


Abstract
Compounds known to modulate P-glycoprotein (P-gp) activity were evaluated in cell monolayers expressing P-gp for their effects on the secretory transport of P-gp substrates paclitaxel, vinblastine, and digoxin. Paclitaxel has been proposed to selectively interact with a binding site on P-gp that is distinct from the vinblastine and digoxin-binding site. Using Madin-Darby canine kidney (MDCK)-multidrug resistance-1 (MDR1), MDCK-wild-type (WT), and Caco-2 cell monolayers, the basal-to-apical (BL-AP) apparent permeability (Papp) of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin in the presence of various concentrations of a series of structurally diverse P-gp substrates and modulators of P-gp function were determined. MDCK-WT cell monolayers demonstrated active secretory transport of all P-gp substrate probes, although the sensitivity to inhibition by verapamil was lower than that demonstrated in MDCK-MDR1 cell monolayers. When evaluated as competitive inhibitors, several known P-gp substrates had no effect or only a slight modulatory effect on the BL-AP Papp of all probe substrates in MDCK-MDR1 cells. The secretory transport of P-gp substrates in MDCK-WT cells was more sensitive to inhibition by known P-gp modulators compared with MDCK-MDR1 cells. Low concentrations of ketoconazole (1–3 μM) activated the BL-AP Papp of [3H]vinblastine and [3H]digoxin in MDCK-MDR1 cells but not in MDCK-WT or Caco-2 cells. Determination of secretory transport in P-gp expressing cell monolayers, such as MDCK-MDR1 and Caco-2, may be complicated by substrate cooperativity and allosteric binding, which may result in the activation of P-gp. In addition, expression of other efflux transporters in these cell lines introduces additional complexity in distinguishing which transporter is responsible for substrate recognition and transport.
P-glycoprotein (P-gp), a member of the ATP-binding cassette superfamily of membrane localized solute transporters, is expressed in a number of tissue compartments, including the luminal membrane of gastrointestinal enterocytes, the canalicular membrane of hepatocytes, the proximal tubular cells of the kidney, and the luminal membrane of brain capillary endothelial cells (Gottesman and Pastan, 1993; Ambudkar et al., 1999). P-gp mediated efflux of many structurally divergent substrate drugs can often limit their intestinal absorption (Benet et al., 1999; Greiner et al., 1999; Chan et al., 2004) and blood-brain barrier penetration (Choo et al., 2000; Sadeque et al., 2000), as well as facilitate their active excretion into bile and urine (Kusuhara and Sugiyama, 2002; Susanto and Benet, 2002). In conjunction with drug-metabolizing enzymes, P-gp provides a protective physiological barrier capable of altering the rate and extent of xenobiotic entry into the systemic circulation.
Drugs that interact with P-gp have been categorized as substrates such as digoxin and paclitaxel, inhibitors such as ketoconazole and GF120918, or inducers such as clotrimazole and rifampin (Gottesman and Pastan, 1993; Ambudkar et al., 1999; Polli et al., 2001; Kim, 2002). Interestingly, certain compounds function as a substrate and inhibitor of P-gp (quinidine, verapamil), an inhibitor and inducer of P-gp (atorvastatin, mefloquine), or possibly a combination of P-gp substrate, inhibitor, and inducer (nelfinavir, ritonavir, saquinavir) (Perloff et al., 2000; Kim, 2002; Marzolini et al., 2004). Clinical drug-drug interactions (DDIs) have been attributed to the modulation of P-gp function following concomitant administration of drugs that fall into one or more of these categories (Gramatte and Oertel, 1999; Greiner et al., 1999; Westphal et al., 2000). For example, a considerable decrease in digoxin exposure (area under the curve and Cmax) as well as a 3.5-fold increase in duodenal P-gp expression was noted after oral coadministration of rifampin in healthy subjects (Greiner et al., 1999). In mice, inhibition of P-gp efflux by LY335979 enhanced the distribution of various HIV-1 protease inhibitors into the brain and testes in a dose-dependent manner (Choo et al., 2000). Therefore, it is important to be able to categorize P-gp substrates, inhibitors, inducers, and combinations thereof in an unambiguous fashion (Litman et al., 2001; Polli et al., 2001).
Various in vitro assays are routinely employed to evaluate and characterize the interaction of drugs, excipients, and drug candidates with human P-gp. Caco-2 monolayers grown on permeable filters express human P-gp and are commonly used to identify P-gp substrates by monitoring their transport properties (Burton et al., 1993; Anderle et al., 1998; Gao et al., 2001). MDCK-MDR1 cells are often used instead of Caco-2 cells and tend to require a relatively short culture period (3–5 days) while displaying interpassage homogeneity and high level of human P-gp expression (Polli et al., 2001; Tang et al., 2002). Accordingly, MDCK-MDR1 cells are frequently used as a model P-gp expression system. Although widely used, there are still unanswered questions regarding the differences in functionally expressed P-gp in the MDCK-MDR1 compared with the MDCK-WT cells, such as the influence of passage number on the expressed P-gp protein (Tang et al., 2002). In addition, these cell lines express other transporters that can potentially make interpretation of efflux transporter activity a complex endeavor. For example, mature Caco-2 cell monolayers express breast cancer resistance protein (BCRP) and multidrug resistance-associated protein 2 (MRP2) (Taipalensuu et al., 2001; Prime-Chapman et al., 2004), and MDCK cell monolayers express canine P-gp (Goh et al., 2002).
Several binding sites within P-gp have been described, although selective probes for each binding site have not yet been identified (Dey et al., 1997; Shapiro et al., 1999; Martin et al., 2000). Additionally, transport and regulatory sites within P-gp are able to modulate between high- and low-affinity conformations (Martin et al., 2000). There is a limited correlation between compounds that stimulate or inhibit ATPase activity, the concentrations at which this occurs, and the corresponding change in P-gp activity (Litman et al., 2001; Polli et al., 2001; Tang et al., 2002). Thus, because of these complex binding and regulatory interactions, it is difficult to adhere to one standardized protocol for determining the interaction of a particular compound with P-gp. Certain groups employ multiple methods to more accurately evaluate the interaction of a compound with P-gp (Polli et al., 2001). In this paper, we describe the effects of a variety of known P-gp substrates and modulators on the active BL-AP transport of vinblastine and paclitaxel, P-gp substrates that have been proposed to interact with distinct P-gp-binding sites in studies using P-gp expressing CHrB30 cells (Martin et al., 2000). We also describe the effects of these compounds on the active BL-AP transport of digoxin, a P-gp substrate that has been proposed to bind to the same site as that of vinblastine as determined using data from various in vitro models and computational analysis of pharmacophore interactions with P-gp (Ekins et al., 2002a,b).
Materials and Methods
Materials. MDCK-MDR1 cells originated in M. M. Gottesman's laboratory (National Institutes of Health, Bethesda, MD) and were licensed for use by Boehringer Ingelheim Pharmaceuticals. MDCK-WT and Caco-2 cells were obtained from the ATCC (Manassas, VA). All cell culture and immunoblotting reagents were purchased from Invitrogen (Carlsbad, CA), unless otherwise noted. [14C]Mannitol and [3H]vinblastine were purchased from (GE Healthcare, Little Chalfont, Buckinghamshire, UK). [3H]Paclitaxel was purchased from Moravek Biochemicals (Brea, CA), and [3H]digoxin was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Hepes and Ultima Gold scintillation fluid were also obtained from Sigma-Aldrich. Polycarbonate Transwells (0.4 μm pore size) were purchased from Corning Life Sciences (Acton, MA). Structures of all of the compounds used in this study are shown in Fig. 1.
Cell Culture. For drug transport experiments, MDCK-MDR1 cells (passage number 22–31) were seeded onto 1.12-cm2 polycarbonate filter Transwells at a density of 104 cells/well. Culture conditions were maintained as has been described previously (Susanto and Benet, 2002). Confluent MDCK-MDR1 monolayers expressing P-gp were obtained 5 to 6 days postseeding. Integrity of the cell monolayers was determined by measuring the trans-epithelial electrical resistance (TEER, Ω · cm2) using an epithelial voltohmmeter (Millicell-ERS; Millipore Corporation, Billerica, MA). After subtraction of background TEER (i.e., the resistance exhibited by the filter alone) mature MDCK-MDR1 cell monolayers exhibited a TEER >1000 Ω · cm2 before use in transport experiments. MDCK-WT cells (passage number 15–30) were seeded at a density of 5.5 × 104 cells/well and, after 6 days, matured into confluent monolayers exhibiting a TEER >200 Ω · cm2 before use in transport experiments. Caco-2 cells (passage number 35–40) were seeded at a density of 0.9 × 104 cells/well and, after 21 days, matured into confluent monolayers exhibiting a TEER >500 Ω · cm2 before use in transport experiments. Cell monolayer integrity was evaluated via either measurement of [14C]mannitol (0.4 μCi/ml) paracellular passive Papp, monitoring the change in TEER over the course of the experiment or a combination of both.
Western Blot Analysis. Western blot was performed using a method described previously (Hosoya et al., 1996). MDCK-MDR1, MDCK-WT, and Caco-2 monolayers were grown as described for transport experiments. Cells were lysed (45 min) in ice-cold phosphate-buffered saline containing SDS (3%), antipain (83 μM), pepstatin A (73 μM), and leupeptin (0.1 μM). Cell lysate was centrifuged for 15 min (12,000g), and the supernatant was saved for analysis. Five micrograms of cell protein was loaded in a 4 to 12% SDS-PAGE gel and run for 50 min. After transfer onto 0.2-μm nitrocellulose membranes (Invitrogen), blots were probed for 90 min (1:25 dilution) with the C219 primary monoclonal P-gp antibody harvested from mouse (Signet Laboratories, Dedham, MA). Reprobing was conducted for 1 h at room temperature using an anti-mouse IgG-alkaline phosphatase conjugate, diluted 1:2000 (Promega, Madison, WI). Bands were developed and visualized using Western Blue Stabilized Substrate for alkaline phosphatase (Promega, Madison, WI).
Transport Experiments: MDCK-WT, MDCK-MDR1, and Caco-2 Monolayers. Prior to the addition of radiolabeled P-gp substrate, growth medium was removed and monolayers were rinsed twice with HBSS at 37°C. The filter inserts containing the cell monolayers were transferred to a separate 12-well cell culture plate containing solutions of the P-gp substrate probe and test compounds. As [3H]paclitaxel and [3H]vinblastine clearly demonstrated nonspecific binding to the cell culture apparatus in our initial studies, a 1-h preincubation step before inserting the filter inserts was developed, which minimized the nonspecific binding and provided a more accurate mass balance assessment at the end of the experiment (BL-AP transport experiments only). All drug transport experiments were performed at 37°C using a 20 or 100 nM solution of each P-gp substrate probe in HBSS at pH 7.4. The AP media volume was 400 μl, and the BL volume was 1200 μl. For most experiments, radiolabeled test compound was added to the BL compartment and its appearance in the AP compartment over time was monitored. The highest concentration of test compound was selected based on the limit of its solubility in HBSS at pH 7.4 or following preliminary experiments to determine the degree of inhibition at various concentrations. After the 1-h preincubation, a 20-μl sample was taken from the donor compartment at t = 0 min to confirm the initial concentration of substrate (Co). At 15, 30, 60, and 90 min, 100 μl of media was removed from the receiver compartment followed by the addition of 100 μl of preheated buffer as replenishment. At 90 min, a 20-μl sample was taken from the donor compartment to determine the concentration of compound remaining in the donor chamber at the end of the experiment. Samples were analyzed using scintillation counting.
Permeability Calculations and Statistical Analysis. The accumulated amount of radiolabeled probe appearing in the AP compartment over time, dQ/dt, was used to calculate the apparent permeability (Papp) using the following equation: Papp = dQ/dt × 1/(A × Co), where A is the area of the filter (1.12 cm2) and C0 is the initial concentration of radiolabeled probe substrate in the donor compartment. In all BL-AP transport experiments, sink conditions were maintained as defined by >80% of compound remaining in the donor compartment at the end of the experiment. Papp values were therefore calculated using the slope of the steady-state rate constant dQ/dt. Statistical analyses were performed by comparison of the means of control BL-AP Papp values versus BL-AP Papp values determined in the presence of each test compound using an unpaired t test (GraphPad InStat, version 3.05; GraphPad Software Inc., San Diego, CA). A p value of <0.05 was taken as the minimum level of statistical significance.
Results
Expression of P-gp. Western blot analysis of MDCK-WT, MDCK-MDR1, and Caco-2 cell monolayer lysates was conducted to determine the expression of human P-gp in these cell lines, as maintained under standardized culture conditions in our laboratory. MDCK-WT cells did not express human P-gp that was immunoreactive with the C219 monoclonal antibody, whereas MDCK-MDR1 and Caco-2 cells did express human P-gp (Fig. 2).
P-gp substrate probes (A), known P-gp substrates and an MRP2 inhibitor (probenecid) evaluated for their potential to inhibit P-gp-mediated secretory transport (B), and a series of compounds evaluated for their potential modulatory effect on the secretory transport of P-gp substrate probes (C).

Western blot of MDCK-WT, MDCK-MDR1, and Caco-2 cell homogenates probed for P-gp expression. Cells were grown as monolayers for 6 days, harvested using phosphate-buffered saline containing 3% SDS and protease inhibitors as described under Materials and Methods, and electrophoresed on an 8% SDS-polyacrylamide gel for 50 min. Lane 1, molecular mass marker; lane 2, MDCK-WT; lane 3, MDCK-MDR1; and lane 4, Caco-2. Each lane contains 5 μg of cell lysate.
P-gp-Mediated BL-AP Transport in MDCK-MDR1 and MDCK-WT Monolayers. The AP-BL and BL-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin (20 nM; Fig. 1A) were evaluated in MDCK-MDR1 and MDCK-WT cell monolayers (Table 1). These studies were conducted in the presence (200 μM) and absence of a known P-gp inhibitor, verapamil (Wacher et al., 1995; Litman et al., 2001; Kim, 2002). Control and verapamil-treated BL-AP and AP-BL Papp values were compared to determine the contribution of verapamil-sensitive efflux for each probe. In experiments using MDCK-WT monolayers, the ratios of control:verapamil-treated BL-AP/AP-BL values were 10.4, 11.8, and 2.6 for [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin, respectively. The presence of 200 μM verapamil reduced the BL-AP/AP-BL values for each probe to near unity (range: 1.6–2.6) compared with untreated control monolayers (range: 5.2–30.7). In experiments using MDCK-MDR1 monolayers, the ratios of control:verapamil-treated BL-AP/AP-BL values were much higher than those calculated for MDCK-WT monolayers: 41.2, 62.9, and 19.6 for [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin, respectively. The presence of 200 μM verapamil reduced the BL-AP/AP-BL value to near unity (range: 1.0–1.4) compared with untreated control monolayers (range: 25.5–88.1).
AP-BL and BL-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin across MDCK-WT and MDCK-MDR1 monolayers in the presence and absence of verapamil (200 μM) a
Effect of P-gp Substrates on BL-AP Papp. Several known P-gp substrates and a known inhibitor of MRP2 activity (Fig. 1B) were evaluated for their potential to competitively inhibit the secretory transport of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin across MDCK-MDR1 monolayers (Table 2). When compared with untreated monolayers as control, colchicine, cortisol, dexamethasone, etoposide, trimethoprim, and probenecid (10 and 100 μM) demonstrated no effect or only a slight modulatory effect on the BL-AP Papp of each probe P-gp substrate.
Known P-gp substrates demonstrating no effect or a slight modulatory effect on the BL-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin in MDCK-MDR1 monolayers a
Effect of P-gp Modulators on BL-AP Papp. Several known modulators of P-gp activity (Fig. 1C) were evaluated for their potential to affect the secretory transport of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin across MDCK-MDR1 and MDCK-WT monolayers. When normalized to untreated monolayers as control, cyclosporine A, ketoconazole, loperamide, verapamil, and nicardipine at various concentrations either inhibited or activated the BL-AP Papp of each probe P-gp substrate (Table 3). All compounds evaluated inhibited the secretory transport of each P-gp substrate probe at high concentration. The BL-AP Papp of all P-gp substrate probes across MDCK-WT cells was more sensitive to the modulators that were evaluated compared with their effect in MDCK-MDR1 cells. The effect of quinidine, a known inhibitor of P-gp activity (Emi et al., 1998; Kim et al., 1999), on the BL-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin was determined in MDCK-WT, MDCK-MDR1, and Caco-2 cell monolayers (Fig. 3). Secretory transport of P-gp substrate probes in MDCK-WT cells was the most sensitive to quinidine, demonstrating >50% inhibition of BL-AP Papp of all substrate probes at concentrations >3 μM (Fig. 3A). MDCK-MDR1 cells were the least sensitive to quinidine, requiring concentrations of 10 to 30 μM to achieve a 50% inhibition of [3H]paclitaxel and [3H]vinblastine BL-AP Papp and a concentration of 30 to 100 μM to achieve a 50% inhibition of [3H]digoxin BL-AP Papp (Fig. 3B). In Caco-2 cell monolayers, [3H]paclitaxel and [3H]digoxin BL-AP Papp was >50% inhibited by quinidine at 3 to 10 μM, whereas [3H]vinblastine transport was not affected by the presence of quinidine at concentrations up to 30 μM (Fig. 3C).
Effect of cyclosporin A, ketoconazole, verapamil, quinidine, and nicardipine on the P-gp-mediated BL-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin across MDCK-WT and MDCK-MDR1 monolayersa
Concentration-Dependent Activation of Substrate Transport. In MDCK-MDR1 cell monolayers, BL-AP Papp of [3H]vinblastine was activated by ketoconazole and loperamide at 1 and 3 μM compared with untreated monolayers, but not at 0.1, 10, or 30 μM (Table 3 and Fig. 4). In contrast, in Caco-2 cell monolayers, BL-AP Papp of [3H]vinblastine was not similarly affected by the presence of low concentrations (1 and 3 μM) of ketoconazole (Fig. 4). BL-AP Papp of [3H]digoxin in MDCK-MDR1 monolayers was activated by ketoconazole (1 and 3 μM) compared with untreated monolayers, but not at 0.1, 10, or 30 μM. BL-AP Papp of [3H]digoxin was activated by loperamide only at 3 μM and not at any other concentrations of loperamide (Table 3). BL-AP Papp of all P-gp probe substrates in MDCK-WT cell monolayers was not activated in the presence of ketoconazole or loperamide (0.1–100 μM) or any of the other modulators of P-gp activity that were evaluated (Table 3).
Discussion
A variety of in vitro techniques for identifying P-gp substrates and modulators, typically using P-gp-expressing cells or cell membranes, have been investigated and described. These methods include determining the ratio of AP-BL to BL-AP Papp across P-gp expressing cell monolayers, measuring P-gp-mediated calcein-AM efflux in intact cells or vesicles, determining the cellular uptake of a radiolabeled P-gp probe in P-gp-expressing cells, and determining intracellular ATPase activity (Burton et al., 1993; Anderle et al., 1998; Martin et al., 2000; Gao et al., 2001; Polli et al., 2001). Radioligand binding studies have demonstrated that at least four drug-binding/transport sites are present on P-gp, and modulatory interactions have been shown to occur in an allosteric manner at various loci on the protein (Dey et al., 1997; Shapiro and Ling, 1997; Shapiro et al., 1999; Martin et al., 2000). Additionally, complex modulatory interactions with P-gp can occur; i.e., certain compounds can function as combinations of substrate, inhibitor, and activator (Kim, 2002). It has been suggested that, to more clearly elucidate the potential for P-gp-mediated DDIs, investigators should evaluate multiple P-gp substrates with affinities for distinct binding sites or a series of assays providing results based on more than one parameter (Gao et al., 2001; Litman et al., 2001; Polli et al., 2001; Yasuda et al., 2002). Using MDCK-MDR1, MDCK-WT, and Caco-2 cell monolayers and binding site-specific radiolabeled P-gp substrate probes, we have investigated the effects of several compounds that are known modulators of P-gp activity for their effect on P-gp-mediated secretory transport.
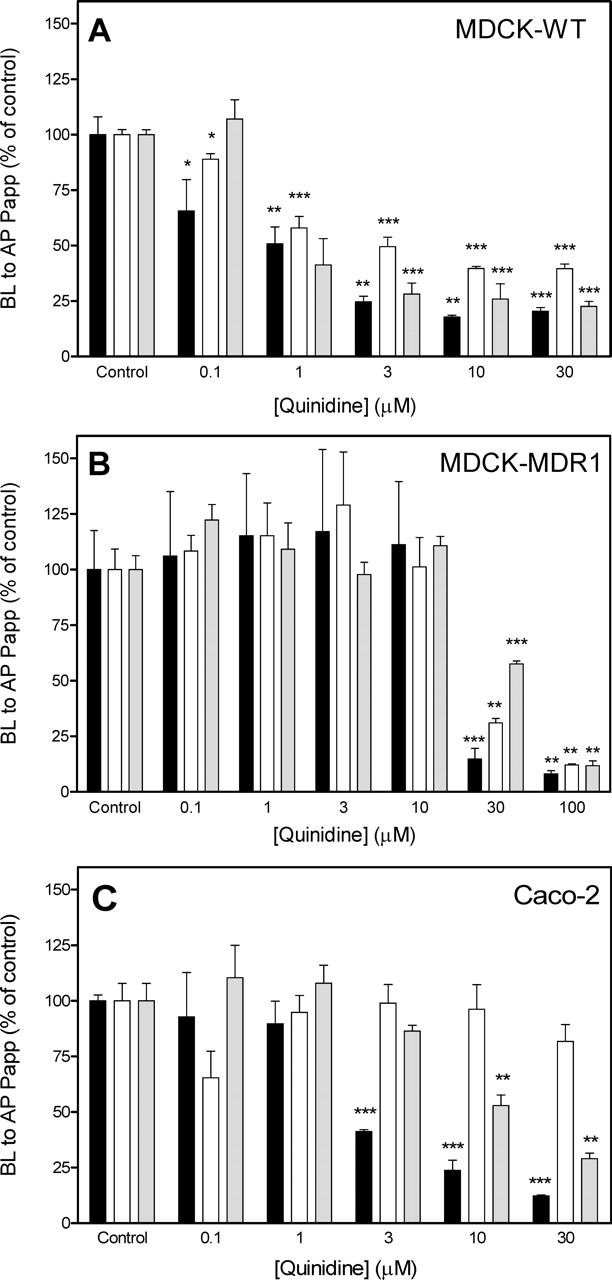
Effect of quinidine on the BL-to-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin across MDCK-WT (A), MDCK-MDR1 (B), and Caco-2 (C) cell monolayers. Each P-gp substrate was added to the donor (BL) compartment at 100 nM, and its BL-AP transport was monitored by sampling media from the AP compartment at various intervals over a 90-min incubation period. The BL-AP transport of [3H]paclitaxel (solid columns), [3H]vinblastine (open columns), and [3H]digoxin (shaded columns) was determined in the absence of quinidine (control) and in the presence of various concentrations of quinidine from 0.1 to 30 μM. Each column represents the mean ± S.D. (n = 3) BL-to-AP Papp expressed as a percentage of treated versus control monolayers. Statistical significance was calculated for the difference between treated and control monolayers for each P-gp substrate probe using a two-tailed unpaired t test. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Mature MDCK-MDR1 cell monolayers are commonly employed as an in vitro model for evaluating human P-gp-mediated secretory transport (Polli et al., 2001; Susanto and Benet, 2002; Tang et al., 2002). It has been reported that MDCK-WT cells express a protein that is immunoreactive with human P-gp, ostensibly the canine ortholog of P-gp (Tang et al., 2002). Using the monoclonal C219 P-gp antibody, we found that P-gp was expressed at a very high level in MDCK-MDR1 cells, expressed at a relatively lesser extent in Caco-2 cells, and was not expressed in MDCK-WT cells (Fig. 2). A sequence alignment analysis of human MDR1 (GenBank accession number M14757) compared with canine MDR1 (GenBank accession number AF045016) revealed that the human and canine orthologs of P-gp share an 81% sequence homology. As such, it is not unreasonable to expect that certain monoclonal antibodies will cross-react with both human and canine P-gp, whereas others will not.
As shown in Table 1, MDCK-WT cell monolayers also possess intrinsic verapamil-sensitive P-gp activity, although the extent of secretory transport of the P-gp substrate probes was much greater in MDCK-MDR1 monolayers. A significantly greater verapamil-sensitive increase in [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin BL-AP Papp (4-, 5.3-, and 7.5-fold, respectively) was demonstrated in MDCK-MDR1 cell monolayers when compared with MDCK-WT monolayers (Table 1). Tang et al. (2002) investigated the polarized transport of [3H]digoxin across MDCK-WT, MDCK-MDR1, and Caco-2 cell monolayers and found that, although MDCK-WT monolayers express canine P-gp, the functional secretory transport of [3H]digoxin was ∼2-fold lower than that for Caco-2 cell monolayers and ∼4-fold lower than that for MDCK-MDR1 cell monolayers. Our data also demonstrate a similar relationship between secretory transport of [3H]digoxin in MDCK-WT and MDCK-MDR1 monolayers; i.e., BL-AP Papp of [3H]digoxin was ∼5-fold lower in MDCK-WT compared with MDCK-MDR1 cell monolayers (Table 1). However, our data suggest that this relationship is not consistent when various P-gp substrates are used as probes of P-gp-mediated secretory transport. For example, when [3H]paclitaxel and [3H]vinblastine were used as probe P-gp substrates, the difference between MDCK-MDR1 and MDCK-WT secretory transport was ∼2.5- and ∼3-fold, respectively (Table 1). This could be because of interspecies differences in binding site affinities on human and canine P-gp that are responsible for recognition and transport or possibly differences in the relative functional activities of human and canine P-gp toward these probe substrates.
We determined the effects of several known P-gp substrates, and one MRP2 substrate (probenecid), used as a control on the secretory transport of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin in MDCK-MDR1 and MDCK-WT monolayers. As shown in Table 2, known P-gp substrates such as cortisol (Ueda et al., 1992) and etoposide (Polli et al., 2001; Kim, 2002) did not significantly alter the BL-AP Papp of any of the P-gp substrate probes. Other compounds that have been described as P-gp substrates did not affect P-gp activity; e.g., colchicine (Kim, 2002), dexamethasone (Ueda et al., 1992), and trimethoprim (Polli et al., 2001; Susanto and Benet, 2002) demonstrated a slight inhibition or activation of BL-AP Papp for one or more P-gp-binding site probe substrates (Table 2). This modulatory effect was only statistically significant in three of 36 experiments: colchicine (100 μM) decreased [3H]vinblastine BL-AP Papp by ∼13% (P < 0.05), dexamethasone (100 μM) increased [3H]digoxin BL-AP Papp by ∼19% (P < 0.05), and trimethoprim increased [3H]digoxin BL-AP Papp by ∼12% (P < 0.01). Gao et al. (2001) have described an assay using Caco-2 monolayers and [3H]paclitaxel as a probe substrate where an apparent KI for P-gp interactions was calculated. Their assay specifically examined the effect of various test compounds on the paclitaxel-binding site of P-gp, and the authors state that this particular system does not differentiate P-gp substrates from inhibitors nor does it reveal any mechanistic information regarding the interaction of test compounds with P-gp. Because the P-gp substrates we evaluated (Table 2) did not elicit a marked change in BL-AP Papp of the P-gp substrate probes compared with control monolayers, this suggests that competitive inhibition of P-gp-mediated secretory transport is not necessarily discernible in monolayer transport experiments using P-gp-expressing cells. Yet, it should be noted that in vitro evaluation of P-gp activity and modulatory interactions can be inconsistent depending upon the inherent complexities of the assay system employed (Polli et al., 2001; Ekins et al., 2002b). For example, conflicting data have been reported for the interaction of verapamil with P-gp interactions when comparing data generated using intact cell monolayers (e.g., MDCK-MDR1 and Caco-2) to data generated using purified plasma membrane vesicles or preparations of membranes from P-gp-expressing cell lines (Polli et al., 2001).

Effect of ketoconazole on the BL to AP transport of [3H]vinblastine across MDCK-MDR1 (solid squares), MDCK-WT (open squares), and Caco-2 (solid circles) cell monolayers. [3H]Vinblastine at 100 nM was added to the donor (BL) compartment, and its BL-AP transport was monitored by sampling media from the AP compartment at various intervals over a 90-min incubation period. The effects of various concentration of ketoconazole on the BL to AP transport of [3H]vinblastine were determined: control (A), 0.1 (B), 1 (C), 3 (D), 10 (E), and 30 μM (F). Each symbol represents the mean ± S.D. (n = 3) monolayers.
Quinidine, a P-gp substrate and inhibitor (Emi et al., 1998), inhibited the BL-AP Papp of [3H]paclitaxel, [3H]vinblastine, and [3H]digoxin in MDCK-MDR1, MDCK-WT, and Caco-2 cell monolayers (Fig. 3). Interestingly, the BL-AP Papp of all probes was inhibited in MDCK-WT monolayers at concentrations ranging from 1 to 30 μM (Fig. 3A), whereas in MDCK-MDR1 monolayers, concentrations of quinidine were 30 μM before any inhibition of BL-AP Papp of substrate probe was noted (Fig. 3B). In Caco-2 monolayers, inhibition of [3H]paclitaxel and [3H]digoxin BL-AP Papp occurred at 3 to 10 μM, whereas quinidine concentrations up to 30 μM did not elicit an effect on [3H]vinblastine secretory transport (Fig. 3C). Unlike ketoconazole (Table 3 and Fig. 4) and loperamide (Table 3), the presence of quinidine at low (1–3 μM) concentrations did not elicit a stimulatory effect on the secretory transport of the P-gp substrate probes. Compounds that modulate P-gp activity (e.g., quinidine, verapamil, and ketoconazole) not only affect P-gp activity but can also affect drug uptake transporters, such as members of the organic anion-transporting polypeptide family (Cvetkovic et al., 1999). Therefore, it is possible that a polarized epithelial cell, such as MDCK, may use not only P-gp-mediated efflux from the AP membrane but it may also use uptake transporters expressed on the BL membrane (Goh et al., 2002), thereby accelerating the clearance of shared substrates for these transporters.
Ketoconazole activated [3H]vinblastine BL-AP Papp in MDCK-MDR1 monolayers at 1 and 3 μM (Table 3 and Fig. 4) yet did not elicit a similar effect on [3H]paclitaxel and caused a much less pronounced effect on [3H]digoxin BL-AP Papp (Table 3). Interestingly, ketoconazole did not elicit a similar stimulatory effect on [3H]vinblastine secretory transport in Caco-2 and MDCK-WT cell monolayers (Fig. 4). Low concentrations of loperamide elicited a similar stimulatory effect on [3H]vinblastine and [3H]digoxin BL-AP Papp in MDCK-MDR1 monolayers (Table 3). Thus, ketoconazole and loperamide (1–3 μM) activated P-gp-mediated secretory transport of [3H]vinblastine and [3H]digoxin in MDCK-MDR1 cell monolayers and inhibited their secretory transport at higher concentrations, whereas the other P-gp modulators that were evaluated (cyclosporine A, verapamil, and nicardipine) inhibited secretory transport of these probe P-gp substrates in a concentration-dependent manner but did not activate the secretory transport in any of the cell types evaluated (Table 3). MDCK-MDR1 monolayers express human P-gp but do not express other human efflux transporters, such as BCRP and MRP2, whereas Caco-2 cells express efflux transporters MRP2 and BCRP (Gutmann et al., 1999; Cummins et al., 2001; Taipalensuu et al., 2001). In Caco-2 cells, it is possible that MRP2 contributes to [3H]vinblastine secretory transport because it has been demonstrated that vinblastine is a substrate for MRP2 as well as P-gp (Evers et al., 1998; Chan et al., 2004). Because ketoconazole is a known inhibitor of P-gp, its presence may alter the mechanism of vinblastine secretory transport in Caco-2 cells, possibly favoring the MRP2-mediated transport pathway over transport via both P-gp and MRP2. It has been proposed that, when two compounds bind to P-gp at two distinct sites, a cooperative interaction can occur, resulting in a stimulation of drug transport (Litman et al., 1997; Shapiro and Ling, 1997; Shapiro et al., 1999). These data reinforce the importance of careful selection of models used to study P-gp-mediated secretory transport. In addition, certain cell lines will express other transporters that may complicate data interpretation, and there is also a potential for cooperative interactions involving P-gp (Litman et al., 1997; Shapiro and Ling, 1997).
Inhibition and/or activation of P-gp can potentially alter the pharmacokinetic profile of a drug, possibly resulting in concurrent changes in pharmacodynamic response. However, altering the tissue distribution of a drug that is a P-gp substrate, via coadministration of a P-gp inhibitor, can be therapeutically advantageous. The therapeutic effect of a drug that is a P-gp substrate can potentially be improved by inhibiting P-gp efflux activity; i.e., eliciting an increase in the level of drug entering a target tissue that is naturally protected by P-gp. This technique has been investigated in mice with the HIV protease inhibitor and P-gp substrate nelfinavir (Choo et al., 2000). In mdr1a wild-type mice, coadministration of the potent P-gp inhibitor LY335979 elicited a marked increase in the penetration of nelfinavir into the testes and brain, tissues that express P-gp and normally function as viral sanctuary sites for HIV. Ketoconazole had little or no effect on nelfinavir penetration into the brain in this study, a finding that may be attributable to its concentration-dependent and binding site-specific effect on P-gp activity, as we have demonstrated (Table 3 and Fig. 4).
To assess the possibility of triggering a P-gp-mediated DDI, it is essential to consider the potential impact of coadministered medications that can modulate P-gp activity. At the blood-brain barrier, the plasma concentration of an orally administered drug will be much lower than the relatively high concentration initially present in the gastrointestinal lumen immediately after oral administration. Thus, drugs that demonstrate activation of P-gp at low concentrations and inhibit P-gp at higher concentrations, such as ketoconazole and loperamide, could inhibit P-gp-mediated efflux in the gastrointestinal tract and activate P-gp-mediated efflux at the blood-brain barrier. We have shown in this study that ketoconazole and loperamide affect the efflux transport of P-gp substrates in not only a concentration-dependent but possibly also a binding site-dependent manner. These results are consistent with other published reports, demonstrating that the MDCK-MDR1 cell line is a useful model for studying the modulatory effects of various compounds on P-gp activity (Polli et al., 2001; Tang et al., 2002). Nevertheless, caution must be exercised when interpreting such results, as we have demonstrated by comparison of studies conducted using MDCK-MDR1 cell monolayers with those conducted using MDCK-WT and Caco-2 cell monolayers.
Acknowledgments
We thank Drs. Donald Tweedie and Richard Kim for insightful advice and critical review of the manuscript.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.005421.
-
ABBREVIATIONS: P-gp, P-glycoprotein; MDCK, Madin-Darby canine kidney cells; Caco-2, human adenocarcinoma cells; DDI, drug-drug interaction; WT, wild type; AP, apical; BL, basolateral; HIV, human immunodeficiency virus; TEER, trans-epithelial electrical resistance; AM, acetoxymethyl ester; HBSS, Hanks' balanced salt solution; MDR1, multidrug resistance protein-1; MRP2, multidrug resistance protein-associated protein-2; BCRP, breast cancer resistance protein; LY335979, zosuquidar trihydrochloride; GF120918, Elacridar.
- Received May 5, 2005.
- Accepted August 10, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}