


Abstract
The study was designed to define the contribution of cytochrome P450 2C19 (CYP2C19) and cytochrome P450 3A4 (CYP3A4) to citalopram N-demethylation and to evaluate the relationship between the disposition of citalopram and CYP2C19 genotype. A single oral 40-mg dose of citalopram was administered to eight extensive metabolizers and five poor metabolizers recruited from 77 healthy Chinese volunteers whose genotypes and phenotypes were predetermined. The plasma concentrations of citalopram and desmethylcitalopram were determined by high-performance liquid chromatography. It was found that the genotype of CYP2C19 had a significant effect on the N-demethylation of citalopram. Poor metabolizers with m1 mutation had higher area under the plasma concentration versus time curve (AUC0→∞) values than did extensive metabolizers. Terminal elimination half-life (t1/2) values of citalopram in poor metabolizers were significantly higher than the values in extensive metabolizers who were either homozygous or heterozygous with CYP2C19*1. The oral clearance (CLoral) of citalopram in poor metabolizers was significantly lower than that of extensive metabolizers. The AUC0→∞ and maximum plasma concentration (Cmax) of desmethylcitalopram in poor metabolizers were significantly lower than the values of extensive metabolizers. The results show that CYP3A4 is not the major enzyme in the N-demethylation of citalopram among extensive metabolizers. The polymorphism of CYP2C19 plays an important role in the N- demethylation of citalopram in vivo. The extensive metabolizers and poor metabolizers of CYP2C19 had significant difference in disposition of citalopram in vivo.
Citalopram (CIT1) is a potent and selective serotonin reuptake inhibitor in the central nervous system and is widely used to treat depression (Hyttel, 1982). It is metabolized mainly through N-desmethylation in the liver to desmethylcitalopram (DCIT) and didesmethylcitalopram (Milne and Goa, 1991). Major problems in the clinical use of this drug are the large interpatient variations in plasma and the difference in clinical response and toxicity. The major source of this variation is considered to be the interindividual difference in the activities of the cytochrome P450 enzymes that catalyze citalopram metabolism.
Some in vitro studies have shown that the formation of desmethylcitalopram was catalyzed by multiple isoforms of P450s in human liver microsomes, including CYP2C19, CYP2D6, and CYP3A4 (Kobayashi et al., 1997; Rochat et al., 1997; Olesen and Linnet, 1999). Nevertheless, there is some confusion as to which P450 isoforms are responsible for citalopram N-demethylation in vivo. Recently, Kobayashi et al. (1997) reported that at least three P450 isoforms are involved in the liver microsomal N-demethylation of citalopram and that the contribution of CYP3A4 is the most important among them. Kobayashi et al. noticed that the results of their study were not consistent with Sindrup et al. (1993), whose in vivo studies revealed that N-demethylation of citalopram was significantly related to CYP2C19 genotype.
Interindividual and ethnic differences in drug metabolism and response are a clinically important issue in the use of many drugs. One of the major causes is the variability of activities of drug-metabolizing enzymes in the liver. CYP2C19 is a clinically important enzyme because of its genetic polymorphism that separates people into two different phenotypes: extensive metabolizers (EMs) and poor metabolizers (PMs) of S-mephenytoin. CYP2C19 activity is genetically determined, and its genetic polymorphism shows marked interracial difference. The incidence of the poor metabolizer phenotype is markedly higher in Asian populations (13–23%) than in white populations (2–5%) (de Morais et al., 1994b). The normal allele and seven defective alleles of CYP2C19 have been designated as CYP2C19*1 (wild-type), CYP2C19*2 (m1), CYP2C19*3 (m2), CYP2C19*4 (m3), CYP2C19*5 (m4), CYP2C19*6 (m5), CYP2C19*7 (m6), and CYP2C19*8 (m7). The combinations of CYP2C19*2 and CYP2C19*3 can account for 100% of Asian poor metabolizers, whereas all the genetic defects are found in white populations (Gonzalez et al., 1990; de Morais et al., 1994a,b; Ferguson et al., 1998; Ibeanu et al., 1998).
Troleandomycin is a macrolide antibiotic that has been shown to effectively inhibit in vitro CYP3A4 and 3A5 in human liver microsomes. It is a selective inhibitor, in that the activity of seven other P450 forms, including CYP1A2 and CYP2C9, was unaffected (Watkins et al., 1989). In addition, Watkins et al. showed that, using the erythromycin breath test, CYP3A activity in vivo was inhibited by 80% by treatment with 250 mg of oral troleandomycin in healthy volunteers (Watkins et al., 1989, 1992; Wang et al., 1997). Our study was designed to determine the effect of CYP2C19 and CYP3A4 in citalopram N-demethylation by testing the effect of troleandomycin on the pharmacokinetics of citalopram in CYP2C19 genotyped subjects.
Materials and Methods
Subjects. Before the pharmacokinetic study of citalopram, 77 unrelated healthy Chinese volunteers were screened for their CYP2C19 genotype. Their phenotypes were identified by omeprazole, a probe drug of CYP2C19. Eight extensive metabolizers (genotyped as CYP2C19*1/*1, CYP2C19*1/*2, or CYP2C19*1/*3) and five poor metabolizers (genotyped as CYP2C19*2/*2 or CYP2C19*2/*3) were randomly selected for participation in the study. The mean age and body weight were 21 ± 1 years (range, 20–22 years) and 64 ± 8 kg (range, 56–81 kg). All subjects were male nonsmokers and healthy as determined by medical history, physical examination, and laboratory screening tests. No medication or ethanol consumption was allowed for at least 2 weeks before and through the clinical study. The protocol of the study was approved by Ethics Committees of Xiang-Ya School of Medicine, Central South University, Hunan, China. Informed written consent was obtained from all subjects.
Determination of CYP2C19 Genotypes and Phenotypes. All subjects belonged to a pool of volunteers with predetermined CYP2C19 genotypes. CYP2C19*2 (m1) and CYP2C19*3 (m2) mutations were determined by conventional restriction fragment length polymorphism and polymerase chain reaction (de Morais et al., 1994b; Ferguson et al., 1998). CYP2C19 phenotypes were determined with the use of omeprazole as a probe drug (Balian et al., 1995). In brief, 13 subjects whose genotypes were known were given a single oral 20-mg dose of omeprazole (Astra, Haässle, Mölndal, Sweden). The concentration of omeprazole and its 5-hydroxymetabolite were measured in plasma 2 h after oral administration of omeprazole, and log omeprazole hydroxylation indices (log10 [omeprazole]/[5-hydroxyomeprazole]) were calculated. An index of a log10 metabolic ratio <1 was taken to correspond to an extensive metabolizer phenotype, whereas an index of ≥1 corresponds to a poor metabolizer phenotype.
Samples Collection. The study was performed in two randomized phases separated by a washout period of 4 weeks. After an overnight fast, all subjects were separated into two groups randomly receiving a single oral dose of 40 mg of citalopram (H. Lundbeck A/S, Copenhagen, Denmark) on two occasions: 1) alone and 2) 2 h after an oral dose of 250 mg of troleandomycin (Pfizer Taiwan, Inc., Tamsui, Taiwan) once daily for 2 days. Food was served 2 h after drug administration. Venous blood samples (8 ml) were collected into heparinized tubes immediately (0 h) and at 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120, 144, and 168 h after drug administration. Plasma was separated after centrifugation and kept frozen at -20°C until analyzed.
Drug Assay. The concentrations of citalopram and desmethylcitalopram in plasma were measured by high-performance liquid chromatography with ultraviolet detector (wavelength, 234 nm) according to the method developed by Olesen and Linnet (1996) with moderate modification. A one-step extraction was used in the study. Phenacetin was used as the internal standard. In brief, 0.5 ml of 0.75 M sodium carbonate/bicarbonate buffer (pH = 10) and 200 μl of phenacetin solution were added to 1 ml of the plasma samples. The samples were then extracted with 4 ml of hexane. The organic phase was transferred to another glass tube and evaporated to dryness at 40°C. The residues were dissolved in 50 μl of mobile phase, 20 μl of which was injected into the high-performance liquid chromatography system equipped with an ultraviolet detector. Chromatography was performed on a reversed-phase Hypersil-ODS C18 (Thermo Hypersil, Keystone Scientific Operations, Bellefonte, PA) column (250 × 4 mm i.d., 5-μm particle size). The detection limits were 1 μg/l for citalopram and desmethylcitalopram. Calibration curves ranged from 2.5 to 500 μg/l for citalopram and desmethylcitalopram. The coefficients of variation for intraday and interday reproducibility ranged from 3.2 to 4.7% and 6.2 to 8.5% for citalopram and desmethylcitalopram, respectively.
Data Analysis. Pharmacokinetic analysis was performed by a noncompartmental approach. Area under the concentration versus time curve (AUC0→t) was calculated by the trapezoidal rule from zero to the last measured concentration point. The elimination rate constant (ke) of citalopram was estimated by least regression analysis. The AUC from zero hour to infinity (AUC0→∞) and the oral clearance of citalopram (CLoral) were calculated with the following equations: 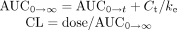 where C is the drug concentration in plasma and CL is the apparent oral clearance of the drug from plasma.
where C is the drug concentration in plasma and CL is the apparent oral clearance of the drug from plasma.
Data were analyzed using the Statistical Package for the Social Sciences (SPSS Inc., Chicago, IL). Variance analysis was used to investigate the effect of administration phase, troleandomycin, and genotype of CYP2C19 on the N-demethylation of citalopram. Then, Student's t test was used to compare the pharmacokinetic parameters of citalopram and desmethylcitalopram between the extensive metabolizers and poor metabolizers. The minimal level of significance accepted was P < 0.05.
Results
Genotype analysis of 77 healthy Chinese young volunteers indicated that the incidence of poor metabolizers and extensive metabolizers was 14.3 and 85.7% (data not shown), respectively, which is consistent with previous reports (Bertilsson et al., 1992; Xiao et al., 1997). Eight extensive metabolizers and five poor metabolizers chosen by the stratified random selection from 66 healthy extensive metabolizers and 11 poor metabolizers, respectively, were enrolled for the pharmacokinetic study of citalopram. Subsequently, CYP2C19 phenotypes of 13 subjects were determined by hydroxylation indices of omeprazole (log10 [omeprazole]/[5-hydroxyomeprazole]). The results were consistent with their previous genotypes.
As shown in Table 1, there was no statistically significant difference in citalopram pharmacokinetic data in EMs, whether they had TAO or not. However, TAO treatment did have a significant effect on citalopram and desmethylcitalopram AUC0→∞ in PMs.
Effects of troleandomycin on pharmacokinetic data (mean ± S.D.) of citalopram and desmethylcitalopram in 13 Chinese subjects
The genotype of CYP2C19 had a significant effect on the N- demethylation of citalopram. The extensive metabolizers and poor metabolizers had distinct plasma concentration-time profiles for citalopram and desmethylcitalopram (Fig. 1). Mean differences in the pharmacokinetic parameters of citalopram and desmethylcitalopram for the extensive metabolizers and poor metabolizers are summarized in Table 2. The poor metabolizers had a distinct increase in citalopram AUC0→∞ (2132.5 ± 242.3 μg · h/l versus 1738.0 ± 166.2 μg · h/l, P < 0.01), and in citalopram terminal elimination half-life (39.1 ± 5.5 h versus 35.6 ± 2.5 h, P < 0.01) compared with extensive metabolizers. The oral clearance (CLoral) of citalopram in poor metabolizers was significantly lower than that in extensive metabolizers whether TAO was used or not (Fig. 2). The AUC0→∞ and Cmax of desmethylcitalopram in poor metabolizers were significantly lower than those in extensive metabolizers (516.7 ± 58.8 μg · h/l versus 855.4 ± 59.9 μg/l, P < 0.01, and 6.1 ± 0.8 nM versus 9.9 ± 0.9 nM, P < 0.05, respectively). The ratio of AUCDCIT/AUCCIT of extensive metabolizers was almost three times higher than that of poor metabolizers (0.5 ± 0.1 versus 0.2 ± 0.1, P < 0.01).

Mean plasma concentrations of citalopram (A) and desmethylcitalopram (B) in Chinese extensive metabolizers (solid circles) and poor metabolizers (open circles) of CYP2C19 after oral administration of 40 mg of citalopram.
Pharmacokinetic data (mean ± S.D.) of citalopram and desmethylcitalopram in CYP2C19 genotyped subjects

The effect of TAO on the apparent oral clearance of citalopram in CYP2C19 extensive metabolizers (solid circles) and poor metabolizers (open circles).
In addition, the values of t1/2 and CLoral of citalopram, as well as AUC, Cmax, and tmax of desmethylcitalopram in poor metabolizers were significantly different from those in homozygous extensive metabolizers and those in heterozygous extensive metabolizers (P < 0.05). Pharmacokinetic parameters of citalopram in heterozygous extensive metabolizers were of intermediate value between poor metabolizers and homozygous extensive metabolizers (Table 2).
The correlation between the CYP2C19 phenotype, as measured by the omeprazole hydroxylation index, and the oral clearance of citalopram and desmethylcitalopram was also determined. The correlation between omeprazole phenotype and citalopram apparent oral clearance yielded a coefficient of r = 0.80, P = 0.000. The correlation between omeprazole phenotype and desmethylcitalopram formation (AUC) was r = 0.50, P = 0.004.
Discussion
The present study was conducted to examine the effect of CYP3A4 and the genetic polymorphism of CYP2C19 on the N-demethylation of citalopram. Previous studies have shown that troleandomycin inhibits the metabolism of a variety of drugs, including carbamazepine (Dravet et al., 1977). All these drugs are substrates of CYP3A. Such inhibitory effects are probably caused in part by the formation of an inactive cytochrome P450-troleandomycin metabolite complex (Pessayre et al., 1981). In the present study, we found that the genotype of CYP2C19 influenced the N-demethylation of citalopram and that TAO treatment had no effect among extensive metabolizers. However, there was a difference in citalopram and desmethylcitalopram AUC0→∞ in PMs whether they had TAO or not. This most likely reflects the effect of TAO on CYP3A when a CYP2C19 contribution is absent.
The 4′-hydroxylase activity of S-mephenytoin is genetically determined and mediated mainly by CYP2C19. The incidence of the phenotype of CYP2C19 for poor metabolizers is much higher in Asian populations (13–23%) than in white populations (2–5%) (Küpfer et al., 1984; Horai et al., 1989; Bertilsson et al., 1992; Xiao et al., 1997). In this study, we identified 66 (85.7%) extensive metabolizers and 11 (14.3%) poor metabolizers in 77 screened Chinese subjects; the results of genotypes analysis are consistent with the previous reports of Bertilsson et al. Our previous study also indicated that two defects in the CYP2C19 gene (CYP2C19*2 and CYP2C19*3) may account for 100% of the Chinese poor metabolizers. The subsequent phenotype analysis of 13 subjects in the study showed concordance with their genotypes.
Citalopram is mainly metabolized to N-desmethylcitalopram and further demethylated to didesmethylcitalopram (Milne and Goa, 1991). In vitro studies have shown that the N-demethylation of citalopram is catalyzed by multiple P450 isoforms in human liver microsomes, including CYP2C19, CYP2D6, and CYP3A4 (Kobayashi et al., 1997; Rochat et al., 1997; Olesen and Linnet, 1999). However, an in vitro study found that CYP2C19 and CYP3A4 might be the major enzymes contributing to the citalopram N-demethylation because of their high affinity for citalopram (Olesen and Linnet, 1999). In addition, Sindrup et al. (1993) reported that the in vivo disposition of citalopram is associated with the polymorphism of CYP2C19. Our results also demonstrate that the CYP2C19 genotype has a significant impact on the metabolism of citalopram. The extensive metabolizers and poor metabolizers exhibited significantly different pharmacokinetic parameters of citalopram and desmethylcitalopram. The poor metabolizers had moderately higher AUC0→∞ and t1/2 values of citalopram than the extensive metabolizers. The CLoral of citalopram in poor metabolizers (17.1 ± 2.0 l/h) was significantly lower than that of extensive metabolizers (24.1 ± 1.6 l/h). In addition, the AUC0→∞ and Cmax of desmethylcitalopram in poor metabolizers were significantly lower than those of extensive metabolizers. The tmax values of desmethylcitalopram were significantly longer in poor metabolizers than in extensive metabolizers.
The fact that gene dose has an effect on drug metabolism has been reported previously. Broly et al (1991) have shown that the metabolic ratio in heterozygous extensive metabolizers of CYP2D6 is significantly higher than that in homozygous extensive metabolizers. Our previous studies also find that gene dose affects the metabolism of S-mephenytoin, diazepam, and sertraline in vivo (Xiao et al., 1997; Feng et al., 1998; Qin et al., 1999; Wang et al., 2001). In this study, we observed that CYP2C19 has a trend of gene dose effect on citalopram N-demethylation. The values of t1/2 and CLoral of citalopram, as well as AUC, Cmax, and tmax, of desmethylcitalopram in poor metabolizers are significantly different than those in homozygous extensive metabolizers (P < 0.05) and heterozygous extensive metabolizers (P < 0.05). Pharmacokinetic parameters of citalopram in heterozygous extensive metabolizers were of medium value between poor metabolizers and homozygous extensive metabolizers, although these parameters have no significant statistical difference between heterozygous extensive metabolizers and homozygous extensive metabolizers (data not shown). These results could in part explain that the genotyped polymorphism of CYP2C19 is likely to be one of the major factors causing the interindividual difference of the steadystate plasma levels of citalopram and desmethylcitalopram. However, because the samples of homozygous extensive metabolizers (n = 4) and heterozygous extensive metabolizers (n = 4) were small, this finding should be further investigated.
In our study, we find that the correlation between omeprazole metabolic ratio and citalopram oral clearance was r = 0.80. This indicates that the CYP2C19 activity accounts for 64% of the observed variability in citalopram apparent oral clearance. The correlation between omeprazole metabolic ratio and desmethylcitalopram AUC was r = 0.505. This indicates that the CYP2C19 activity may only account for 28% of the observed variability in desmethylcitalopram formation. One possible reason for a low contribution is that desmethylcitalopram was further metabolized to its didesmethyl metabolite.
During the experiment, all subjects received a single oral dose of 40 mg of citalopram, according to previous reports by Milne and Goa (1991) and the clinically therapeutic dose used in Chinese patients with depression. We found that one poor metabolizer with a homozygous mutant of CYP2C19 genotype had some severe side effects of gastrointestinal disturbances including nausea, vomiting, and nervous symptoms such as drowsiness 1 h after administration of citalopram. One of the causes of those side effects is probably due to the fact that poor metabolizers of CYP2C19 cannot immediately metabolize citalopram to inactive desmethylcitalopram, thus causing an accumulation of citalopram. Another factor is that the poor metabolizer may have a weak tolerance for citalopram. Thus, poor metabolizers appear to be at increased risk for accumulation of citalopram and the possible development of citalopramassociated toxicity. Therefore, when citalopram is clinically used for the treatment of depression, it is necessary to properly decrease its clinical dose for patients who are poor metabolizers of CYP2C19.
In summary, we conclude that CYP3A is not the major enzyme in the N-demethylation of citalopram. The polymorphism of CYP2C19 plays an important role in the N-demethylation of citalopram in vivo. The extensive metabolizers and poor metabolizers of CYP2C19 had significant differences in disposition of citalopram in vivo.
Acknowledgments
We thank H. Lundbeck A/S, Copenhagen, Denmark for the kindly donating the standard agents of citalopram and desmethylcitalopram.
Footnotes
-
↵1 Abbreviations used are: CIT, citalopram; P450, cytochrome P450; EM, extensive metabolizer; PM, poor metabolizer; TAO, troleandomycin; AUC, area under the plasma concentration versus time curve; CLoral, oral clearance.
-
Project supported by China Medical Board Grants 99-697 and 01-755 and National Natural Science Foundation Grant F30130210.
- Received March 20, 2002.
- Accepted June 17, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}