with eq. 5.
Inhibitor Metabolism Studies.
No evidence of ketoconazole consumption was observed with increasing microsomal protein concentration. Ketoconazole concentrations remaining in incubates containing no microsomal protein were essentially identical to those in incubates containing protein, regardless of the actual protein concentration. The same relationship was obtained with coaddition of diazepam to the incubation mixtures.
Inactive Protein Studies.
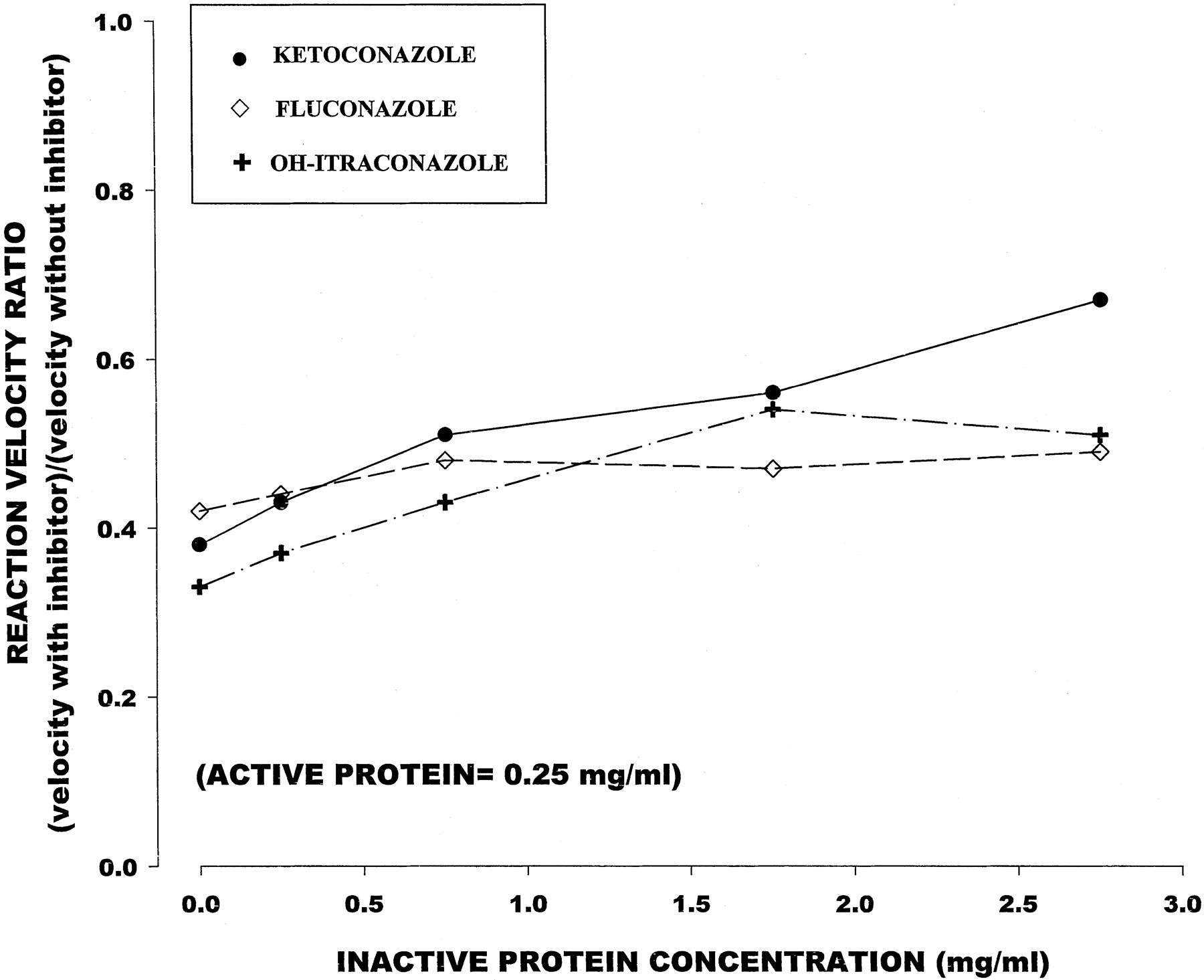
Temazepam formation in the presence of a constant active protein concentration (0.25 mg/ml) decreased with increasing concentrations heat-inactivated microsomal protein (0–2.75 mg/ml). The inhibitory capacity of 0.05 μM ketoconazole and 0.25 μM OH-itraconazole also declined with increasing inactive protein concentration (Table 2; Fig. 3); however, the effect was less pronounced than that in the active protein study. Fluconazole, on the other hand, showed little change in its inhibitory capacity with increasing inactive protein concentration.
Effect of inactive microsomal protein concentration on inhibitory effect of CYP3A inhibitors at a constant active protein concentration (0.25 mg/ml)

Mean (± S.E.) fractional decrement in reaction velocity relative to inhibitor-free control (y-axis) in relation to inactive microsomal protein concentrations (x-axis).
Active protein concentration was fixed at 0.25 mg/ml.
Equilibrium Dialysis Studies.
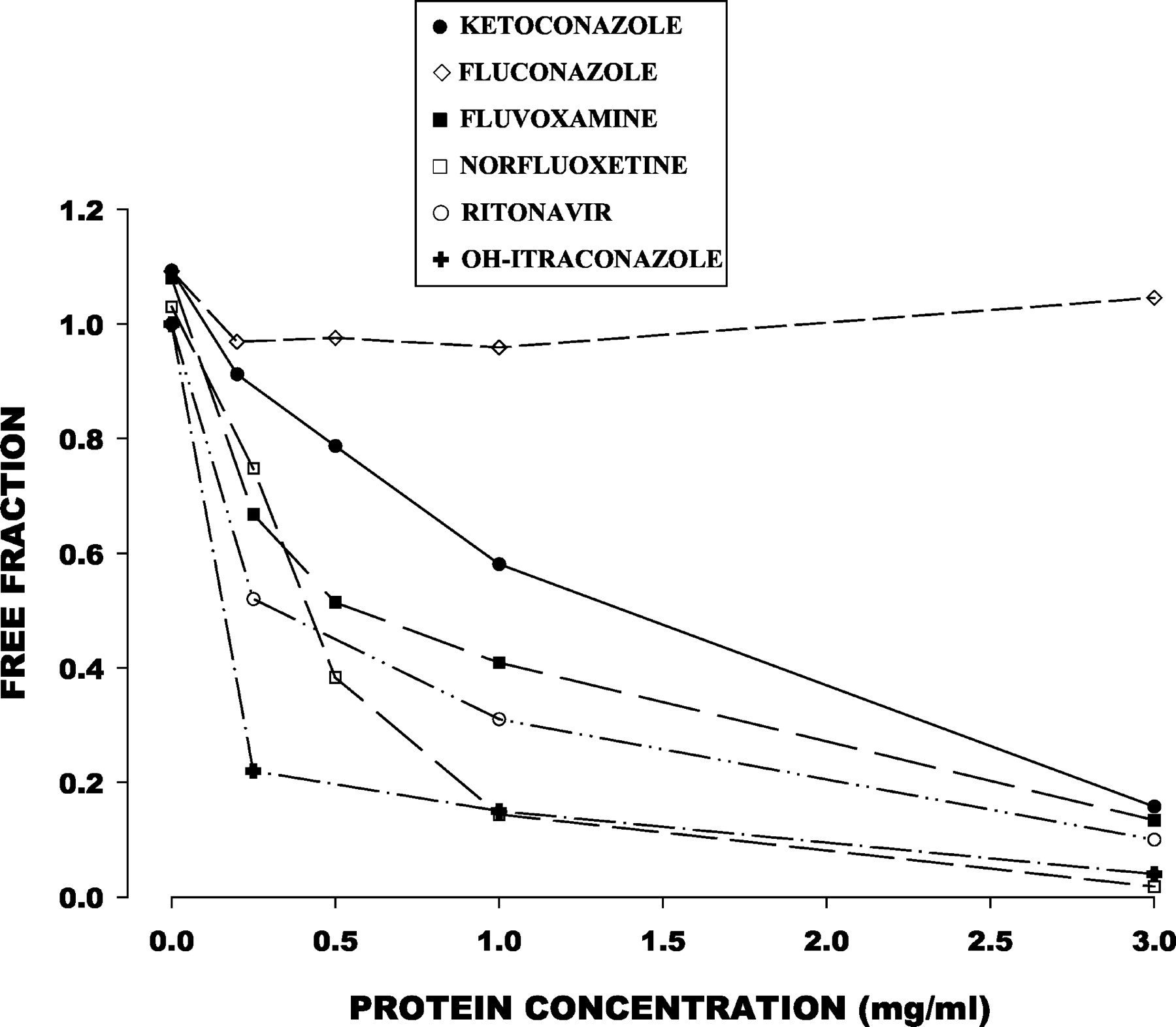
The free fraction of all inhibitors except fluconazole declined as the microsomal protein concentration increased (Fig.4). The free fraction for fluconazole, however, was close to 1.0 regardless of microsomal protein concentration.

Free fraction of six CYP3A inhibitors in relation to microsomal protein concentration.
Discussion
Consistent with previous reports (Gibbs et al., 1999a), the CYP3A inhibitory effect of ketoconazole was substantially reduced with increasing concentrations of active microsomal protein. Because some substrate depletion also occurred at higher protein concentrations, the effect of protein concentration on ketoconazole inhibition was, if anything, underestimated. Metabolic consumption of ketoconazole in the microsomal system was evaluated as a possible contributor to the decrement in inhibitory action of ketoconazole. Erve et al. (2000)reported substantial metabolic consumption of 1 μM ketoconazole by liver microsomes at a protein concentration of 2 mg/ml and an incubation duration of 30 min. However, we did not observe evidence of ketoconazole consumption by microsomes at 5 μM ketoconazole, protein concentrations up to 3 mg/ml, and an incubation duration of 10 min. In contrast to ketoconazole, inhibition by fluconazole was minimally dependent on microsomal protein concentration. The differences between ketoconazole and fluconazole apparently are explained by their differing affinity for nonspecific binding sites. Equilibrium dialysis demonstrated decreased ketoconazole free fraction with increasing microsomal protein concentration, whereas fluconazole free fraction was close to 1.0 regardless of microsomal protein concentration.
A number of other established CYP3A inhibitors, including the azole derivative itraconazole (and its principal metabolite), the selective serotonin reuptake inhibitors norfluoxetine and fluvoxamine, and the viral protease inhibitor ritonavir, exhibited decreases in inhibitory potency with increasing active microsomal protein in a manner similar to that observed with ketoconazole. All of these compounds are lipophilic agents with moderate-to-extensive binding to human plasma proteins, and equilibrium dialysis studies indicated that microsomal binding in vitro apparently explains the decrement in inhibitory activity with increasing microsomal protein. The magnitude and rank order of estimated unbound IC50 values for these agents parallel previously reported data (Venkatakrishnan et al., 2000b, 2001b; von Moltke et al., 1994b, 1995, 1996a,b, 1998a,b). We did not evaluate actual inhibitor consumption by microsomes for these other inhibitors.
Previous studies using diazepam 3-hydroxylation as an index reaction have demonstrated the functional CYP3A content of liver samples used in the present study to be approximately 125 pmol/mg protein (Venkatakrishnan et al., 2001a). At 0.05 μM (50 pmol/ml) ketoconazole and 0.5 mg/ml active protein (63 pmol/ml CYP3A), diazepam 3-hydroxylation activity was reduced to approximately 50% of control, implying occupancy by ketoconazole of a corresponding fraction of CYP3A binding sites (Table 1). Under the assumption of a single diazepam active site per molecule of CYP3A, 32 pmol/ml ketoconazole is anticipated to be bound to active sites, with the remaining 18 pmol/ml either unbound or bound to other sites. At higher concentrations of microsomal protein, available active binding sites would exceed the availability of ketoconazole in the in vitro mixture, and the decrement in inhibitory capacity could be explained mainly by specific microsomal binding. Similar estimations would hold for ritonavir. However, for the less potent inhibitors norfluoxetine and fluvoxamine, 50% inhibition is achieved at concentrations that greatly exceed the availability of specific binding sites, and the decrement in inhibition would be explained mainly by nonspecific binding.
The present study indicates that microsomal binding may have a major impact on estimation of inhibitory potency using in vitro systems. Decrements in inhibitory activity with increasing microsomal protein concentration may be observed with strong as well as weak inhibitors. The mechanism of binding may be either specific, nonspecific, or a combination of the two. It is likely that the decrease in inhibitory potency observed for relatively weaker inhibitors at high microsomal concentrations is attributable mainly to nonspecific binding. The findings suggest that large variability among laboratories in estimated inhibitory potency of a single inhibitor versus a single substrate (such as ketoconazole versus midazolam) could in part be explained by variations in protein and/or enzyme concentration (Gascon and Dayer, 1991; Hargreaves et al., 1994; Wrighton and Ring, 1994; Ghosal et al., 1996; von Moltke et al., 1996b; Gibbs et al., 1999a,b; Wang et al., 1999; Perloff et al., 2000; Venkatakrishnan et al., 2000b). In general, the impact of microsomal binding, both specific and nonspecific, may be minimized by use of the lowest possible microsomal concentration.
Acknowledgments
This work was supported by Grants MH-58435, DA-13209, DK/AI-58496, DA-05258, DA-13834, AG-17880, MH-34223, MH-01237, and RR-00054 from the Department of Health and Human Services.
Footnotes
- Abbreviations used are::
- GC
- gas chromatography
- HPLC
- high-performance liquid chromatography
- Rt
- retention time
- Received July 3, 2002.
- Accepted September 11, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}