


Abstract
Critical elements from studies that have led to our current understanding of the factors that cause the observed primary deuterium isotope effect, (kH/kD)obs, of most enzymatically mediated reactions to be much smaller than the “true” or intrinsic primary deuterium isotope effect, kH/kD, for the reaction are presented. This new understanding has provided a unique and powerful tool for probing the catalytic and active site properties of enzymes, particularly the cytochromes P450 (P450). Examples are presented that illustrate how the technique has been used to determine kH/kD, and properties such as the catalytic nature of the reactive oxenoid intermediate, prochiral selectivity, the chemical and enzymatic mechanisms of cytochrome P450-catalyzed reactions, and the relative active site size of different P450 isoforms. Examples are also presented of how deuterium isotope effects have been used to probe mechanisms of the formation of reactive metabolites that can cause toxic effects.
Historically, the determination of deuterium isotope effects has been a powerful tool to help unravel the intricacies of carbon-hydrogen bond cleavage and define the mechanism of specific chemical reactions. The intrinsic primary isotope effect, kH/kD, for a reaction is the magnitude of the isotope effect on the rate constant for the bond-breaking step (C–H versus C–D) of the reaction and is related to the symmetry of the transition state for that step. The larger the isotope effect, the more symmetrical the transition state, with the theoretical limit being 9 at 37°C in the absence of tunneling effects (Bell, 1974). The chemical events leading to the transition state and subsequent formation of products are the descriptors of a chemical mechanism. It is the relationship of kH/kD to transition state that provides mechanistic insight. Thus, kH/kD is the quantity that needs to be known, and for homogenous chemical reactions, the experimentally observed deuterium isotope effect, (kH/kD)obs, where (kH/kD)obs is defined as the ratio of a kinetic parameter such as Vmax or Vmax/Km obtained from a nondeuterated substrate to a deuterated substrate, is identical to kH/kD.
This is generally not true of enzymatically mediated reactions and is the reason for the much less successful application of deuterium isotope effects to such reactions until the system was better understood. The experimentally observed isotope effect, (kH/kD)obs, was invariably found to be much smaller than kH/kD, the intrinsic isotope effect for that reaction, thereby obscuring both meaning and mechanism. Even more perplexing was the observation that (kH/kD)obs for the same enzymatic reaction could vary with different experimental designs. For example, (kH/kD)obs determined at saturating substrate concentrations could differ from values determined at decreasing substrate concentrations. The disparity between kH/kD and (kH/kD)obs is a consequence of the multistep nature (substrate binding, debinding, product release, etc.) of the catalytic cycle of enzymatically mediated reactions. If deuterium isotope effects are to be successfully applied to such reactions, then the relationship between kH/kD and (kH/kD)obs must be known, since experiment can only yield (kH/kD)obs. Fortunately, the relationship between kH/kD and (kH/kD)obs has been largely clarified (Northrop, 1975, 1978, 1981a; Cleland, 1982); and how partially rate-limiting steps exclusive of the bond-breaking step can mask kH/kD by lowering the magnitude of (kH/kD)obs is now clearly understood.
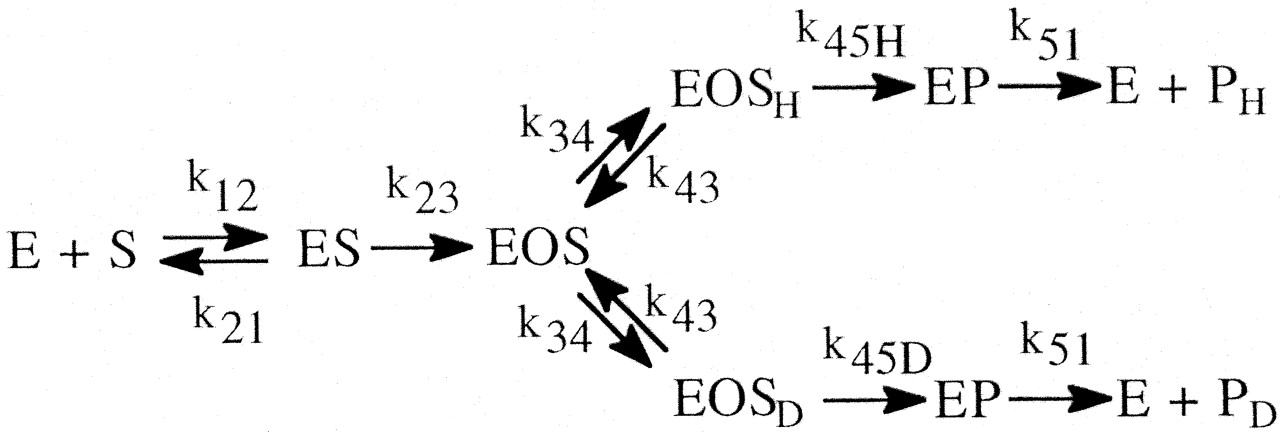
Even the simplest of enzymatic reactions is composed of three components (Northrop, 1981b). The first involves substrate combining with enzyme to form an enzyme-substrate complex. Rate constants k12 and k21 govern the process. The second component is the catalytic component that transforms the enzyme-substrate complex into an enzyme-product complex with rate constant k23 (Reaction 1). In this simplest of cases the catalytic step is considered to be irreversible. The third component is the product release step that entails dissociation of the complex to free enzyme and product with rate constant k31. Equations 1 to 3 define the kinetic expressions for the maximum velocity, V, the Michaelis constant, K, and the ratio of the two, V/K (Northrop, 1975, 1981b). In analyzing the kinetic 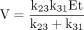
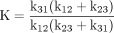
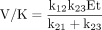 parameters, Northrop (1975) demonstrates that the expression of deuterium isotope effects on V and V/K should be the most revealing and important as they depend on the fewest variables and on rate constants for different parts of the kinetic scheme. Since V is determined at saturating concentrations, the binding component is eliminated and its value reflects the catalytic and product release components. V/K, which is reflective of the kinetics at low substrate concentration, is dependent on all rate constants up to and including the first irreversible step. In this case it is also the catalytic step. Isotope effects on V and V/K for this example are described by eqs. 4 and 5.
parameters, Northrop (1975) demonstrates that the expression of deuterium isotope effects on V and V/K should be the most revealing and important as they depend on the fewest variables and on rate constants for different parts of the kinetic scheme. Since V is determined at saturating concentrations, the binding component is eliminated and its value reflects the catalytic and product release components. V/K, which is reflective of the kinetics at low substrate concentration, is dependent on all rate constants up to and including the first irreversible step. In this case it is also the catalytic step. Isotope effects on V and V/K for this example are described by eqs. 4 and 5. 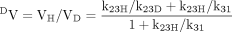
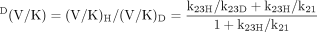 DV and D(V/K) are short-form terms introduced by Northrop (1975) for VH/VD and (V/K)H/(V/K)D, respectively. If eqs. 4 and 5 are rewritten in general form as eqs. 6 and 7, as proposed by Northrop (1975), the factors
DV and D(V/K) are short-form terms introduced by Northrop (1975) for VH/VD and (V/K)H/(V/K)D, respectively. If eqs. 4 and 5 are rewritten in general form as eqs. 6 and 7, as proposed by Northrop (1975), the factors 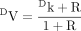
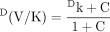 that can lead to the observed isotope effect, DV or D(V/K), having a much lower value than the intrinsic isotope effect, Dk (Northrop's nomenclature), become clear. In the equation for the isotope effect on V, R, termed the “ratio of catalysis,” is a measure of rate of the catalytic step relative to the rate of the other forward steps contributing to maximal velocity. In the present example, the other forward step would be the product release step. The value of DV [the value of (kH/kD)obs for Vmax conditions] will only be close to Dk (intrinsic isotope effect, kH/kD) when product release is fast relative to the catalytic step. In the equation for the isotope effect on V/K, C, termed “commitment to catalysis,” is a measure of the enzyme-substrate complex tendency to proceed through the first irreversible step versus reversing to free enzyme and substrate. The value of D(V/K) [the value of (kH/kD)obs for V/K conditions] will only be close to Dk if the first irreversible step is also the catalytic step and the reverse step to free enzyme and substrate is fast relative to the catalytic step. What makes Northrop's presentation of isotope effects in this way so powerful is that steady-state equations for much more complex enzyme mechanisms can all be reduced to this form. R and C simply become more complex expressions of a collection of rate constants. In more complicated schemes involving a greater number of reversible steps, an additional grouping of rate constants, Cr, termed the “commitment to reverse catalysis,” can be factored from both DV and D(V/K) experiments. Cr is a measure of the substrates tendency to return to E + S. In such schemes DV experiments now contain both R and Cr, while D(V/K) experiments will contain Cr and Cf, where Cf is a collection of rate constants termed the “commitment to forward catalysis” that measures the substrate's tendency to move forward to E + P. Despite the greater complexity, the general forms of the equations still indicate that modification of (kH/kD)obs relative to kH/kD can be readily understood in the interplay of a collection of specific rate constants.
that can lead to the observed isotope effect, DV or D(V/K), having a much lower value than the intrinsic isotope effect, Dk (Northrop's nomenclature), become clear. In the equation for the isotope effect on V, R, termed the “ratio of catalysis,” is a measure of rate of the catalytic step relative to the rate of the other forward steps contributing to maximal velocity. In the present example, the other forward step would be the product release step. The value of DV [the value of (kH/kD)obs for Vmax conditions] will only be close to Dk (intrinsic isotope effect, kH/kD) when product release is fast relative to the catalytic step. In the equation for the isotope effect on V/K, C, termed “commitment to catalysis,” is a measure of the enzyme-substrate complex tendency to proceed through the first irreversible step versus reversing to free enzyme and substrate. The value of D(V/K) [the value of (kH/kD)obs for V/K conditions] will only be close to Dk if the first irreversible step is also the catalytic step and the reverse step to free enzyme and substrate is fast relative to the catalytic step. What makes Northrop's presentation of isotope effects in this way so powerful is that steady-state equations for much more complex enzyme mechanisms can all be reduced to this form. R and C simply become more complex expressions of a collection of rate constants. In more complicated schemes involving a greater number of reversible steps, an additional grouping of rate constants, Cr, termed the “commitment to reverse catalysis,” can be factored from both DV and D(V/K) experiments. Cr is a measure of the substrates tendency to return to E + S. In such schemes DV experiments now contain both R and Cr, while D(V/K) experiments will contain Cr and Cf, where Cf is a collection of rate constants termed the “commitment to forward catalysis” that measures the substrate's tendency to move forward to E + P. Despite the greater complexity, the general forms of the equations still indicate that modification of (kH/kD)obs relative to kH/kD can be readily understood in the interplay of a collection of specific rate constants.

The simplest formulation for a cytochrome P450 (P4501)-catalyzed reaction involves one extra step over the general scheme presented above. This is the single irreversible step prior to substrate transformation in which the enzyme-substrate complex, ES, is irreversibly transformed into the substrate-bound active oxygenating perferryl oxene enzymatic species, EOS, which then proceeds to the enzymeproduct complex, EP, with the rate constant k34 (the isotopically sensitive step) (Korzekwa et al., 1989) (Reaction 2). The kinetics of the system have been described (Northrop, 1978, 1981; Cleland, 1982) and the following equations for the isotope effect derived. The term k34H(1/k23 + 1/k41) in 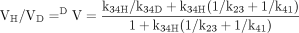
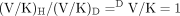 eq. 8 is termed the V ratio and is the factor that will increasingly mask the intrinsic isotope effect, k34H/k34D; the larger k34 is relative to k23 and k41. So if product formation is faster than product release, the formation of the perferryl oxene species, the magnitude of the intrinsic isotope effect, will be suppressed; i.e., (kH/kD)obs will be smaller than kH/kD. Thus, the faster product formation is, the greater the suppression of kH/kD. Equation 9 indicates that for V/K conditions [(DV/K)], no isotope effect should be observed, i.e., kH/kDobs must equal 1. This is because the first irreversible step in the scheme, conversion of ES to EOS, is not isotopically sensitive. Once EOS is formed, it is committed to continue on to product irrespective of whether or not substrate contains deuterium. Under V/K conditions, when an irreversible step precedes the isotopically sensitive step, the decreased rate constant, k34D, generated by deuterium substitution, will be compensated for by an equal and opposite increase in the concentration of EOSD, i.e., (EOSH)k34H = (EOSD)k34D. Thus, for cytochrome P450 reactions, deuterium isotope effects on V/K should in general never be observed (Korzekwa et al., 1989). Isotope effects can only be observed if some mechanism or some special experimental condition exists that diminishes the increase in EOSD that occurs in response to the decreased value of k34D relative to k34H. Fortunately, there are both mechanisms and experimental conditions that will tend to equalize (EOSH) and (EOSD) and thereby unmask the intrinsic isotope effect, i.e., cause (kH/kD)obs to approach kH/kD. One mechanism that can achieve this goal is the presence of a branched reaction pathway from the substrate-bound active oxygenating perferryl oxene species, EOSD, to a nonisotopically sensitive alternate product and free enzyme. A second and similar mechanism is reduction of the perferryl oxene EOSD to free substrate, free enzyme, and water (Atkins and Sligar, 1987, 1988; Korzekwa et al., 1989). A special experimental condition that can achieve the same end is an isotope effect experiment of symmetrical intramolecular design (Hjelmeland et al., 1977; Miwa et al., 1980; Gelb et al., 1982; Lindsay Smith et al., 1984).
eq. 8 is termed the V ratio and is the factor that will increasingly mask the intrinsic isotope effect, k34H/k34D; the larger k34 is relative to k23 and k41. So if product formation is faster than product release, the formation of the perferryl oxene species, the magnitude of the intrinsic isotope effect, will be suppressed; i.e., (kH/kD)obs will be smaller than kH/kD. Thus, the faster product formation is, the greater the suppression of kH/kD. Equation 9 indicates that for V/K conditions [(DV/K)], no isotope effect should be observed, i.e., kH/kDobs must equal 1. This is because the first irreversible step in the scheme, conversion of ES to EOS, is not isotopically sensitive. Once EOS is formed, it is committed to continue on to product irrespective of whether or not substrate contains deuterium. Under V/K conditions, when an irreversible step precedes the isotopically sensitive step, the decreased rate constant, k34D, generated by deuterium substitution, will be compensated for by an equal and opposite increase in the concentration of EOSD, i.e., (EOSH)k34H = (EOSD)k34D. Thus, for cytochrome P450 reactions, deuterium isotope effects on V/K should in general never be observed (Korzekwa et al., 1989). Isotope effects can only be observed if some mechanism or some special experimental condition exists that diminishes the increase in EOSD that occurs in response to the decreased value of k34D relative to k34H. Fortunately, there are both mechanisms and experimental conditions that will tend to equalize (EOSH) and (EOSD) and thereby unmask the intrinsic isotope effect, i.e., cause (kH/kD)obs to approach kH/kD. One mechanism that can achieve this goal is the presence of a branched reaction pathway from the substrate-bound active oxygenating perferryl oxene species, EOSD, to a nonisotopically sensitive alternate product and free enzyme. A second and similar mechanism is reduction of the perferryl oxene EOSD to free substrate, free enzyme, and water (Atkins and Sligar, 1987, 1988; Korzekwa et al., 1989). A special experimental condition that can achieve the same end is an isotope effect experiment of symmetrical intramolecular design (Hjelmeland et al., 1977; Miwa et al., 1980; Gelb et al., 1982; Lindsay Smith et al., 1984).

Symmetrical Intramolecular Design
In an isotope effect experiment of intramolecular design, a substrate is chosen that is susceptible to enzymatic attack at either of two symmetrically equivalent sites (Hjelmeland et al., 1977; Miwa et al., 1980; Gelb et al., 1982; Lindsay Smith et al., 1984). One site contains deuterium and the other retains its normal complement of hydrogen. The enzyme then has the choice of attacking a deuterated or an equivalent protio site within the same molecule. It can be viewed as a special case of branching and can be modeled as shown. The observed isotope effect, (kH/kD)obs, measured as the ratio of product resulting from attack at the protio site versus product resulting from attack at the deutero site, PH/PD, reflects the intramolecular competition between these two sites, i.e., PH/PD = kH[EOSH]/kD[EOSD] (Reaction 3). Kinetically, the experimental conditions correspond to a V/K isotope effect (Korzekwa et al., 1989). Thus, (kH/kD)obs is independent of all kinetic steps following branching of EOS and can be described by eq. 10. The equation reveals that the faster the rate of reorientation of the substrate is in the active site of the enzyme, k43, relative to the 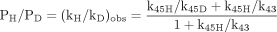 commitment to catalysis (catalytic step), k45H, i.e., k43 > k45H, the more the concentrations of EOSD and EOSH will equalize and the closer (kH/kD)obs will be to k45H/k45D. Conversely, the higher the commitment to catalysis relative to the rate of reorientation, the more the intrinsic isotope effect will be suppressed. An early, if the not first, example of the power of an intramolecular isotope effect experiment to unmask the intrinsic isotope effect is provided by the work of Foster et al. (1974). These investigators measured the deuterium isotope effect associated with the oxidative O-demethylation of p-methoxyanisole using two different experimental designs. The first was the traditional experiment of intermolecular design in which the rate of demethylation of the hydrogen-containing substrate was measured and compared with the rate of demethylation of the deuterium-containing substrate determined in a separate experiment (Fig. 1a). The observed isotope, (kH/kD)obs, was found to be 2. The second experiment was of intramolecular design in which the hydrogens of one of the O-methyl groups were replaced with deuterium. Incubation of the substrate with a microsomal preparation of cytochrome P450 followed by measurement of the p-methoxyphenol to p-trideuteromethoxyphenol product ratio gave a (kH/kD)obs of 10 (Fig. 1b). The observed products, p-methoxyphenol and formaldehyde, are consistent with at least two distinct mechanisms that might be operative. The first would be an insertion mechanism in which an oxygen atom inserts between a carbon-hydrogen bond of one of the methoxy groups to generate a hemiacetal intermediate. The hemiacetal is then hydrolyzed to generate the observed products. This mechanism should be accompanied by a primary deuterium isotope effect of low magnitude, approximately 2, since the transition state for oxygen insertion would necessarily be nonlinear (Shea et al., 1983). The second mechanism would involve initial hydrogen atom abstraction from the methyl group to form methylene and hydroxy radicals followed by recombination to generate the intermediate hemiacetal. This is the mechanism first postulated by Groves et al. (1978) to account for the cytochrome P450-catalyzed hydroxylation of norbornane, a mechanism that is now generally accepted as the mechanism for all cytochrome P450-catalyzed hydroxylations of aliphatic carbon-hydrogen bonds. Since direct hydrogen abstraction would, if possible, involve a linear transition state, the magnitude of the primary deuterium isotope effect could approach the theoretical value of approximately 9, depending upon the symmetry of the transition state. The (kH/kD)obs of 2 given by the intermolecular isotope effect experiment is supportive of a mechanism involving oxygen insertion. In contrast, the (kH/kD)obs of 10 from the intramolecular isotope effect experiment is only consistent with the abstraction recombination mechanism. Since (kH/kD)obs from the intramolecular isotope effect experiment is much closer to kH/kD than is (kH/kD)obs from the intermolecular experiment, the choice between possible mechanisms is clear. Indeed the (kH/kD)obs of 10, which is a composite value for one primary and two secondary isotope effects, i.e., PS2 (Hanzlik et al., 1985) is still large enough to suggest that the transition state is close to symmetrical and that the primary isotope effect is not too far from its maximum value.
commitment to catalysis (catalytic step), k45H, i.e., k43 > k45H, the more the concentrations of EOSD and EOSH will equalize and the closer (kH/kD)obs will be to k45H/k45D. Conversely, the higher the commitment to catalysis relative to the rate of reorientation, the more the intrinsic isotope effect will be suppressed. An early, if the not first, example of the power of an intramolecular isotope effect experiment to unmask the intrinsic isotope effect is provided by the work of Foster et al. (1974). These investigators measured the deuterium isotope effect associated with the oxidative O-demethylation of p-methoxyanisole using two different experimental designs. The first was the traditional experiment of intermolecular design in which the rate of demethylation of the hydrogen-containing substrate was measured and compared with the rate of demethylation of the deuterium-containing substrate determined in a separate experiment (Fig. 1a). The observed isotope, (kH/kD)obs, was found to be 2. The second experiment was of intramolecular design in which the hydrogens of one of the O-methyl groups were replaced with deuterium. Incubation of the substrate with a microsomal preparation of cytochrome P450 followed by measurement of the p-methoxyphenol to p-trideuteromethoxyphenol product ratio gave a (kH/kD)obs of 10 (Fig. 1b). The observed products, p-methoxyphenol and formaldehyde, are consistent with at least two distinct mechanisms that might be operative. The first would be an insertion mechanism in which an oxygen atom inserts between a carbon-hydrogen bond of one of the methoxy groups to generate a hemiacetal intermediate. The hemiacetal is then hydrolyzed to generate the observed products. This mechanism should be accompanied by a primary deuterium isotope effect of low magnitude, approximately 2, since the transition state for oxygen insertion would necessarily be nonlinear (Shea et al., 1983). The second mechanism would involve initial hydrogen atom abstraction from the methyl group to form methylene and hydroxy radicals followed by recombination to generate the intermediate hemiacetal. This is the mechanism first postulated by Groves et al. (1978) to account for the cytochrome P450-catalyzed hydroxylation of norbornane, a mechanism that is now generally accepted as the mechanism for all cytochrome P450-catalyzed hydroxylations of aliphatic carbon-hydrogen bonds. Since direct hydrogen abstraction would, if possible, involve a linear transition state, the magnitude of the primary deuterium isotope effect could approach the theoretical value of approximately 9, depending upon the symmetry of the transition state. The (kH/kD)obs of 2 given by the intermolecular isotope effect experiment is supportive of a mechanism involving oxygen insertion. In contrast, the (kH/kD)obs of 10 from the intramolecular isotope effect experiment is only consistent with the abstraction recombination mechanism. Since (kH/kD)obs from the intramolecular isotope effect experiment is much closer to kH/kD than is (kH/kD)obs from the intermolecular experiment, the choice between possible mechanisms is clear. Indeed the (kH/kD)obs of 10, which is a composite value for one primary and two secondary isotope effects, i.e., PS2 (Hanzlik et al., 1985) is still large enough to suggest that the transition state is close to symmetrical and that the primary isotope effect is not too far from its maximum value.
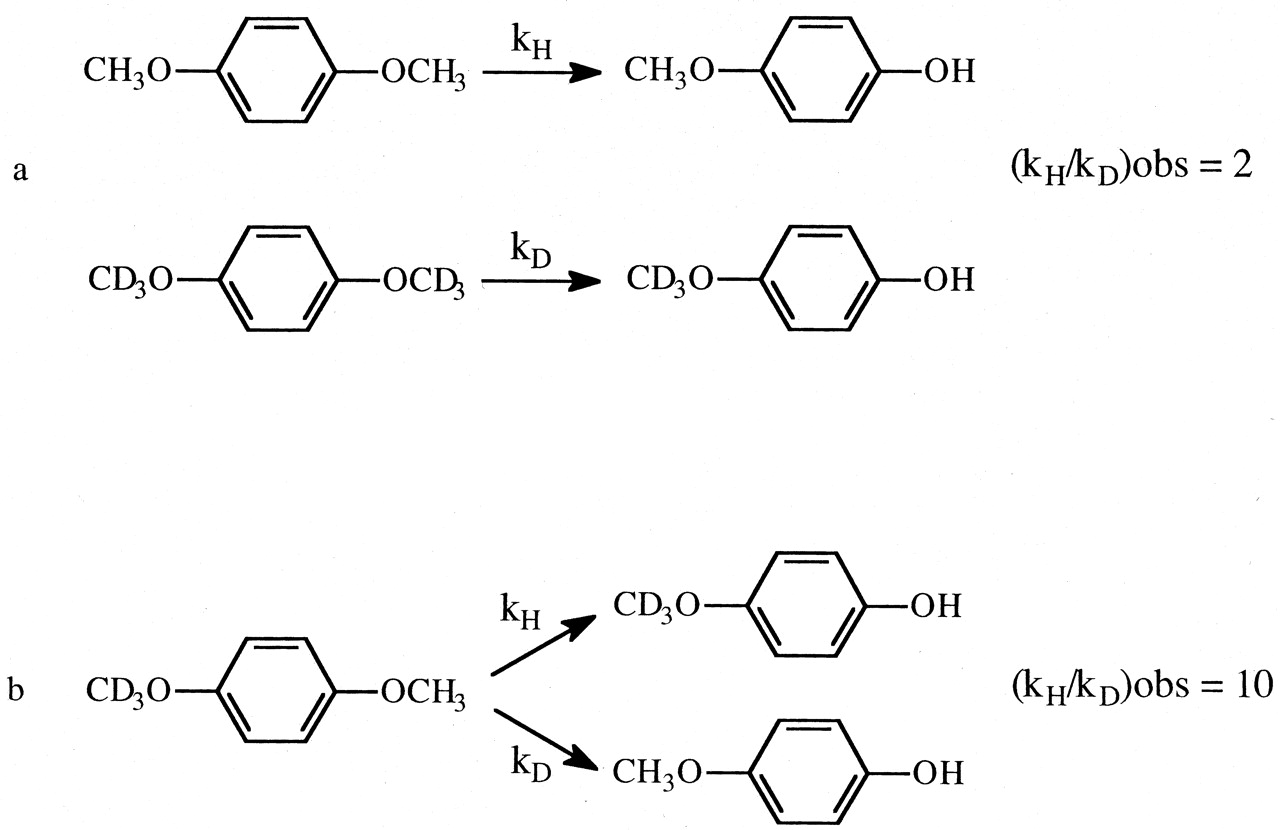
The (kH/kD)obs value for the O-demethylation of p-methoxyanisole determined from an experiment of intermolecular design (a) and intramolecular design (b).
Although the ratio of the concentration of [EOSH] to the concentration of [EOSD] must equal 1 for (kH/kD)obs to be equal to kH/kD, rapid equilibration of equivalent but isotopically distinct intramolecular oxidation sites is not the only means by which this condition can be met. As indicated above, the presence of a branched reaction pathway from the substrate-bound active oxygenating perferryl oxene species, EOSD, to a nonisotopically sensitive alternate product and free enzyme or reduction of the perferryl oxene EOSD to free substrate, free enzyme, and water can also equalize [EOSH] and [EOSD] and lead to (kH/kD)obs being equal to kH/kD. A model for a branched reaction pathway in competition with the isotopically sensitive step follows (Harada et al., 1984; Jones et al., 1986). In the model, substrate can reversibly reorient in the active site of the enzyme with rate constants k34 and k43 (Reaction 4). This allows it to present either the deutero site, EOSD, or the protio site, EOSH, for catalysis. EOSH and EOSD then go on to form products. P1 is formed from EOSH and EOSD with rate constants, k45H or k45D, respectively, whereas a single rate constant, k46, characterizes the formation of P2 from EOSH and EOSD. This is because P2 arises from oxidative attack at a molecular site remote from the site of deuterium substitution. The observed isotope effect for the model is given by eq. 11. Equation 11 reveals that the rates of substrate reorientation in the active site, k43, and formation of P2, k46, relative to the rate of formation of isotopically sensitive P1 are the factors that govern the magnitude of (kH/kD)obs and define the degree of masking of k45H/k45D. The larger k43 and/or k46 are relative to k45H, the closer (kH/kD)obs will be to k45H/k45D. 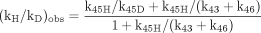 The values of (kH/kD)obs found for cytochrome CYP2B1-catalyzed ω-hydroxylation of the dideuterated analog of n-octane, n-octane-1,82H2, the heptadeuterated analog, n-octane-1,2,3-2H7, and the trideuterated analog, n-octane-1-2H3, of 16.11, 4.0, and 11.77, respectively, nicely illustrate the effects of k43 and k46 (Fig. 2). Before comparing values, it is first necessary to determine kH/kD for the reaction. Of the three substrates, the ω-hydroxylation of n-octane-1,8-2H2 can best be expected to provide kH/kD. Both terminal methyl groups are isotopically equivalent, and it can be assumed that the inherent rate of rotation of a methyl group is a much faster process than the rate of cleavage of a carbon-hydrogen bond via hydrogen atom abstraction. Thus, the enzyme always has the choice of oxidizing a carbon-hydrogen or carbon-deuterium bond irrespective of which methyl group is oriented for catalysis. The value of (kH/kD)obs = 16.11 for this substrate corresponds to an intrinsic primary isotope effect of 9.18 once it has been corrected for the contribution of secondary isotope effects and the fact that the methyl group contains two hydrogens but only one deuterium (Jones and Trager, 1987). As indicated above, an intrinsic primary isotope effect of 9 is the theoretical limit for cleavage of a carbon-hydrogen bond in the absence of tunneling effects (Bell, 1974). This provides compelling evidence that the cytochrome CYP2B1-catalyzed ω-hydroxylation of n-octane involves a highly symmetrical transition state and is consistent with the abstraction-recombination mechanism for aliphatic hydroxylation (Groves et al., 1978). In the case of n-octane-1,2,3-2H7, the enzyme has the choice of oxidizing either a carbon-hydrogen bond or a carbon-deuterium bond, depending on which terminal methyl group of the substrate is properly oriented for catalysis. According to eq. 11, if (kH/kD)obs is to be close to kH/kD, the rate of methyl group interchange, k43, must be much faster than the rate of bond breaking, k45H. The (kH/kD)obs of 4 indicates that this is clearly not true for this substrate and suggests that the distance between terminal methyl groups is large enough so that the rate of interchange is slow enough to prevent the concentrations of [EOSH] and [EOSD] from equalizing. For the case of n-octane-1-2H3, the enzyme also has the choice of oxidizing either a carbon-hydrogen bond or a carbon-deuterium bond, depending on which terminal methyl group of the substrate is properly oriented for catalysis. What is different about this substrate relative to n-octane-1,2,3-2H7 is that it also has the choice of a branched reaction pathway, (ω-1)-hydroxylation, that is blocked by deuteration in the case of n-octane-1,2,3-2H7. Although both substrates will form the (ω-1)-product, it will form at a much faster rate when n-octane-1-2H3 is the substrate. The value of 11.77 for (kH/kD)obs for n-octane-1-2H3 corresponds to a primary isotope effect of 9.14, when the secondary isotope effect contribution is removed, indicating that k46 is much larger than k45H, so that the sum (k43 + k46) is now big enough to overwhelm k45H and allow full expression of the intrinsic isotope effect. The dominance of k46 over k45H is substantiated by the product ratio. 2-Octanol is formed 23 times faster than 1-octanol.
The values of (kH/kD)obs found for cytochrome CYP2B1-catalyzed ω-hydroxylation of the dideuterated analog of n-octane, n-octane-1,82H2, the heptadeuterated analog, n-octane-1,2,3-2H7, and the trideuterated analog, n-octane-1-2H3, of 16.11, 4.0, and 11.77, respectively, nicely illustrate the effects of k43 and k46 (Fig. 2). Before comparing values, it is first necessary to determine kH/kD for the reaction. Of the three substrates, the ω-hydroxylation of n-octane-1,8-2H2 can best be expected to provide kH/kD. Both terminal methyl groups are isotopically equivalent, and it can be assumed that the inherent rate of rotation of a methyl group is a much faster process than the rate of cleavage of a carbon-hydrogen bond via hydrogen atom abstraction. Thus, the enzyme always has the choice of oxidizing a carbon-hydrogen or carbon-deuterium bond irrespective of which methyl group is oriented for catalysis. The value of (kH/kD)obs = 16.11 for this substrate corresponds to an intrinsic primary isotope effect of 9.18 once it has been corrected for the contribution of secondary isotope effects and the fact that the methyl group contains two hydrogens but only one deuterium (Jones and Trager, 1987). As indicated above, an intrinsic primary isotope effect of 9 is the theoretical limit for cleavage of a carbon-hydrogen bond in the absence of tunneling effects (Bell, 1974). This provides compelling evidence that the cytochrome CYP2B1-catalyzed ω-hydroxylation of n-octane involves a highly symmetrical transition state and is consistent with the abstraction-recombination mechanism for aliphatic hydroxylation (Groves et al., 1978). In the case of n-octane-1,2,3-2H7, the enzyme has the choice of oxidizing either a carbon-hydrogen bond or a carbon-deuterium bond, depending on which terminal methyl group of the substrate is properly oriented for catalysis. According to eq. 11, if (kH/kD)obs is to be close to kH/kD, the rate of methyl group interchange, k43, must be much faster than the rate of bond breaking, k45H. The (kH/kD)obs of 4 indicates that this is clearly not true for this substrate and suggests that the distance between terminal methyl groups is large enough so that the rate of interchange is slow enough to prevent the concentrations of [EOSH] and [EOSD] from equalizing. For the case of n-octane-1-2H3, the enzyme also has the choice of oxidizing either a carbon-hydrogen bond or a carbon-deuterium bond, depending on which terminal methyl group of the substrate is properly oriented for catalysis. What is different about this substrate relative to n-octane-1,2,3-2H7 is that it also has the choice of a branched reaction pathway, (ω-1)-hydroxylation, that is blocked by deuteration in the case of n-octane-1,2,3-2H7. Although both substrates will form the (ω-1)-product, it will form at a much faster rate when n-octane-1-2H3 is the substrate. The value of 11.77 for (kH/kD)obs for n-octane-1-2H3 corresponds to a primary isotope effect of 9.14, when the secondary isotope effect contribution is removed, indicating that k46 is much larger than k45H, so that the sum (k43 + k46) is now big enough to overwhelm k45H and allow full expression of the intrinsic isotope effect. The dominance of k46 over k45H is substantiated by the product ratio. 2-Octanol is formed 23 times faster than 1-octanol.


The (kH/kD)obs values for the formation of n-octanol from n-octane-1,8-2H2, n-octane-1-2H3, and n-octane-1,2,3-2H7.
Establishing that the ω-hydroxylation of n-octane proceeds with a maximum intrinsic isotope effect of 9, and therefore a highly symmetrical transition state, provides an important limit for the mechanistic interpretation of all cytochrome P450 reactions that involve cleavage of a carbon-hydrogen bond. It means that cleavage of a carbon-hydrogen bond of any saturated carbon atom having a different substitution pattern (secondary, tertiary, substituted with other atoms in addition to hydrogen) must proceed with a lower intrinsic isotope effect and a less symmetrical transition state. The degree to which the intrinsic isotope effect departs from maximum and the transition state from symmetrical depends on the difference that that specific substitution pattern engenders relative to a primary aliphatic carbon-hydrogen bond. This is because the magnitude of kH/kD varies as a bell-shaped curve and correlates with transition state symmetry. The magnitude of kH/kD is maximum when the transition state is most symmetrical and declines when the transition state is either more reactant-like or more product-like (Melander, 1960; Westheimer, 1961; Hammond, 1965). It is therefore not surprising that the intrinsic primary isotope effect associated with the benzylic hydroxylation of o- and p-xylene by various microsomal preparations and purified P450s is less than maximal, falling within the range of 5.2 to 7.4 (Fig. 3) (Hanzlik and Ling, 1993; Iyer et al., 1997). Because the benzylic radical can be resonance stabilized, the less than maximal value for the intrinsic isotope effect for benzylic hydroxylation of xylene indicates that the transition state for the reaction is more reactant-like, and the value of the isotope effect lies on the ascending portion of the bell-shaped curve.

The intrinsic isotope effect, kH/kD, for the cytochrome P450-catalyzed hydroxylation of (1,2)-dideuteromethyl-o-xylene (a) and (1,2*)-dideuteromethyl-p-xylene (b).
Another factor, besides substrate structure, that could affect the symmetry of the transition state for hydrogen atom abstraction is the active site structure of the enzyme itself. This raises the question of whether or not different P450s can catalyze the same oxidative reaction with different kH/kD values. If they can, it must mean that either different P450s can catalyze the same reaction with different mechanisms or the symmetry of the transition state can vary as a function of different active site topographies by modulating the stability of the activated oxygen-substrate complex. The activated oxygen atom of cytochromes P450 is an extremely reactive species that is capable of oxidizing virtually any carbon-hydrogen bond with which it comes in contact. These enzymes are different from most other enzymes in that the energy cost for catalysis is in generating the active oxygen species rather than in orienting the substrate toward a transition state-like structure by specific binding. This suggests that the mechanism of a given reaction catalyzed by different P450s is likely to be unchanged. In this regard, considerable evidence exists indicating that aliphatic hydroxylation reactions, irrespective of enzyme, proceed by a hydrogen atom abstraction-recombination mechanism. Thus, if differences are found, they are likely to be caused by differences in active site topographies. To probe this question, the isotope effects for the ω-hydroxylation of n-octane by three very different P450s, CYP1A1, CYP2B1, and CYP2B4, were determined (Jones et al., 1990). All three enzymes gave virtually identical intrinsic primary isotope effects of approximately 9, even though the ω- to (ω-1)-hydroxylation product ratio was very different for each isoform and the individual P450s were from different species (rat and rabbit) and different enzyme families. The N-demethylation of a series of para substituted (H, Cl, CN, NO2) dimethylanilines (Fig. 4) catalyzed by CYP1A2, CYP2B1, CYP4B1, and CYP101 gave similar findings (Karki et al., 1995). Whereas the magnitude of the intrinsic isotope effect varied from substrate to substrate, it was virtually identical across enzymes. The variation in the magnitude of the intrinsic isotope effect between substrates is expected because the transition state for the N-demethylation of this series of substrates would fall on the ascending slope of the bell-shaped curve that defines the relationship between transition state and the magnitude of the isotope effect. Different aromatic substituents would differ in their ability to stabilize the radical intermediate. The picture that emerges from these data is that any given cytochrome P450-catalyzed reaction is likely to proceed by the same mechanism with the same value for the intrinsic isotope effect, irrespective of the isoform catalyzing the reaction.

The cytochrome P450-catalyzed N-demethylation of various (X = H, Cl, CN, and NO2) p-substituted N,N-dimethylaniline analogs.
The lack of sensitivity to individual P450s and different active site architectures of the intrinsic primary isotope effect for aliphatic hydroxylation suggests that the masking effect might be used to explore effective active site dimensions. As has been indicated above, the concentrations of [EOSH] and [EOSD] for isotopically nonequivalent but otherwise identical catalytic sites within a substrate must be equal for (kH/kD)obs to be equal to kH/kD. The larger and less impeded an active site and/or the smaller the intramolecular distance between protium and deuterium sites, the less likely the catalytic event will occur before equilibration of [EOSH] and [EOSD]. A set of substrates for aliphatic hydroxylation with a range of known fixed distances between protium and deuterium sites might be expected to display a range of (kH/kD)obs values for different P450 isoforms reflecting different active site topographies. The concept was tested and verified with CYP101, a P450 with a known active site, using the benzylic hydroxylation of o- and p-xylene-α-2H3 and 4-2H3,4′-dimethylbiphenyl, in which the protio and deuterio catalytic sites vary by 2.5 Å, 6.6 Å, and 11 Å, respectively (Audergon et al., 1999). Results from CYP2B1, several microsomal preparations, and the same set of substrates indicated that an intramolecular distance of less than 7 Å between catalytic sites allowed [EOSH] and [EOSD] to equalize prior to catalysis, whereas a distance greater than 11 Å led to complete suppression of kH/kD (Iyer et al., 1997). In a subsequent study, 2-2H3, 6-dimethylnaphthalene (Fig. 5), was added to the set of additional fixed distance probes (methyl versus trideuteromethyl groups) to explore the active site topography of CYP4B1 relative to CYP2B1 and CYP102 (Henne et al., 2001). The results of the study led to the conclusion that the active site of CYP4B1 is considerably restricted relative to CYP2B1. In a similar study, p-xylene-α-2H3 and 4-2H3,4′-dimethylbiphenyl were used to demonstrate that if phenylalanine 87 of cytochrome P450BM-3 is replaced by alanine, a much more open active site results, as indicated by freer substrate motion. The results indicate that F87 in the wild-type enzyme occupies active site space that effectively leads to a smaller active site that is more restrictive toward larger substrates (Rock et al., 2002).

The fixed intramolecular distance between methyl and trideuteromethyl groups of o-xylene-α-2H3, p-xylene-α-2H3, 2-2H3-6-dimethylnaphthalene, and 4-2H3-4′-dimethylbiphenyl.
Nonsymmetrical Intramolecular Design
More than a single product can sometimes be formed by the action of a single P450 on a single substrate. If one of the intramolecular product-forming sites is deuterated, the possibility of formation of an alternate product from a different site could be in competition with the deuterated site and thereby modulate the value of (kH/kD)obs. A model describing this possibility is shown below (Korzekwa et al., 1989, 1995). The model assumes that the substrate is free to reorient within the enzyme-substrate complex, EOS, rapidly, relative to the rate of substrate oxidation, k34 and k35 (Reaction 5). When P1 is the isotopically sensitive step while P2 is in competition with P1 but is not itself isotopically sensitive, DVP1 and D(V/K)P1 are given by eqs. 12 and 13, respectively. It is apparent from the model that an isotope effect on k34, k34D, will alter the ratio of products, P1 and P2. Equation 12 also indicates that the larger k35 is relative to k34, the closer (kH/kD)obs will approach the intrinsic isotope effect, k34H/k34D. Furthermore, if product release from the isotopically sensitive pathway, k41, is slow, k34H/k34D will tend to be masked. In contrast, if product release from the alternate pathway, k51, is slow, k34H/k34D will tend to be unmasked. 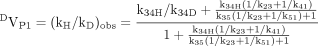
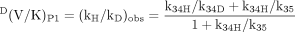 Finally, if formation of the active oxygenating species, k23, is slow and product release, k41, is fast, DVP1 will equal D(V/K)P1. For D(V/K)P1 the intrinsic isotope effect is masked solely by the branching ratio, k34H/k35. The presence of a second product pathway allows [EOSH](k34H + k35) to remain equal to [EOSD](k34D + k35) by increasing the flux through k35 as the flux through k34 decreases, because of the presence of deuterium.
Finally, if formation of the active oxygenating species, k23, is slow and product release, k41, is fast, DVP1 will equal D(V/K)P1. For D(V/K)P1 the intrinsic isotope effect is masked solely by the branching ratio, k34H/k35. The presence of a second product pathway allows [EOSH](k34H + k35) to remain equal to [EOSD](k34D + k35) by increasing the flux through k35 as the flux through k34 decreases, because of the presence of deuterium.

The increased flux through k35 that offsets the decreased flux through k34 because of deuteration results in an increase in the relative amount of P2 formed. The effect of deuteration at catalytic site 1, while resulting in a normal isotope effect on P1, also leads to an inverse isotope effect on P2 even though catalytic site 2 is remote from the deuterated site. Since the relative amounts of P1 and P2 formed depend entirely on the ratio of k34H/k35, the intrinsic isotope effect can be obtained directly from the product ratios of the protio and deuterio substrates according to eq. 14. Substrates that are metabolized to 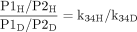 at least two products arising from oxidation of different parts of the molecule by the catalytic action of a single P450 conform to this model (Korzekwa et al., 1989, 1995).
at least two products arising from oxidation of different parts of the molecule by the catalytic action of a single P450 conform to this model (Korzekwa et al., 1989, 1995).
A subclass of substrates within this general class is the class of prochiral substrates. An example is cumene, a substrate that is prochiral by virtue of its two methyl groups (Sugiyama and Trager, 1986). The enzyme has the choice of oxidizing either of the chemically equivalent pro (R) or pro (S) methyl groups, Fig. 6. However, the chemical equivalence of the two methyl groups does not mean that the enzyme will oxidize each to equal extents. On the contrary, the selective oxidation of one methyl group over the other would be expected. Because the active site of any P450 is chiral, the relationship between the activated complexes for catalysis of each of the methyl groups, EOSproR and EOSproS, is diastereomeric, i.e., the energy contents of EOSproR and EOSproS are different. Therefore, the concentrations and the relative amounts of products formed by EOSproR and EOSproS will be different. In accord with expectations, three different cytochrome P450 rat liver microsomal preparations catalyzed selective methyl group hydroxylation of cumene. Normal and phenobarbital-induced preparations selectively hydroxylated the pro (S) methyl group by ratios of 2.2 and 1.65, respectively. In contrast, a β-naphthoflavone preparation favored pro (R) methyl group hydroxylation by a ratio of 1.12 (Sugiyama and Trager, 1986).

The cytochrome P450-catalyzed formation of (S)- and (R)-2-methyl-2-phenylethanol.
The discussion above establishes that the general effect of an alternate pathway to the isotopically sensitive step is to unmask the isotope effect. If additional pathways are added, they only add to the unmasking of the isotope effect (Korzekwa et al., 1989). Similarly, the effect of reduction of the perferryl oxene EOS to free substrate, free enzyme, and water rather than conversion to product is equivalent to the presence of an alternate pathway (Atkins and Sligar, 1987, 1988; Korzekwa et al., 1989). Although an alternate pathway will always tend to unmask the intrinsic isotope effect at the deuterated site, the assumption that the rate of substrate reorientation for catalysis at the deuterated versus the alternate pathway site is fast relative to either bond-breaking step must hold for eqs. 12 to 14 to apply.
Kinetic Mechanisms
One of at least three distinct kinetic mechanisms could be operative when multiple products are formed from a single substrate by a single cytochrome P450. Gillette has defined the three as 1) the parallel pathway mechanism, 2) the dissociative mechanism, and 3) the nondissociative mechanism, and has developed methodology to unambiguously determine which mechanism might be operative (Gillette and Korzekwa, 1990; Gillette, 1991; Gillette et al., 1994). The parallel pathway mechanism is defined as one in which the EOS complexes for the formation of the different products are stable and do not dissociate or interconvert. In the dissociative mechanism the EOS complexes cannot interconvert while the substrate remains within an EOS complex, but substrate can dissociate from EOS and then recombine in a different or the same orientation. The nondissociative mechanism is defined by the ability of substrate to interconvert within the EOS complex. The mechanisms can be distinguished by monitoring the isotope effect on the alternate pathway (nondeuterated site) when two isotope effect experiments are conducted (Gillette et al., 1994). One experiment is of noncompetitive design. The product ratio values given by the hydrogen-containing substrate are first determined and then compared with the product ratio values obtained from the deuterated substrate. In the second experiment, the isotope effect value at the alternate pathway site is determined from a 50:50 mixture of the hydrogen- and deuterium-containing substrates. The unique solutions that result for each of the three mechanisms from this experimental design are presented in Table 1.
Value of the isotope effect expected for the cytochrome P450 hydroxylation of a substrate at a nondeuterated site by parallel pathway, nondissociative, and dissociative mechanisms determined from competitive and noncompetitive experiments
The method was used to determine the kinetic method by which CYP2C11 is able to hydroxylate both the A-ring and D-ring of testosterone to convert the steroid to either 2α-hydroxytestosterone or 16α-hydroxytestosterone, respectively (Fig. 7) (Darbyshire et al., 1994). Given the intramolecular distance between the A- and D-rings, one might expect the kinetic mechanism to be either a parallel pathway or a dissociative mechanism. As it turns out, the results clearly establish the dissociative mechanism, indicating that the perferryloxoenzyme species is stable enough to allow the association-disassociation-reassociation of substrate. In contrast, when the intramolecular distance between oxidative sites is shorter, as found in the CYP2D6-catalyzed formation of 2,3- and 5,6-didehydrosparteine from sparteine (Fig. 8), the nondissociative mechanism is found to be operative (Ebner et al., 1995).
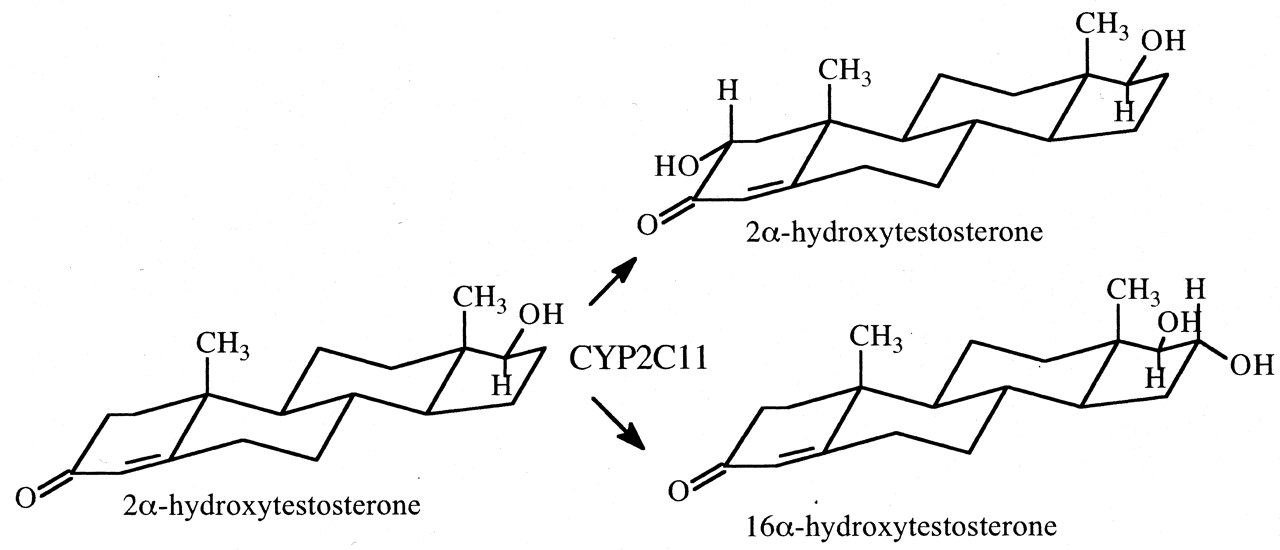
The CYP2C11-catalyzed hydroxylation of testosterone to 2α-hydroxytestosterone and 16α-hydroxytestosterone.

The CYP2D6-catalyzed formation of 2,3-didehydrosparteine and 2S-[2H]-5,6-didehydrosparteine from 2S-[2H]-sparteine.
Use of Deuterium Isotope Effects to Probe Mechanisms of Metabolically Dependent Toxicity
An extension of the use of deuterium isotope effects to probe mechanisms of enzyme-catalyzed reactions is their use to probe mechanisms of the formation of reactive metabolites that can cause toxicity. Drugs and other chemicals are usually eliminated from the body by multiple pathways, and deuterium substitution for hydrogen at sites of metabolism may decrease, increase, or have no effect on the toxicity of a compound.
As a simple example, assume that compound A is metabolized by pathways k1 and k2 and excreted unchanged by pathway k3, all by first order processes. As described by Pohl and Gillette (1985), the fraction of a dose of compound A eliminated through each pathway is expressed as a ratio of the rate constants for each pathway, divided by the sum of the rate constants for all pathways of elimination (eq. 15): 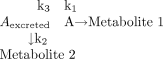
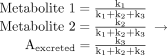 If Metabolite 1 is formed by cleavage of a C–H bond and is the toxic species, substitution of deuterium for hydrogen at that carbon can decrease the toxicity of compound A, the degree of which will be dependent both on the magnitude of the expression of the intrinsic primary deuterium isotope effect in the enzymatic reaction, as described previously in this review, and on the relative contributions of k1, k2, and k3 to the elimination of compound A. Large deuterium isotope effects would be observed when the expression of the intrinsic isotope effect was high and the fraction of the dose of compound A converted to Metabolite 1 was low, i.e., the ratio of k1/(k1 + k2 + k3) approaches zero.
If Metabolite 1 is formed by cleavage of a C–H bond and is the toxic species, substitution of deuterium for hydrogen at that carbon can decrease the toxicity of compound A, the degree of which will be dependent both on the magnitude of the expression of the intrinsic primary deuterium isotope effect in the enzymatic reaction, as described previously in this review, and on the relative contributions of k1, k2, and k3 to the elimination of compound A. Large deuterium isotope effects would be observed when the expression of the intrinsic isotope effect was high and the fraction of the dose of compound A converted to Metabolite 1 was low, i.e., the ratio of k1/(k1 + k2 + k3) approaches zero.
If Metabolite 2 is the toxic species, deuterium substitution for hydrogen in compound A that would be expected to decrease the formation rate of Metabolite 1 will either increase toxicity or have little effect on the toxicity associated with compound A. In this case, large effects on toxicity would be observed when the expression of the intrinsic primary deuterium isotope effect in the formation of Metabolite 1 was high, and the fraction of the dose of compound A converted to Metabolite 1 was high, i.e., the ratio of k2/(k1 + k2 + k3) will increase more because the denominator will decrease as a result of a greater decrease in k1 upon deuterium substitution.
It should be emphasized that the magnitude of a deuterium isotope effect on toxicity is dependent on several other variables that can affect either the enzyme and transporters involved in metabolism and disposition of the compound and its metabolites, or the response of the cell, tissue, or animal to the tissue damage. Furthermore, dose-response studies are important, since there often are dose-thresholds for toxicity so that above a certain dose, enough reactive metabolite may be formed, even with the deuterated compound, to cause toxicity equivalent to that of the protiated compound.
What follows are examples of several compounds that have elicited deuterium isotope effects on reactive metabolite formation and toxicity. In no case has there been a thorough examination of the kinetics of the formation and disposition of metabolites of a drug or other chemical in vivo as it relates to the isotope effects on toxicity because of the reactive nature of some of these metabolites. However, in a few cases, studies in vivo, coupled with studies in vitro comparing unlabeled drug and its deuterium-labeled analogs, have provided significant mechanistic insights into initiating events in toxic reactions. Note that due to space limitations, this section will only describe effects of deuterium substitution for hydrogen in reactions that lead to an overt toxicity. For other examples, see Baillie (1981).
Nitrosamines and Related Compounds.N-Nitrosodimethylamine (NDMA) is a hepatocarcinogen in rats, and Keefer et al. (1973) found that overall tumor incidence in NDMA-treated rats was approximately 3-fold greater than in NDMA-d6-treated rats at low doses, with only one hepatoma found in the NDMA-d6-treated group (29 rats) versus eight hepatomas in the NDMA-treated group (30 rats). At 5-fold higher doses, 18 hepatomas were detected in the NDMA-d6-treated group and 24 hepatomas in the NDMA-treated group. Similarly, at low doses, NDMA-d6 forms significantly fewer 7-methylguanine DNA adducts in rat liver than does NDMA, whereas there is no significant difference at high doses (Swann et al., 1983).
These results are consistent with results of other studies both in vivo (Mico et al., 1985) and in vitro (Wade et al., 1987) that showed larger isotope effects on metabolism when low doses or concentrations of NDMA and NDMA-d6 were used (Vmax/Km conditions), but small isotope effects when large doses or concentrations were used (Vmax conditions). There was a negligible deuterium isotope effect on Vmax, but an approximately 5-fold effect on Km (Wade et al., 1987; Yang and Ishizaki, 1992). Since CYP2E1, the P450 isoform mostly responsible for oxidation of NDMA, is known to be a “leaky” enzyme, the results have been interpreted as a low commitment to catalysis wherein C–H bond-breaking and product release are slow relative to formation of EO2S and EOS complexes and their reduction back to the ES complex with the formation of peroxide and water, respectively (Yang and Ishizaki, 1992).
An unanswered question is how the isotope effect expresses itself on carcinogenicity, since over 90% of doses of either NDMA or NDMA-d6 are cleared by oxidative metabolism in the liver. The two major pathways of oxidation of NDMA are described in Fig. 9. Pathway a leads to the generation of the DNA-methylating agent, whereas pathway b generates methylamine, formaldehyde. and nitrite. Streeter et al. (1990) have observed an approximately 3-fold increase in the denitrosation pathway after NDMA-d6 administration to rats compared with NDMA. For this to occur, either enzymes other than CYP2E1 can cause denitrosation of NDMA, or a switch in mechanism in the denitrosation of NDMA takes place upon deuteration.

Major pathways of metabolism of N-nitrosodimethylamine via oxidative demethylation (a) and oxidative denitrosation (b).
Nu represents an anionic nucleophile.
represents an anionic nucleophile.
Deuterium isotope effects on the toxicities of other nitrosamines have been reported, but the reasons for their expression have not been as thoroughly investigated as they have with NDMA. Deuteration of the 3- and 5-positions (α-) on 4-nitrosomorpholine reduced both the liver tumor incidence in rats and mutagenicity in Salmonella typhimurium TA 1535 by 5-fold (Lijinsky et al., 1976; Charnley and Archer, 1977). α-Deuteration of 4-nitroso-2,6-dimethylmorpholine also decreased esophageal tumorigenesis in rats, whereas β-deuteration enhanced it (Lijinsky et al., 1980). This same nitrosamine primarily causes pancreatic tumors in hamsters and α-deuterationenhanced pancreatic tumorigenesis in hamsters, whereas β-deuteration decreased it (Rao et al., 1981). In both species, deuterium substitution apparently leads to switching of the site of metabolism away from the deuterated site, but a major β-hydroxylated pancreatic procarcinogenic metabolite of the cis-isomer of 4-nitroso-2,6-dimethylmorpholine is formed much faster in hamsters than in rats (Kokkinakis et al., 1984). Interestingly, β-deuteration of the ethyl group of N-nitrosoethylmethylamine shifted both carcinogenesis and DNA methylation in rats from the liver to the esophagus (Lijinsky et al., 1982; von Hofe et al., 1991).
In contrast to the results with nitrosamines, substitution of deuterium for hydrogen on carbons α to nitrosoureas, nitrosourethanes, and nitrosoguanidines has little or no effect on mutagenicity (Elespuru, 1978; Thurst et al., 1985) or carcinogenicity (Lijinsky, 1982). However, deuterium substitution in some of these structures, such as the antitumor agent 1,3-bis(2-chloroethyl)-1-nitrosourea, has revealed multiple mechanisms of elimination and hydrolysis in generating reactive metabolites (Brundrett et al., 1976; Weinkam and Lin, 1979).
A combination of N- and C-oxidations converts 1,2-disubstituted hydrazines to the same reactive alkylating species as that formed from N-nitroso compounds (Fig. 10) (Fiala, 1975; Dipple et al., 1985). The relative risk of colon tumor incidence in male CBA mice over their lifetime after treatment for 8, 16, or 32 weeks with 1,2-dimethylhydrazine was nearly 6 times greater than that in cohorts treated with equimolar doses of 1,2-dimethylhydrazine-d6 (Turusov et al., 1988). These results are consistent with results of studies with deuterated azoxymethane procarcinogenic metabolites, which were found to be significantly less active as colon carcinogens compared with azoxymethane itself (Lijinsky et al., 1984).

Major pathway of oxidation of 1,2-disubstituted hydrazines to reactive alkylating metabolites.
The initial azo metabolites can apparently be formed by monoamine oxidases, whereas the azoxy and metabolites of α-carbon oxidation arise from cytochrome P450 oxidations.
The antitumor drug, procarbazine (Table 2), is a 1,2-disubstituted hydrazine that is metabolized to reactive metabolites like other such hydrazines (Fig. 10) (Moloney and Prough, 1983; Prough et al., 1984). In humans it causes testicular damage leading to azoospermia (Chapman, 1984). It caused a similar sterility in mice that was reversed by deuterium substitution for hydrogen at the benzylic carbon atom, but not on the N-methyl group (Table 2) (Yost et al., 1985). Consistent with these results, a deuterium isotope effect (∼4) was observed on genotoxicity in rat liver and testes after administration of either procarbazine or its N-CD3 analog versus its CD2-benzylic analog (Holme et al., 1989). The results suggest that oxidative debenzylation occurs with concomitant formation of either methyldiazene or N1-hydroxy-N2-methyldiazene as the toxic or proximate toxic species (Fig. 10).
Effect of procarbazine and its deuterated analogs on sperm count 18 days after administration of 200 mg/kg procarbazine HCl and equimolar amounts of the deuterated analogs to male BDF2mice ( Yost et al., 1985 )
Allylic Compounds. Deuterium substitution for hydrogen in the methylene carbon of allyl alcohol decreases by 1.5- to 2-fold periportal liver necrosis in rats and, to the same extent, the rate of oxidation of allyl alcohol by alcohol dehydrogenases to its major reactive metabolite, acrolein (Patel et al., 1983).
Genotoxicity of the widely used antitumor drug, tamoxifen, was decreased 2- to 3-fold in vivo in rats and in vitro in a MCL-5 human cell line that retains cytochrome P450 activity by deuterium substitution for hydrogen in the allylic ethyl group (Table 3) (Phillips et al., 1994). These and other results suggest that liver carcinogenicity in rats caused by tamoxifen involves allylic α-carbon oxidation that may generate a reactive quinone methide.
Effect of deuterium substitution on rat hepatic DNA adduct formation and micronucleus formation in MCL-5 human cells
Acute hepatocellular injury is caused by an allylic monoterpene, pulegone, in rodents (Gordon et al., 1982) and humans (Anderson et al., 1996). A dose-response study showed that deuterium substitution for hydrogen in the allylic methyl groups decreased the extent of hepatocellular injury in mice by 2- to 3-fold (Gordon et al., 1987). An intrinsic deuterium isotope effect was observed in vitro for oxidation (kH/kD ∼7–8) of the allylic methyl groups of pulegone to an E-allylic alcohol and menthofuran, which arises from intramolecular cyclization of a Z-allylic alcohol and subsequent dehydration (Table 4 from Nelson et al., 1992). Isotopically sensitive branching of the metabolism of pulegone to other oxidation products also occurs (Nelson et al., 1992). The relatively low isotope effect observed for pulegone-d3 appears to reflect the ability of the isopropylidene group to topomerize within the cytochrome P450 active site after initial hydrogen atom abstraction (McClanahan et al., 1988).
Kinetic deuterium isotope effects on the cytochrome P450-mediated oxidation of deuterated pulegones to an E-allylic alcohol and menthofuran
a

Halogenated Alkyl Compounds. Oxidative dehalogenation is a major reaction catalyzed by cytochromes P450 and can lead to different kinds of reactive metabolites, depending on the alkyl halide structure. Di- and trihalomethanes can yield carbon monoxide as one potentially toxic product, and deuterium substitution for hydrogen in these structures has been shown to decrease CO production in vivo in rats and in vitro by 2- to 3-fold (Anders et al., 1978; Kubic and Anders, 1978). A detailed kinetic study of the conversion of dichloromethane and its mono- and dideuterated analogs to carbon monoxide and carbon dioxide in mice revealed a (kH/kD)obs of ∼7 for disproportionation to CO with large effects on the Km for the dideuterated substrate (Anderson et al., 1994). Thus, the kinetics were complex. The authors suggested either rate-limiting product release after the isotopically sensitive step, or a limiting oxygen activation step followed by a second-order reaction between the EO complex and substrate as possible reasons for their results.
The major toxicities caused by chloroform are hepatotoxicity and nephrotoxicity, which result from its oxidation to phosgene formed by oxidative dehalogenation. Deuterium substitution for the hydrogen of chloroform decreases phosgene formation (Fig. 11) in mice by approximately 2- to 3-fold, and decreases hepatotoxicity and nephrotoxicity to a similar extent (Pohl and Krishna, 1978; Ahmadizadeh et al., 1981; Branchflower and Pohl, 1981).

Oxidation of chloroform to phosgene and its further acylation of nucleophiles (Nu ).
).
Halogenated anesthetics are metabolized by cytochromes P450 to yield two different toxic species, depending on the substrate structures (McCarty et al., 1979). Methoxyflurane is oxidatively demethylated, and the resulting difluorocarbinol rapidly decomposes via dehydrodefluorination and hydrolysis to generate two fluoride ions that can cause renal toxicity (Fig. 12a). Perdeuteration of methoxyflurane resulted in a 33% decrease in the amount of fluoride ion produced (McCarty et al., 1979). Burke et al. (1980) found that deuterium substitution for hydrogen in the chlorofluoromethyl carbon of enflurane (Fig. 12b) produced approximately 5-fold less fluoride ion than either unlabeled enflurane or enflurane labeled with deuterium at the difluoromethyl carbon. Finally, studies with deuterated halothane (Fig. 12c) have suggested that under conditions of low oxygen tension in rats, a mild liver toxicity may be initiated by reductive metabolism of halothane to a radical since no deuterium isotope effect was observed (Holaday, 1977). In contrast, the idiosyncratic severe hepatotoxicity observed in humans after re-exposure to halothane is most likely mediated by oxidative debromination to form trifluoracetyl chloride as a hapten that reacts with lysine groups on proteins, some of which become antigenic (Fig. 12c). Deuterated halothane substantially decreases formation of these antigens (Satoh et al., 1985).

Formation of reactive metabolites of halogenated anesthetics.
Methoxyflurane primarily causes renal toxicity by oxidative demethylation and subsequent elimination of fluoride ion from the difluorocarbinol (a). Enflurane also yields the fluoride ion, primarily from oxidation of the chlorofluoromethyl group (b). Reductive metabolism of halothane forms a reactive radical that may cause mild liver injury, whereas evidence suggests that severe idiosyncratic drug sensitization and liver injury is caused by oxidative debromination of halothane to trifluoroacetyl chloride, which reacts with nucleophilic groups (Nu ) on proteins to form antigens (c).
) on proteins to form antigens (c).
Ethylene dibromide (EDB) is a 1,2-dihaloalkyl carcinogenic compound that can undergo either oxidative debromination to form bromoacetaldehyde (Hill et al., 1978) or conjugation with glutathione, which then reacts intramolecularly to form a reactive episulfonium ion (Fig. 13) (Rannug and Beije, 1979). The tetradeutero analog of EDB was shown to be more genotoxic (approximately twice as many DNA strand breaks in rats) than EDB (White et al., 1983). An inverse isotope effect was observed because of a decreased rate of oxidation of EDB-d4 compared with EDB, which resulted in the metabolism of a greater fraction of the dose via glutathione conjugation. Consistent with these results is that DNA adducts of EDB in rats are formed by reaction of an EDB-GSH episulfonium ion with the N7-position of guanine (Peterson et al., 1988).

Formation of reactive metabolites of ethylene dibromide via cytochrome P450 oxidation and glutathione S-transferase-catalyzed GSH conjugation.
The latter pathway appears to be responsible for DNA damage.
Both oxidative and glutathione-mediated bioactivation pathways of 1,2- and 1,2,3-halopropyl compounds may be involved in various toxicities associated with these structures (Anders and Pohl, 1985; Dybing et al., 1989). Deuterium substitution for hydrogen at the terminal bromomethyl carbon atom of tris(2,3-dibromopropyl)phosphate (Tris-BP) and 1,2-dibromo-3-chloropropane (DBCP) resulted in 5- to 6-fold decreased rates of mutagenicity and formation of the highly mutagenic metabolite, 2-bromoacrolein (Nelson et al., 1984; Omichinski et al., 1988). However, evidence from investigations with deuterated and methylated analogs suggested that tissue organ damage in rats caused by Tris-BP and DBCP resulted from GSH conjugation as the major rate-limiting reaction (Dybing et al., 1989; Pearson et al., 1990). Both pathways may be involved in organ toxicity since sequential metabolism of Tris-BP and DBCP by oxidation and glutathione conjugation forms reactive metabolites of these compounds as well (Fig. 14). Evidence for the importance of this sequential pathway comes from studies that showed a substantial deuterium isotope effect (∼8) for perdeuterated Tris-BP on the clastogenicity of this compound in rat liver (Van Beerendonk et al., 1994), and a similar isotope effect was observed on the formation of GSH conjugates of oxidized Tris-BP metabolites (Van Beerendonk et al., 1995). Studies with DBCP showed the formation of several mercapturic acid conjugates of oxidative metabolites of DBCP that formed alternate products with selectively deuterated analogs of DBCP, most likely as a result of isotopically sensitive branching in the oxidation step (Weber et al., 1995).

Pathways of vicinal 1,2-dibromopropanes to reactive, toxic metabolites.
X represents a leaving group such as the phosphate ester group in Tris-BP or chlorine in DBCP. Initial cytochrome P450 oxidation yields an α-bromoaldehyde that undergoes β-elimination to the mutagen and carcinogen 2-bromoacrolein, known to react with DNA. This acrolein can also undergo conjugation with GSH and formation of an episulfonium ion that reacts with nucleophiles (Nu ). Alternatively, the 1,2-dibromopropanes can react with GSH directly in a glutathione S-transferase-catalyzed reaction to form episulfonium ions.
). Alternatively, the 1,2-dibromopropanes can react with GSH directly in a glutathione S-transferase-catalyzed reaction to form episulfonium ions.
Other Compounds.Cyclophosphamide. Deuterium isotope effects on both metabolism and antitumor activity of the widely used antitumor drug, cyclophosphamide, helped establish its mechanism of activation (Cox et al., 1976). Although a deuterium isotope effect (∼2) was observed for the formation of 4-ketocyclophosphamide and carboxyphosphamide (Fig. 15), there was no effect of deuterium substitution at C-4 on antitumor activity. However, deuterium substitution at C-5 led to a significant isotope effect (∼5) on formation of acrolein and phosphoramide mustard, presumably via β-elimination of 4-aldocyclophosphamide (Fig. 15), and this was paralleled by a marked decrease (∼10-fold) in antitumor potency.

Pathways of oxidation of cyclophosphamide via 4-hydroxylation to its 4-keto and carboxy metabolites, and β-elimination to acrolein and the major antitumor metabolite, phosphoramide mustard.
N-Methylformamide (NMF). NMF was regarded as a potential antitumor agent but caused hepatoxicity in humans (Eisenhauer et al., 1986). NMF is also hepatotoxic in mice, and deuterium substitution of the formyl hydrogen increases the threshold for hepatotoxicity by ∼3-fold (Threadgill et al., 1987). Since the formation of S-(N-methylcarbamoyl)glutathione is decreased by ∼7-fold after comparable doses of deuterated versus undeuterated NMF in mice (Threadgill et al., 1987), there is apparently a relatively large primary deuterium isotope effect on the cytochrome P450 oxidation of NMF to a reactive, toxic isocyanate (Fig. 16).
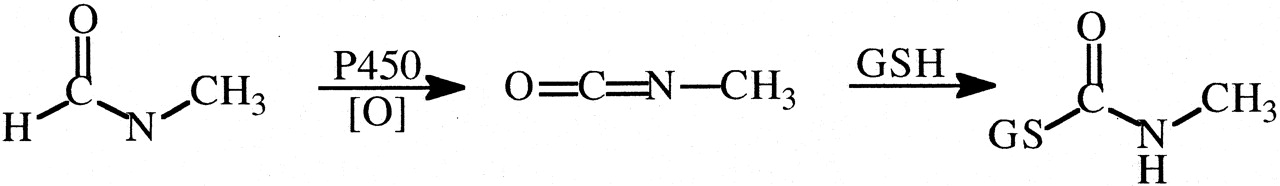
Oxidation of N-methylformamide to a reactive isocyanate and its conjugation with glutathione.
3,3′-Iminodipropionitrile. 3,3′-Iminodipropionitrile (Fig. 17) is a neurotoxicant in rats, and deuterium substitution for hydrogen at sites α to the nitrile groups decreases the incidence of axonal lesions, whereas substitution at carbon atoms β to the nitrile (α to the amine nitrogen) increases the incidence of lesions (Denlinger et al., 1992). This suggests that a product or products of oxidation at C-2 initiate damage.

Structures of 3,3′-iminodipropropionitrile and its deuterated analogs.
N-(3,5-Dichlorophenyl)succinimide (NDPS). The fungicide NDPS causes nephrotoxicity that is markedly reduced by deuterium substitution for hydrogen in the succinimide ring methylene bridge carbon atoms (Rankin et al., 1986). At 1.0 mmol/kg doses of NDPS and NDPS-d4 in rats, NDPS caused significant proteinuria, glucosuria, hematuria, elevated blood urea nitrogen and kidney weight, and proximal tubular necrosis, whereas none of these parameters was significantly affected by the deuterated analog. Evidence has accumulated (Henesey and Harvison, 1995; Rankin et al., 2001) that a sulfate conjugate of a succinimide ring-hydroxylated product is involved in the toxic pathway (Fig. 18).
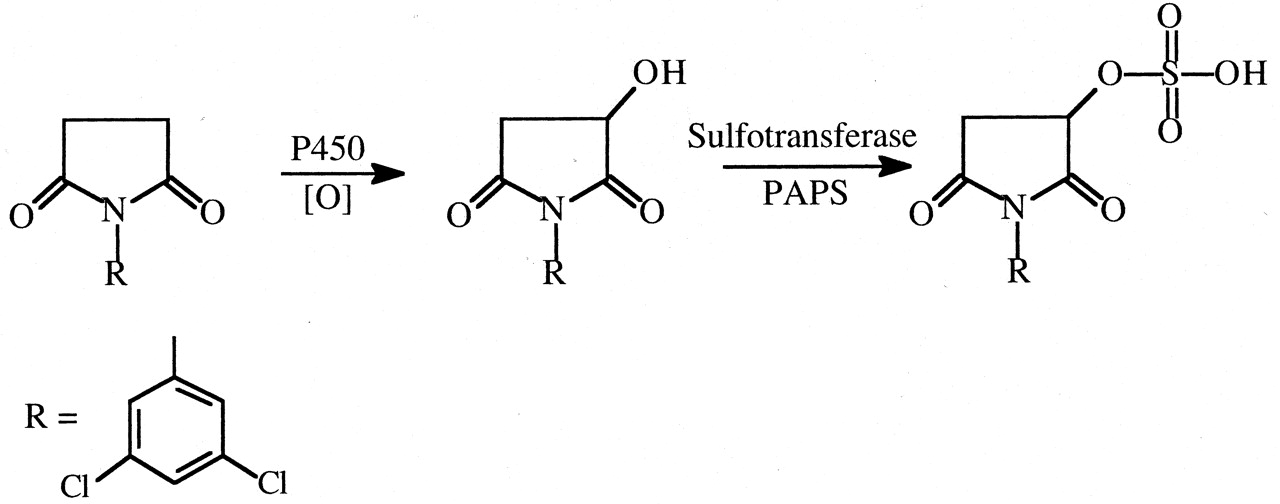
Proposed scheme for the formation of a reactive sulfate metabolite of N-(2,3-dichlorophenyl)succinimide.
A deuterium isotope effect would be observed in the first oxidation step.
3-Methylcholanthrene (3-MC). 3-MC labeled with deuterium in the 1- and 5-positions (Fig. 19) was found to be significantly less tumorigenic to mouse skin than unlabeled 3-MC (Cavalieri et al., 1975). Tumors appeared 12 to 13 weeks later in 3-MC-d3-treated versus 3-MC-treated animals, and overall incidence and mortality were decreased by 20 to 30%. The authors suggest that oxidation at C-1 is a critical step in tumorigenesis.
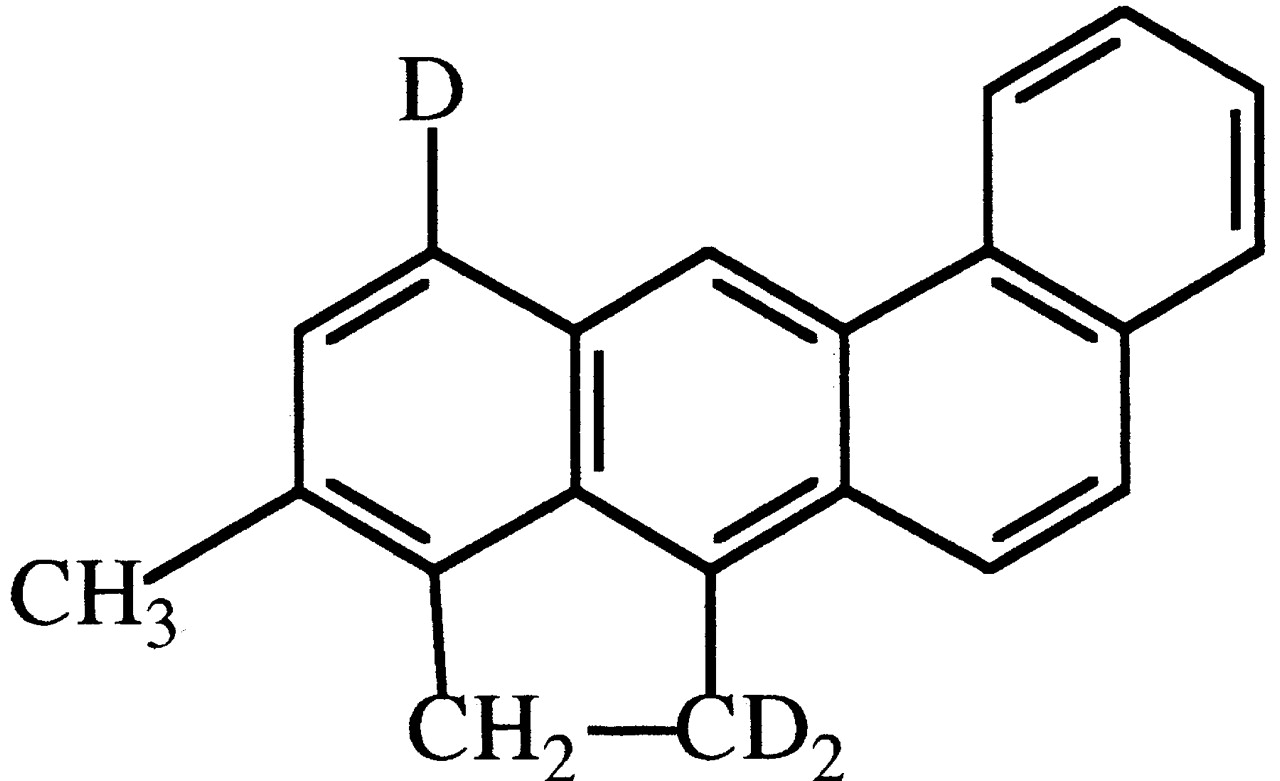
Structure of 3-MC-d3used in carcinogenesis studies as described in the text.
Butylated Hydroxytoluene (BHT). BHT labeled with deuterium in the 4-methyl group was found to be significantly less lung-toxic in mice than unlabeled BHT (Mizutani et al., 1983). Lung/body weight ratios were increased 100% more in mice treated with BHT versus BHT-d3. Studies in vitro showed that BHT-d3 was oxidized to its reactive quinone methide metabolite ∼40% more slowly than BHT. It should be noted that oxidative metabolism of BHT is complex (Fig. 20) and that subsequent studies have implicated a hydroxyquinone methide as the major ultimate lung toxin (Bolton et al., 1990).

Partial scheme for the metabolism of BHT to its quinone methide and a hydroxyquinone methide.
Substitution of deuterium for hydrogen in the 4-methyl group of BHT would be expected to decrease the rates of formation of both quinone methides and increase the fraction of a dose of BHT excreted as the hydroxylated metabolite.
Phenacetin. Whereas deuterium substitution for hydrogen in the ethoxymethylene carbon of phenacetin significantly decreases (∼3-fold) the incidence and extent of hepatic necrosis in hamsters caused by this drug, it significantly increased blood methemoglobin concentrations by ∼50% (Nelson et al., 1978). A deuterium isotope effect (∼2) was observed for hamster liver microsomal oxidation of phenacetin to acetaminophen (Fig. 21), the major proximate hepatotoxic metabolite of phenacetin in hamsters. Recent investigations with human CYP1A2 and some mutants revealed similar inter- and intramolecular deuterium isotope effects (∼2–3) for oxidative O-deethylation of phenacetin, to acetaminophen, indicating that C–H bond cleavage is only partially rate-determining in the process (Yun et al., 2000). Increased concentrations of methemoglobin observed with deuterated phenacetin analogs suggest that there is a shift in metabolism from oxidative O-deethylation to hydrolysis to p-phenetidine (Fig. 21), which is known to be oxidized to products that can cause methemoglobinemia and hemolysis (Heymann et al., 1969).

Partial scheme for the metabolism of phenacetin via oxidative O-deethylation to acetaminophen, which is further oxidized to its reactive toxic quinone imine metabolite, and via amide hydrolysis to p-phenetidine, which can be further oxidized to a hydroxyl-amine that can itself, or via its nitroso oxidation product, cause methemoglobinemia.
Deuterium substitution at the position shown decrease the fraction of the dose oxidatively O-deethylated to acetaminophen, which would increase the fraction of the dose deacetylated to p-phenetidine.
Valproic Acid (VPA). VPA is a widely used anticonvulsant drug that can cause severe hepatotoxicity (Levy and Penry, 1991). The S-CoA ester of 3-oxo-Δ2,4-VPA (Fig. 22) has been proposed as an ultimate hepatotoxic species (Baillie, 1988; Baldwin et al., 1996), and both Δ4-VPA and Δ2,4-VPA are hepatotoxins in rats (Kesterson et al., 1984). An intramolecular deuterium isotope effect (∼6) has been observed on the cytochrome P450-mediated oxidation of VPA to Δ4-VPA using 4,4-2H-VPA as a substrate (Rettie et al., 1988). However, studies have not been carried out to determine whether deuteration at the 4-and 4′-positions of VPA decreases the incidence or extent of hepatotoxicity versus VPA in rats.

Metabolism of VPA to a possible ultimate hepatotoxin 3-oxo-Δ4-VPA via its unsaturated metabolites Δ4-VPA and Δ2,4-VPA.
3-Methylindole (3MI). 3MI is a pneumotoxin, and damage to lungs in mice was found to be significantly decreased by deuteration of the methyl group, as was the rate of glutathione depletion (Huijzer et al., 1987; Yost, 1989). Mechanistic studies on the cytochrome P450 oxidation of 3MI and its CD3 and CD2H analogs showed noncompetitive intermolecular isotope effects of DV = 3.3 and D(V/K) = 1.1, and an intramolecular isotope effect of ∼5 (Skiles and Yost, 1996). The results suggest rate-limiting hydrogen abstraction from the methyl group that was masked by high forward commitment to catalysis. These and other results on the characterization of DNA adducts of 3MI and its deuterated analogs (Regal et al., 2001) suggest P450 dehydrogenation to 3-methyleneindolenine (Fig. 23) as a major pathway in the initiation of toxicity by 3MI.

Oxidation of 3-methylindole to its proposed reactive, toxic metabolite, 3-methyleneindolenine, and further reaction with nucleophiles (Nu598).
Summary
An understanding of factors that affect deuterium isotope effects in enzymatic reactions is providing new insights into mechanisms of catalysis and active site properties of drug-metabolizing enzymes, particularly the cytochromes P450. Deuterium isotope effects have also provided insights into metabolite-mediated toxic reactions, the structures of reactive metabolites, and their mechanisms of formation and disposition. However, the kinetics of the formation and disposition of reactive metabolites, particularly in vivo, have not been well characterized in most cases.
Footnotes
-
↵1 Abbreviations used are: P450, cytochrome P450; NDMA, N-nitrosodimethylamine; EDB, ethylene dibromide; GSH, glutathione; Tris-BP, tris(2,3-dibromopropyl)phosphate; DBCP, 1,2-dibromo-3-chloropropane; NMF, N-methylformamide; NDPS, N-(3,5-dichlorophenyl)succinimide; 3-MC, 3-methylcholanthrene; BHT, butylated hydroxytoluene; VPA, valproic acid; 3MI, 3-methylindole.
-

-
Sid Nelson is a Professor of Medicinal Chemistry and Dean of the School of Pharmacy, University of Washington, Seattle, WA. He received his B.S. degree in Pharmacy from the University of Washington in 1968, and after serving 2 years in the U.S. Army Medical Corps., he enrolled at the University of California, San Francisco, where he received his Ph.D. degree in Medicinal Chemistry in 1974. After 2 years as a Pharmacology Research Associate (PRAT) fellow and 1 year as a staff research associate in the Laboratory of Chemical Pharmacology at the National Institutes of Health (NIH), he returned to the University of Washington as an Assistant Professor in 1977, where he was promoted to the rank of Professor of Medicinal Chemistry in 1983. At the University of Washington, Dr. Nelson teaches basic organic medicinal chemistry and a specialized course in clinical chemistry and diagnostic products to undergraduate pharmacy students. He teaches graduate student courses in medicinal chemistry and drug metabolism.
-
His research interests include mechanisms of formation and disposition of reactive drug metabolites and the design and synthesis of enzyme inhibitors. He has over 190 peer-reviewed research publications and over 40 reviews or book chapters. He has been awarded two patents and has received the John J. Abel Award from the American Society of Pharmacology and Experimental Therapeutics (ASPET), and shared the Frank R. Blood Award in Toxicology from the Society of Toxicology. Dr. Nelson has been continuously funded by NIH for his research since 1977. He was elected a Fellow of the American Association for the Advancement of Science (AAAS) in 1986 and served as Member-at-Large and Councilor for AAAS. In 1990, Dr. Nelson received the Alumnus of the Year Award from the University of Washington School of Pharmacy for excellence in teaching and research, and in 1992 the Sato Memorial International Award from the Pharmaceutical Society of Japan for excellence in research. In 1995, he received the Gibaldi Excellence in Teaching Award from the graduating class of Pharmacy students at the University of Washington.
-
Dr. Nelson is a Councilor for the International Society for the Study of Xenobiotics (ISSX) and is a member of the Society of Toxicology for which he served as Vice President and President of the Mechanisms section. He also has served as Councilor for ASPET. Dr. Nelson was a regular member of the Pharmacology NIH Study Section Review Committee from 1986 to 1990, and a regular member of the Pharmacological Sciences NIH Program Review Committee from 1991 to 1996. In addition, he is serving as an Editorial Board Member for Drug Metabolism and Disposition.
-
-
Bill Trager is currently Emeritus Professor of Medicinal Chemistry of the School of Pharmacy, University of Washington, Seattle, Washington. He is a naturalized citizen from Winnipeg, Canada. In 1960, he received his B.S. degree in Chemistry from the University of San Francisco, San Francisco, California and in 1965 his Ph.D. degree in Medicinal Chemistry from the University of Washington under the direction of Professor Alain C. Huitric. The subject of his thesis work was the conformational analysis of substituted cyclohexanes by NMR. After receiving his Ph.D. he spent two years as a Postdoctoral Fellow with Professor Arnold H. Beckett at the Chelsea School of Science and Technology in London, England. His postdoctoral studies involved the structural analysis of mitragyna alkaloids of unknown structure by spectral means (NMR, CD, IR, and UV).
-
In 1967, Dr. Trager joined the faculty of the School of Pharmacy, University of California at San Francisco as an Assistant Professor of Medicinal Chemistry. While at the University of California, he was responsible for the High Resolution Mass Spectral Facility and developed his ongoing interest in drug metabolism. In 1972, he returned to the University of Washington as an Associate Professor of Medicinal Chemistry. In 1977, he was promoted to full Professor and received a Career Development Award from the National Institutes of Health. In 1978, Dr. Trager spent 3 months at the NIH working in the Laboratory of Chemical Pharmacology with Dr. James Gillette. In 1980, he was appointed as Science Advisor to the U.S. Food and Drug Administration (FDA), Seattle District, Chairman of the Department of Medicinal Chemistry in the School of Pharmacy, and Adjunct Professor of Chemistry in the Department of Chemistry. In 1984, he left the Chair and returned to his academic positions. From 1983 to 1987, Dr. Trager served as a committee member of the Pharmacological Sciences Review Committee of the National Institutes of General Medical Sciences, and in subsequent years, he frequently served as an ad hoc member of various study sections and site visit teams.
-
Dr. Trager's primary research interests are in drug metabolism and he was the Principle Investigator of an NIH Program Project Grant for the past 20 years investigating the molecular basis of the phenomena of drug interactions. More specific interests include quantitative mass spectrometry using stable isotopes, stereoselective metabolism, and the use of deuterium isotope effects to study mechanisms of cytochrome P450-catalyzed reactions and active site structure. In 2002, Dr. Trager retired from the University and converted his full-time position to that of a part-time faculty member.
- Received April 2, 2003.
- Accepted July 1, 2003.


- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
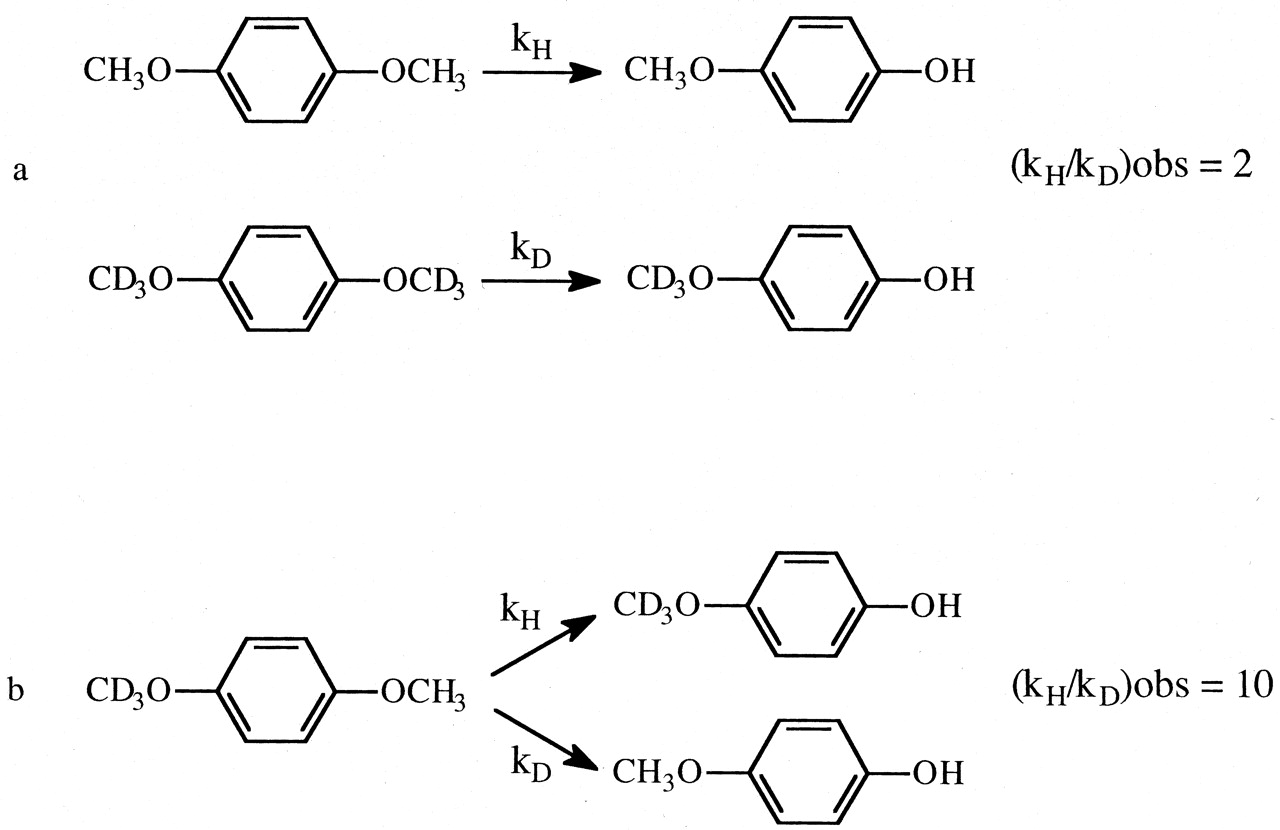{kind=link}
{kind=link}
{kind=link}
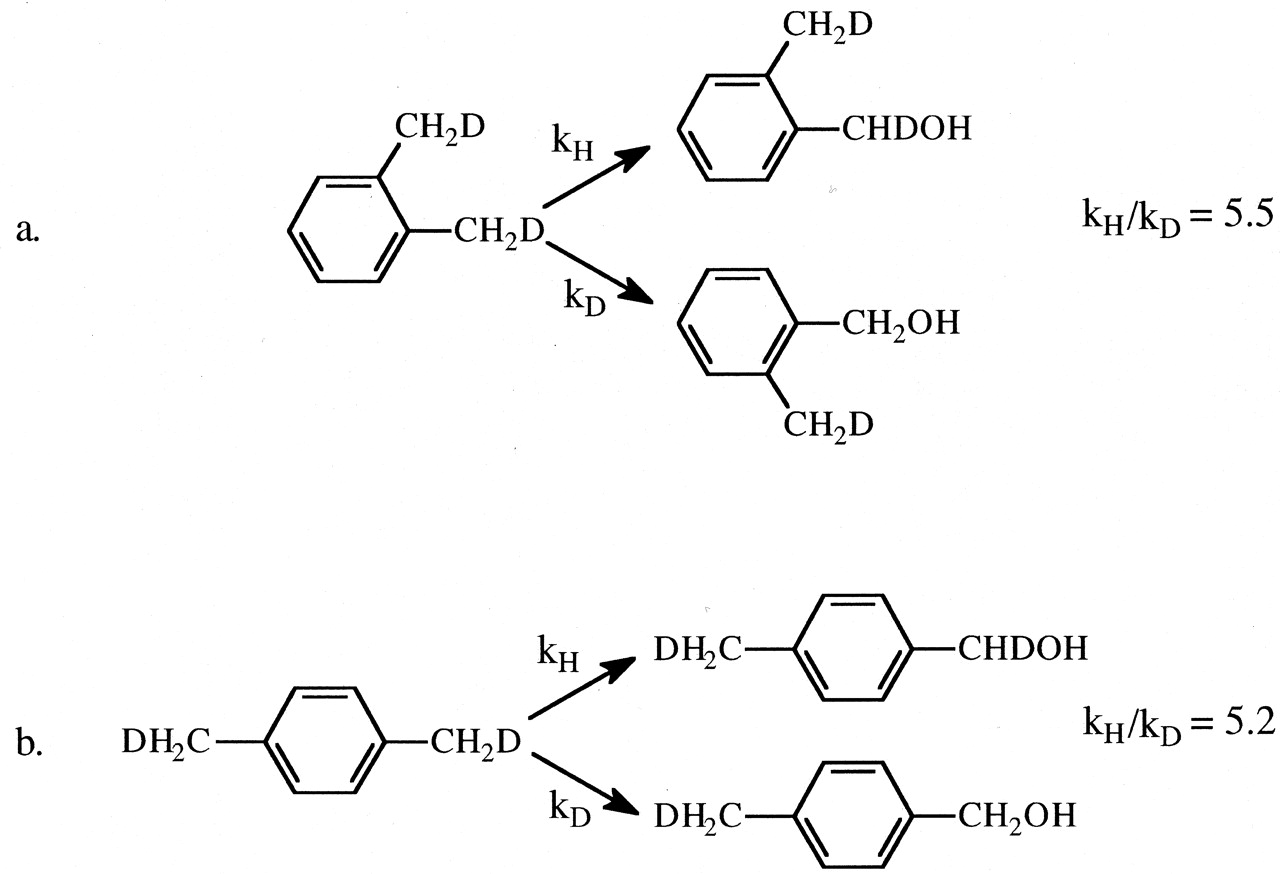{kind=link}
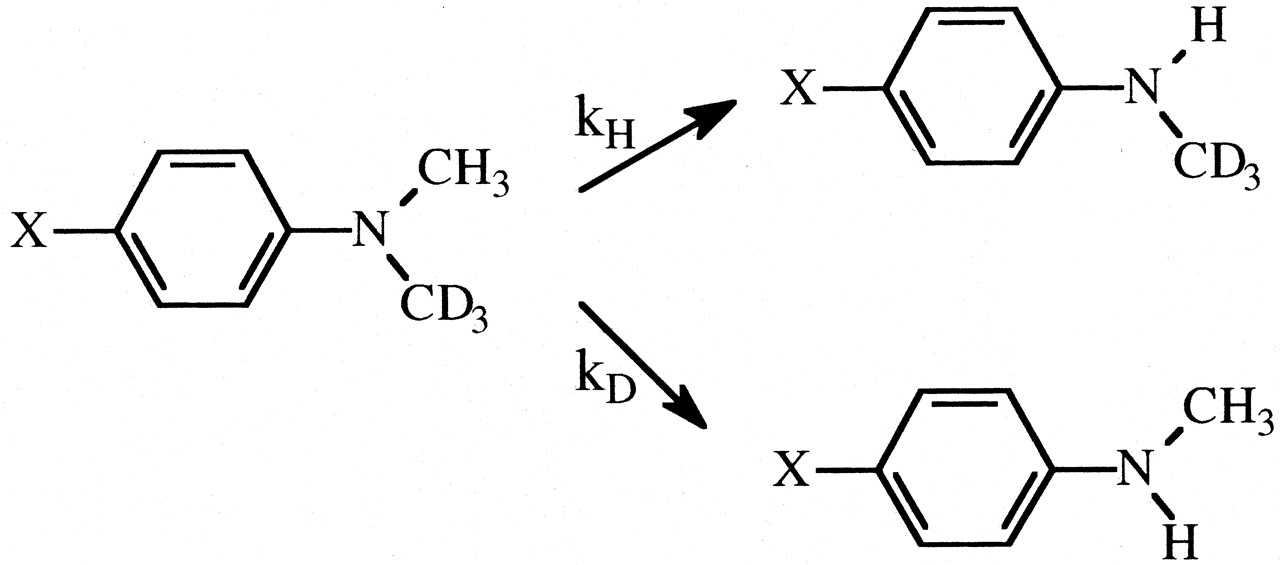{kind=link}
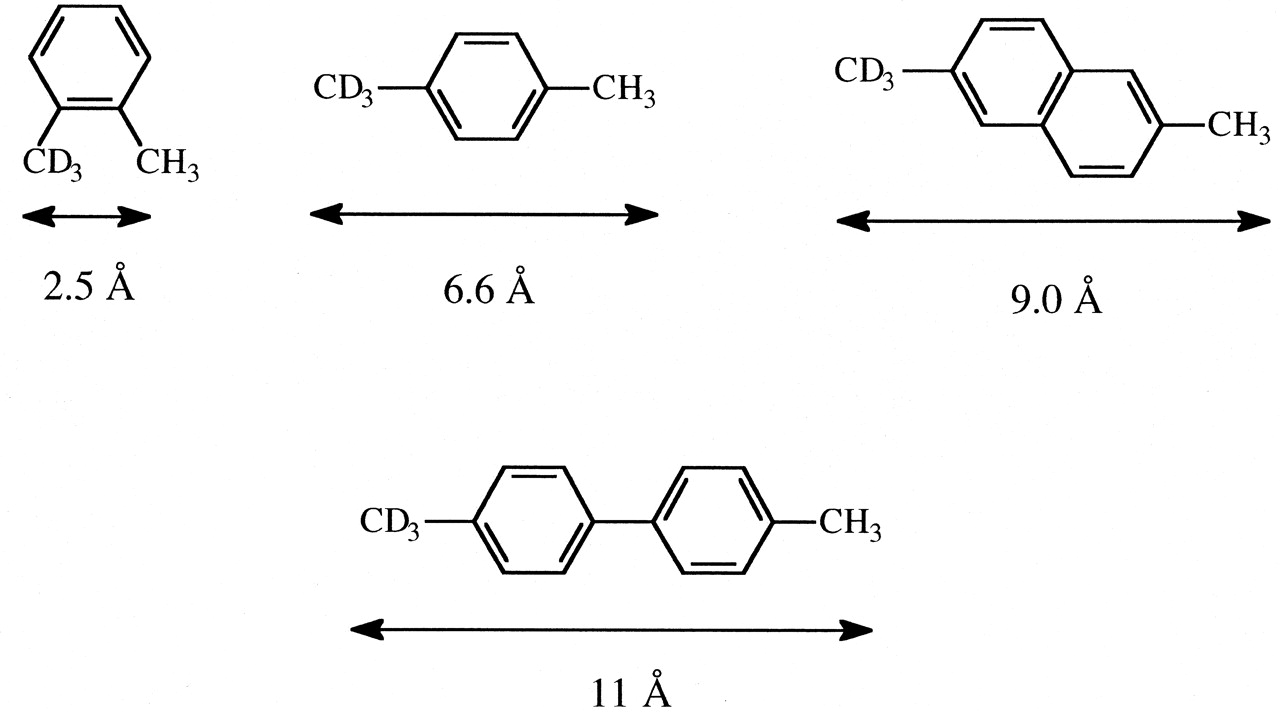{kind=link}
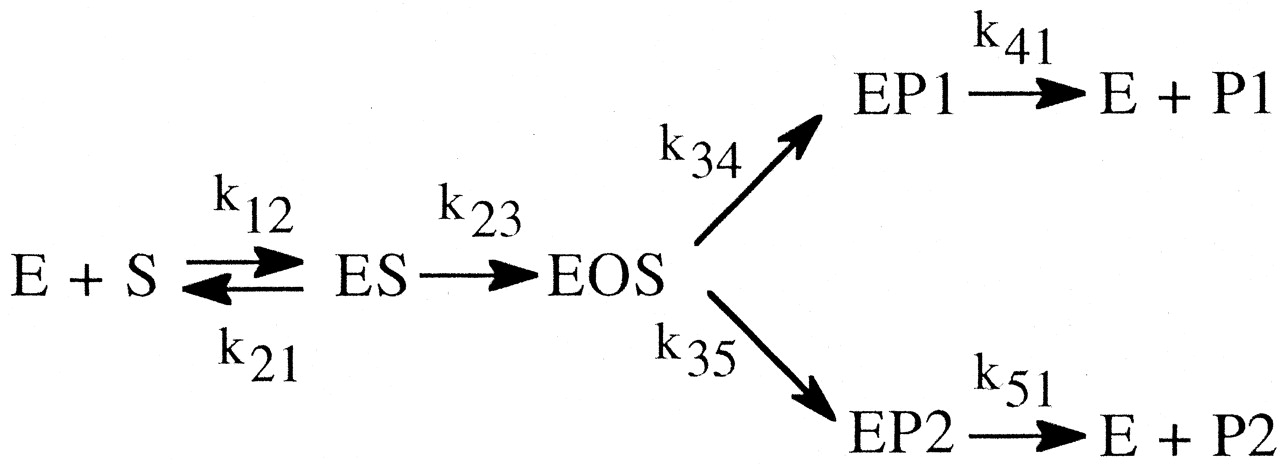{kind=link}
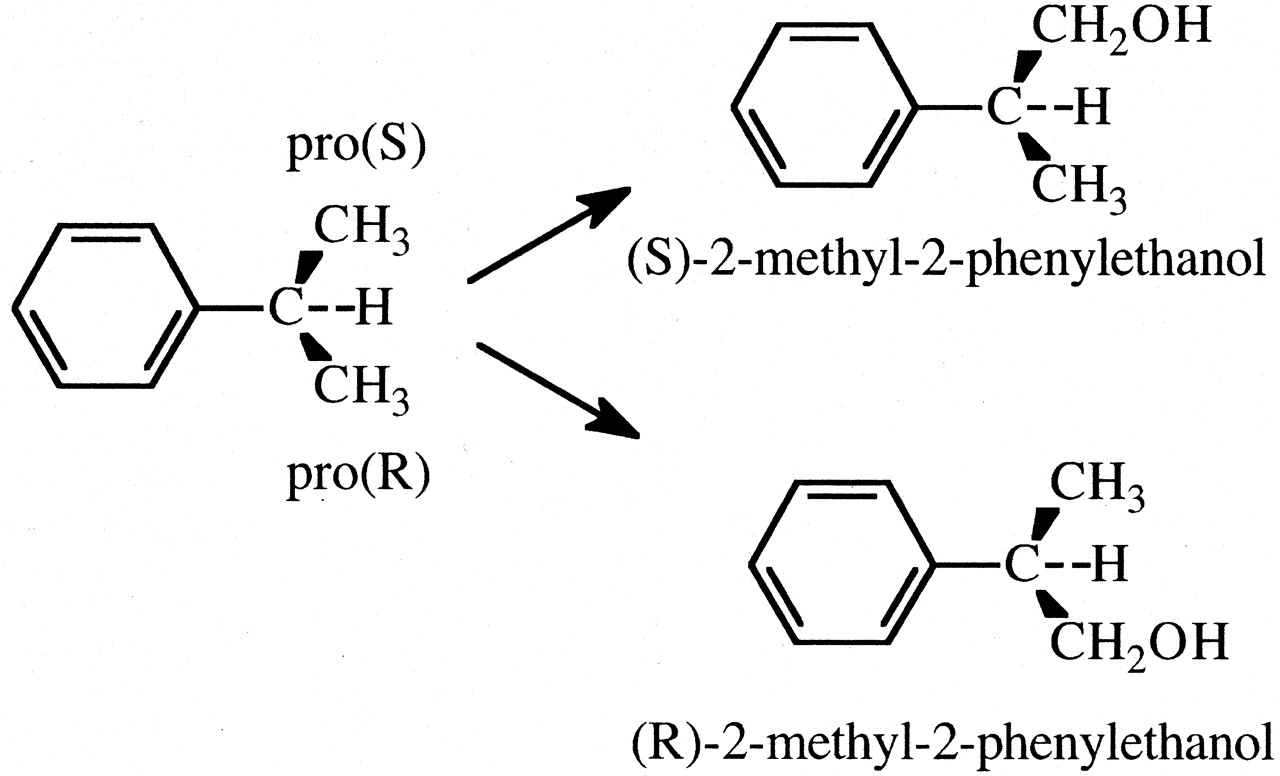{kind=link}
{kind=link}
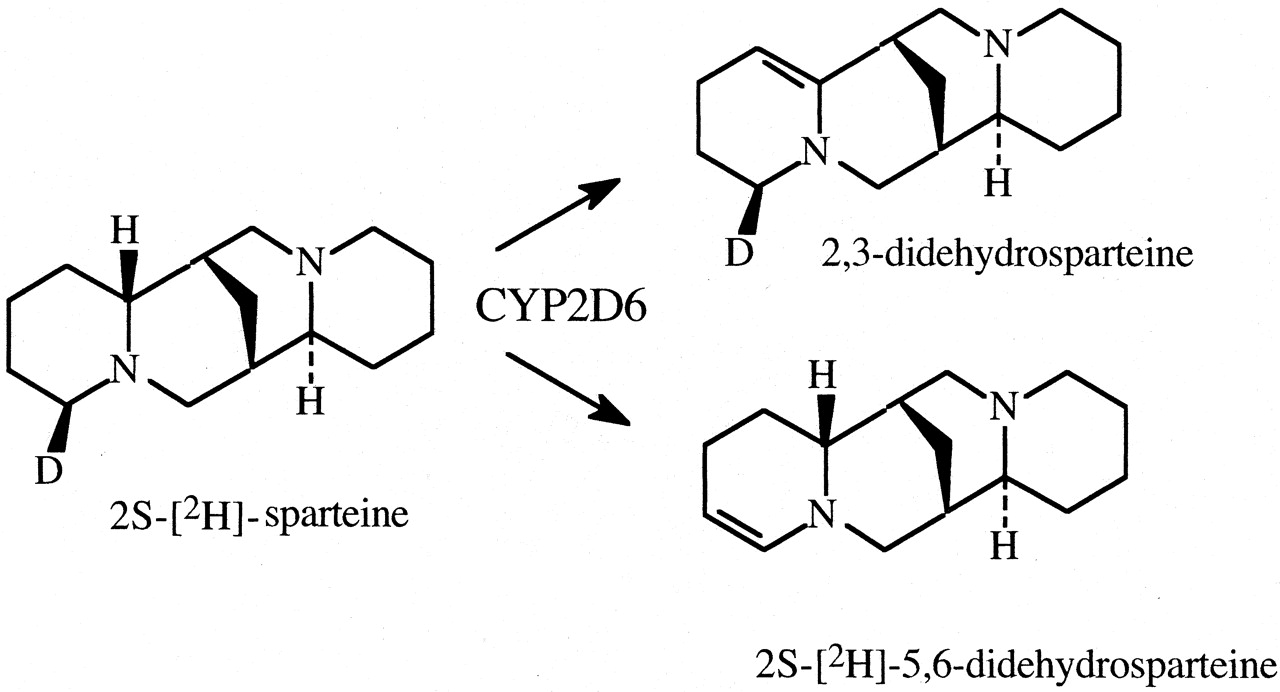{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
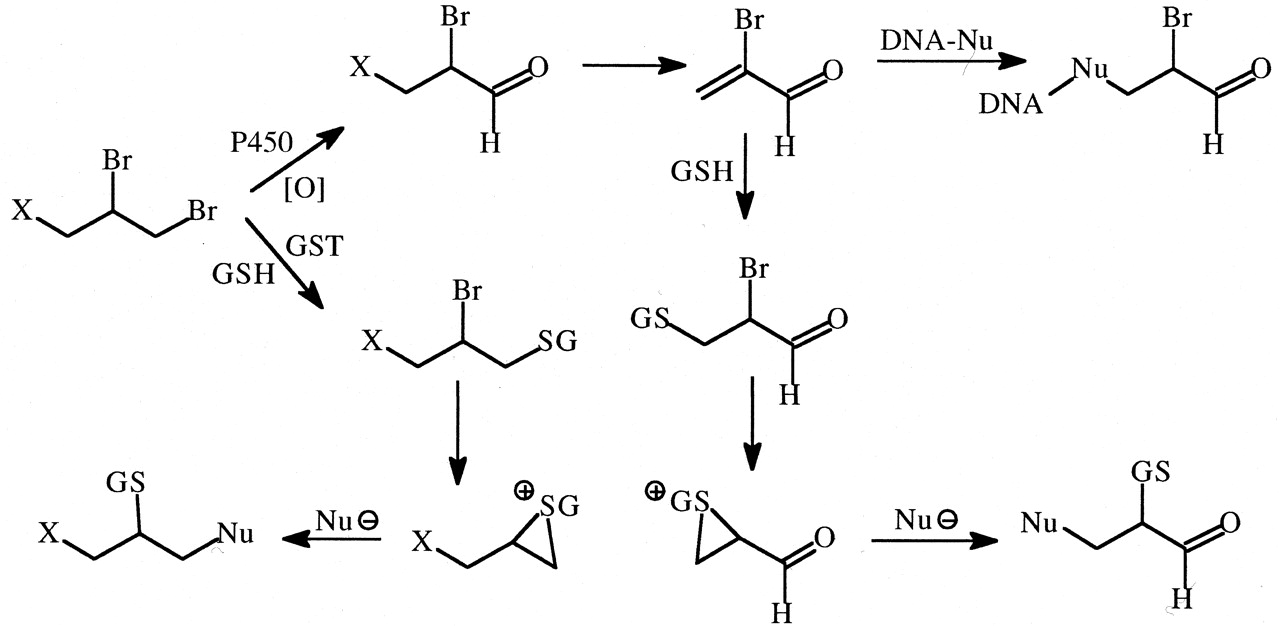{kind=link}
{kind=link}
{kind=link}
{kind=link}
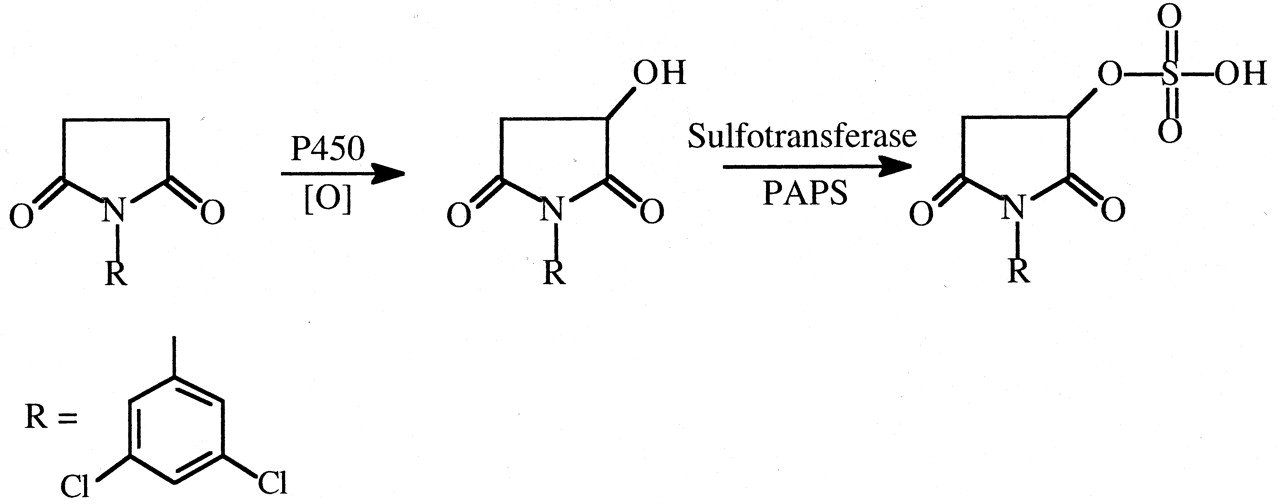{kind=link}
{kind=link}
{kind=link}
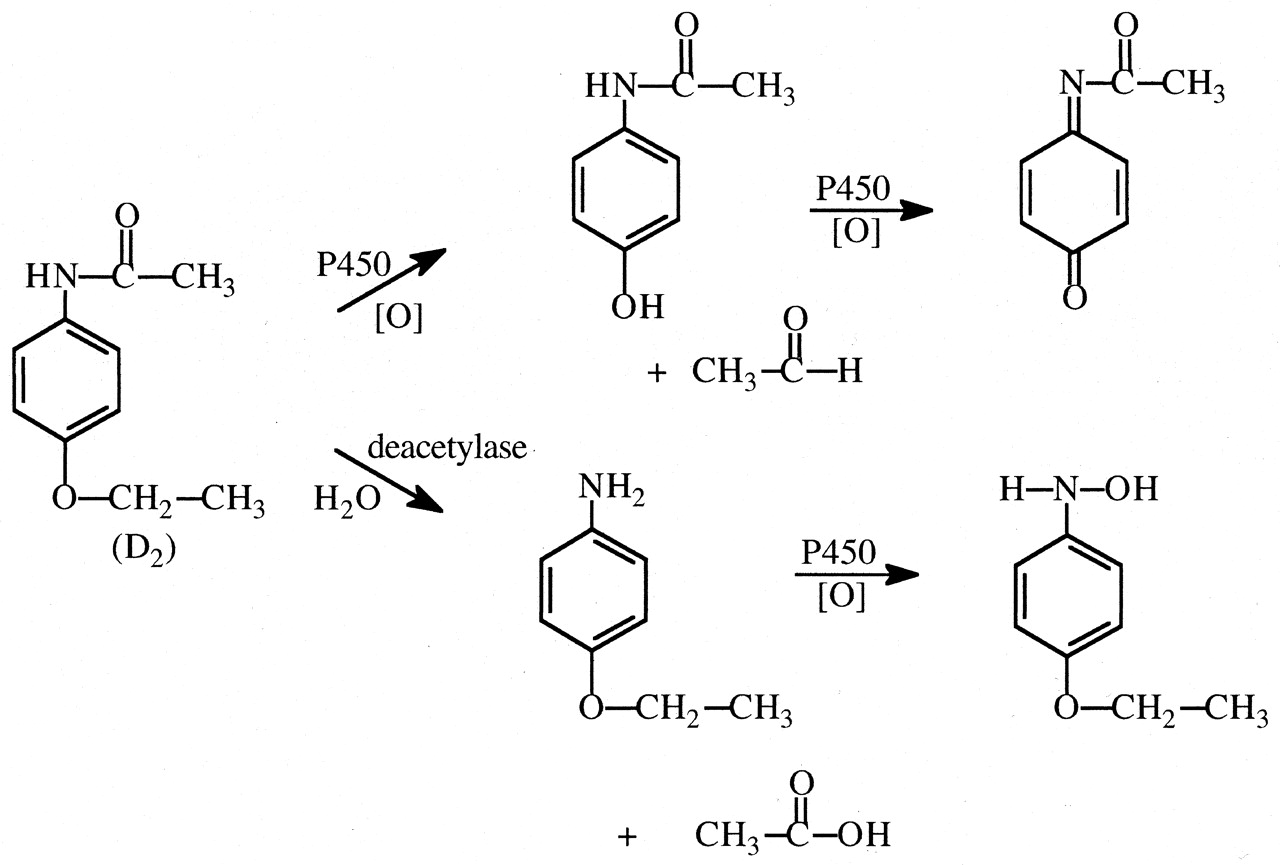{kind=link}
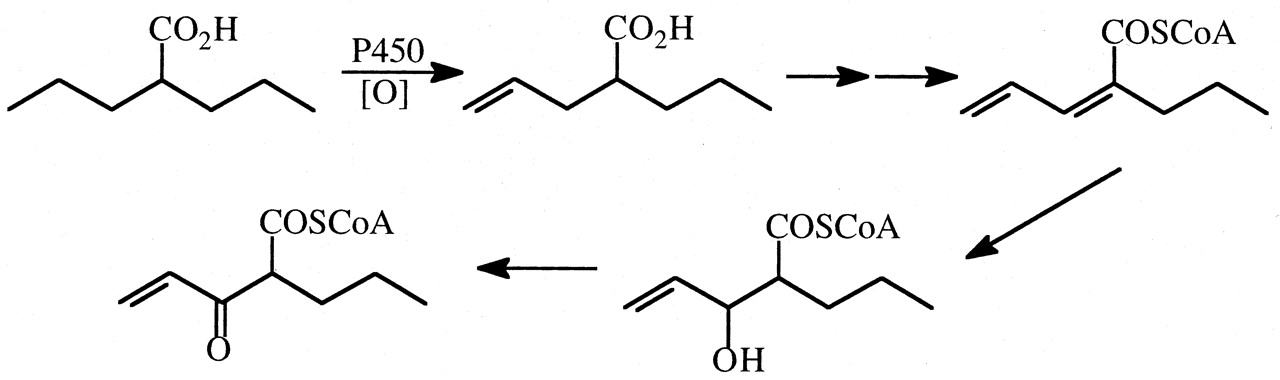{kind=link}
{kind=link}