


Abstract
Hydrolase activity from human liver and small intestine microsomes was compared with that of recombinant human carboxylesterases, hCE-1 and hCE-2. Although both hCE-1 and hCE-2 are present in human liver, the dominant component was found to be hCE-1, whereas the hydrolase activity of the human small intestine was found to be predominantly hCE-2. hCE-2 has a limited ability to hydrolyze large acyl compound substrates. Interestingly, propranolol derivatives, good substrates for hCE-2, were easily hydrolyzed by substitution of the methyl group on the 2-position of the acyl moiety, but were barely hydrolyzed when the methyl group was substituted on the 3-position. These findings suggest that hCE-2 does not easily form acylated intermediates because of conformational interference in its active site. In contrast, hCE-1 could hydrolyze a variety of substrates. The hydrolytic activity of hCE-2 increased with increasing alcohol chain length in benzoic acid derivative substrates, whereas hCE-1 preferentially catalyzed the hydrolysis of substrates with short alcohol chains. Kinetic data showed that the determining factor for the rate of hydrolysis of p-aminobenzoic acid esters was Vmax for hCE-1 and Km for hCE-2. Furthermore, the addition of hydrophobic alcohols to the reaction mixture with p-aminobenzoic acid propyl ester induced high and low levels of transesterification by hCE-1 and hCE-2, respectively. When considering the substrate specificities of hCE-1, it is necessary to consider the transesterification ability of hCE-1, in addition to the binding structure of the substrate in the active site of the enzyme.
The hydrolase activity of various tissues is increasingly used as the basis for drug design, particularly of prodrugs containing functional groups such as carboxylic acid esters (Bodor and Buchwald, 2002; Buchwald and Bodor, 2002). Introduction of an ester linkage generally improves the bioavailability of therapeutic agents, because of increased passive transport following oral administration. The requirements for a better oral prodrug are that it is stable to hydrolytic breakdown in its absorptive stage and that it is easily hydrolyzed to an active drug once it enters the systemic circulation (Beaumont et al., 2003). Carboxylesterases (CESs; EC.3.1.1.1) play an important role in biotransformation of a variety of ester-containing drugs and prodrugs such as angiotensin-converting enzyme inhibitors (e.g., temocapril, cilazapril, quinapril, and imidapril; Takai et al., 1997), antitumor drugs (CPT-11 and capecitabin; Humerickhouse et al., 2000; Tabata et al., 2004), and narcotics (cocaine, heroin, and meperidine; Pindel et al., 1997; Zhang et al., 1999).
CESs are members of the α/β-hydrolase family and show ubiquitous tissue expression profiles with high levels in the liver, small intestine, and lung (Satoh and Hosokawa, 1998; Satoh et al., 2002). CESs use a catalytic triad (Ser-His-Glu) for catalysis, which is located at the base of a deep catalytic gorge (Bencharit et al., 2002, 2003a,b). CES cleaves esters via a two-step process that involves the formation and degradation of an acyl-enzyme intermediate. First, the acyl carbonyl group of the substrate binds to the hydroxyl group of serine to produce an acyl-enzyme complex while the alcohol moiety is released. Then the acid component is released by an attack of histidine-activated water on this acyl-enzyme complex. The microenvironment surrounding the catalytic triad is important, not only for binding of substrate but also for the ease with which alcohol and/or acyl components can be released.
Mammalian CESs comprise a multigene family, in which the isozymes are classified into four main groups and several subgroups according to the homology of the amino acid sequence (Satoh and Hosokawa, 1998; Satoh et al., 2002). The majority of CESs belong to the CES1 and CES2 families, and are differentiated on the basis of substrate specificity, tissue distribution, immunological properties, and gene regulation. For example, hCE-1 (CES1A1, HU1), a human CES1 family isozyme, is widely distributed in many tissues but is only found at very low levels in the intestine, whereas hCE-2 (CES2A1, hiCE), a human CES2 family isozyme, is widely distributed in the intestine, liver, and kidney (Xu et al., 2002). Whereas hCE-1 preferentially catalyzes the hydrolysis of compounds esterified with a small alcohol group, hCE-2 hydrolyzes compounds with a relatively small acyl group and large alcohol group (Pindel et al., 1997; Takai et al., 1997; Satoh et al., 2002). In the case of cocaine, hCE-1 catalyzes the hydrolysis of the methyl ester of cocaine, producing benzoylecgonine and methanol, whereas hCE-2 catalyzes the hydrolysis of the benzoyl ester (Pindel et al., 1997). hCE-1 has also been found to catalyze the transesterification of cocaine with ethanol to generate cocaethylene (Brzezinski et al., 1994). Furthermore, hCE-1 has been reported to possess acyl coenzyme A:cholesterol acyltransferase activity, which generates cholesterol esters from fatty-acyl coenzyme A and free cholesterol (Becker et al., 1994). Therefore, bioconversion of orally administered ester-containing drugs is affected by the expression level of hCE-1 and hCE-2 in the liver and small intestine. Some reports have described greater expression of hCE-1 than hCE-2 in human liver and little expression of hCE-1 in human small intestine (Satoh et al., 2002; Xie et al., 2002). However, the extent to which the overall hydrolase activity of the liver and small intestine is dependent upon hCE-1 and hCE-2 activities has not been reported, and differences of substrate specificity between hCE-1 and hCE-2 have not been systematically demonstrated using structurally related compounds.
The purpose of the present study is to demonstrate the relative contributions of CES isozymes in the hydrolase activity of microsomes from human liver and small intestine, and to investigate differences in substrate specificity between hCE-1 and hCE-2 expressed in V79 and Sf9 cells using several structurally related compounds. In addition, we examine whether or not the transesterification activity of enzymes via acyl-CES intermediates affects substrate specificity for hCE-1 and hCE-2.
Materials and Methods
Materials.O-Acyl-propranolol hydrochloride was synthesized from propranolol hydrochloride (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and an appropriate acyl chloride (Tokyo kasei, Tokyo, Japan), according to previously described methods (Shameem et al., 1993). Flurbiprofen derivatives were synthesized from the appropriate alcohol and 2-(2-fluoro-4-biphenylyl)-propionyl chloride, which had been synthesized by flurbiprofen and SOCl2, according to a previously reported method (Imai et al., 1993). The identity and purity of the propranolol derivatives and the flurbiprofen derivatives were confirmed by IR, NMR, atomic analysis, and HPLC. Temocapril and temocaprilat were kindly provided by Sankyo Co., Ltd. (Tokyo, Japan). Betamethasone valerate, betamethasone, and 1-naphthylbutyrate were purchased from Wako Pure Chemical Industries (Tokyo, Japan). Benzoic acid derivatives were purchased from Tokyo Kasei Kogyo Co. Ltd. (Tokyo, Japan). Fast Red TR was purchased from Sigma (St. Louis, MO). Pooled human liver microsomes from 10 subjects (5 males and 5 females, 21–61 years old, of mixed Caucasian, Hispanic, and African-American races) and pooled small intestine microsomes from 10 subjects (4 males and 6 females, 24–63 years old, of mixed Caucasian, Hispanic, and African-American races) were obtained from BD Gentest (Woburn, MA). All other chemicals and reagents were of analytical grade.
Expression of hCE-1 and hCE-2. Expression of hCE-1 (GenInfo Identifier: 34740321) and hCE-2 (GenInfo Identifier: 37622885) in V79 cells was carried out as described previously (Mori et al., 1999; Hosokawa et al., 2001). The parental V79 cells [V79-4; American Type Cell Culture Collection (CL93), Manassas, VA] were cultured at cell densities of 1 × 106 cells/100-mm plate with D-MEM (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), penicillin/streptomycin (Invitrogen), and l-glutamine (Invitrogen) in an atmosphere of 5% CO2 and 95% air at 37°C. The next day, 10 μg of vector DNA (CES/pTARGET or pTARGET) was transfected into the cells using Lipofectamine Transfection Reagent (Invitrogen) with OPTI-MEM medium (Invitrogen). On day 3, the V79 cells were spread over 100-mm plates with media containing the selective agent, G418 (Invitrogen), at 400 to 800 μg/ml. The medium was changed every 2 to 3 days, and the cells were maintained for at least 3 weeks to obtain stable expression colonies. Ten colonies were identified and homogenized in SET buffer (0.25 M sucrose, 1 mM EDTA, 100 mM Tris-HCl buffer, pH 7.4), and the expression of hCE-1 or hCE-2 was determined by measurement of hydrolase activity, immunoblot analysis, and the molecular weight of a subunit. The background esterase activity in the mock V79 cells (V79 cells transfected by pTARGET), which were used as a control, was extremely low, comparable with the hydrolysis activity in buffer solution. These G418-resistant colonies could be stored in liquid N2 for several months without loss of expression level.
Recombinant hCE-1 was expressed in Sf9 cells using a BAC-TO-BAC Baculovirus Expression System (Invitrogen) according to a previously described procedure (Furihata et al., 2003). The cDNA in the cloning vector was subcloned into the pFAST BAC1 vector using EcoRI and alkaline phosphatase. The pFAST BAC1 vector containing hCE-1 was transformed into DH10Bac cells, and this was followed by transposition of the inserts into bacmid DNA. Likewise, nonrecombinant bacmid DNA (mock) was also prepared. The recombinant and mock bacmid DNAs were separately transfected into Sf9 cells with CELL FECTIN Reagent (Invitrogen), and the virus was harvested 72 h later. The cells were centrifuged at 1700g for 10 min to separate cells and virus. The supernatant containing the virus was stored at 4°C in the dark with 5% fetal bovine serum until required for use. Cells were routinely infected with the virus and were harvested 72 h after infection, washed twice with phosphate-buffered saline, and stored at –80°C until used. Lysates were prepared by disrupting the cells with a sonicator until the cells were completely lysed as determined by microscopy. Cytosol from Sf9 cells expressing hCE-1 and from Sf9 cells infected with mock virus were prepared by subjecting the cell lysate to centrifugation (105,000g for 60 min at 4°C). The expression of hCE-1 was identified by molecular weight of a subunit, immunoblot analysis, and hydrolase activity. The background esterase activity in the mock cells was extremely low, comparable to hydrolysis activity in buffer solution.
The recombinant hCE-1 and hCE-2 showed the same substrate specificity as purified human hCE-1 and hCE-2 in the preliminary experiment (data not shown), indicating expression of actual CES isozyme. Although the recombinant hCE-1 expressed in V79 cells showed the same pattern as hydrolysis of propranolol derivatives by hCE-1 expressed in Sf9 cells, the activity of the hCE-1 expressed in V79 cells was very low because of its low expression. Therefore, the activity of recombinant hCE-1 was evaluated using the recombinant hCE-1 in Sf9 cells.
Hydrolysis Experiments. Hydrolysis experiments were performed using tissue microsomes and the homogenates or cytosol of cells expressing CES. The initial hydrolytic activity was measured under reaction conditions such that less than 25% of substrate was hydrolyzed. Enzyme solutions were diluted to the appropriate concentration with HEPES buffer (50 mM, pH 7.4). The hydrolysis reaction was initiated by the addition of test compounds dissolved in dimethyl sulfoxide after preincubation of each subcellular fraction (200 μl) for 5 min. The final concentration of dimethyl sulfoxide was less than 1%, which has no effect on hydrolase activity. The reaction of benzoic acid derivatives was terminated by the addition of acetonitrile. After centrifugation, the supernatant was injected onto an HPLC column. For betamethasone valerate, propranolol derivatives, and flurbiprofen derivatives, the reaction was terminated by the addition of 5 ml of ethyl acetate. Saturated NaCl (0.5 ml), adjusted to pH 1 by phosphoric acid, was added to reaction samples of flurbiprofen derivatives, whereas 0.5 ml of saturated NaCl, adjusted to pH 4 by phosphate buffer, was added to the reaction samples of propranolol derivatives. After each sample had been shaken for 10 min, the isolated organic phase was evaporated and the residue was redissolved in HPLC mobile phase and injected onto an HPLC column. The rates of hydrolysis of aspirin and methyl salicylate were determined by the increase in fluorescence intensity of salicylic acid at ex 296 nm and em 405 nm over 7 min (Hitachi F-4500; Hitachi, Tokyo, Japan). The rate of hydrolysis of p-nitrophenylacetate was determined by the initial linear increase in absorbance of p-nitrophenol at 405 nm over 3 min (JASCO V530; JASCO Co., Tokyo, Japan). The rate of hydrolysis was determined by subtraction of the hydrolysis rate in buffer from that in tissue microsomes, and by subtraction of the hydrolysis rate in mock/cell preparations from that in CES/cell preparations. Hydrolytic activity was represented as the concentration of hydrolysis product per milligram of protein. Protein content was determined using the method described by Bradford (1976), with bovine serum albumin as standard.
HPLC Analysis. Concentrations of betamethasone, flurbiprofen, propranolol, and benzoic acid derivatives were determined by HPLC. The HPLC system comprised a JASCO PU-980 pump, a JASCO 980-UV detector, a JASCO AS950 autosampler, a JASCO CO-965 column oven, and a JASCO FP-1520S fluorescence detector, and a Shimadzu Chromatopac C-R7A plus (Shimadzu Co., Ltd., Kyoto, Japan). The column and composition of the mobile phase used for each assay are listed in Table 1. All substrates and hydrolysates were clearly separated. Each hydrolysate was measured in a quantitatively linear range.
Conditions for HPLC analysis
Polyacrylamide Gel Electrophoresis. Polyacrylamide gel electrophoresis (PAGE) was performed as described by Mentlein et al. (1980). Polyacrylamide gels (7.5% w/w) containing 1% w/v Nonidet P-40 for solubilization of proteins were used for the separation of native enzymes. After electrophoresis of the microsomes and S9 samples (5–15 μg of protein), the gels were stained for esterase activity with 1-naphthylbutyrate, through coupling of the liberated 1-naphthol with Fast Red TR-salt.
Inhibition Experiments for Hydrolysis of Valeryl-Propranolol Using Anti-hCE-1 Polyclonal Antibody. Anti-hCE-1 polyclonal IgG (Yamada et al., 1994) was purified from anti-hCE-1 rabbit serum using a Protein D column (Funakoshi, Tokyo, Japan). Human liver microsomes (1 mg protein/ml in pH7.4 HEPES buffer) were incubated with 0.5% cholic acid for 30 min on ice. After centrifugation at 10,000g for 30 min at 4°C, the supernatant was diluted to 25 μg protein/ml by 50 mM HEPES buffer (pH7.4) and then incubated with anti-hCE-1 IgG for 30 min at 37°C. The mixtures (200 μl) were allowed to stand for 24 h at 4°C before adding 30 μl of Protein A Sepharose 4 Fast Flow (GE Healthcare, Little Chalfont, Buckinghamshire, UK). After centrifugation at 20,000g for 5 min, the resulting supernatant was used to assay hydrolase activity for valeryl-propranolol. The control activity was measured using control rabbit IgG (Sigma).
Results
Hydrolase Activity of Human Liver and Small Intestine Microsomes.Figure 1 shows the hydrolase activity of human liver and small intestine microsomes for several substrates. In a preliminary study, bis(p-nitrophenyl)phosphate, a CES-specific inhibitor (Brandt et al., 1980), was found to inhibit the hydrolysis of a variety of compounds tested in this study by more than 90% (data not shown). In comparison with the liver microsomes, the small intestine microsomes showed lower activity for substrates with a large acyl group, for example, temocapril, p-nitrobenzoic acid methylester, and methyl salicylate. In contrast, betamethasone valerate and aspirin, substrates with a small acyl group, were hydrolyzed to either the same or a greater extent in the small intestine microsomes than in the liver microsomes. Furthermore, the substrate with the largest alcohol group, betamethasone valerate, was preferentially hydrolyzed in the small intestine microsomes.
Figure 2 shows the native PAGE gel stained by esterase activity using 1-naphthylbutyrate. The small intestine microsomes showed only one band corresponding to hCE-2, whereas the liver microsomes showed two bands, a strong upper band and a lower weak band, corresponding to hCE-1 and hCE-2, respectively. Therefore, the hydrolase activity of small intestine microsomes is due principally to hCE-2 activity, whereas that in the liver microsomes is attributable to both hCE-1 and hCE-2.

Hydrolysis of several ester-containing compounds by the human liver and small intestine microsomes. Concentrations in parentheses show the initial concentrations of substrate in the hydrolysis experiment.
Hydrolysis of Flurbiprofen Derivatives in Human Liver and Small Intestine Microsomes. Flurbiprofen ethylene glycol ester and trimethylene glycol ester were used as model compounds with large acyl groups. As shown in Fig. 3, both flurbiprofen derivatives were barely hydrolyzed in the small intestine microsomes, while being rapidly hydrolyzed in the liver microsomes. Moreover, the R-isomers were preferentially hydrolyzed in the liver microsomes. The recombinant hCE-1 showed a hydrolysis pattern similar to that of the human liver microsomes, although the latter expressed both hCE-1 and hCE-2. In common with the small intestine microsomes, the recombinant hCE-2 showed only low activity. A significant difference in the enantioselectivity of hydrolysis was not observed in recombinant hCE-2 or the small intestine microsomes due to the low activity observed.

Polyacrylamide gel electrophoresis of human liver (25 μg of protein) and small intestine (9.5 μg of protein) microsomes followed by staining for esterase activity using 1-naphthylbutyrate.

Structure of flurbiprofen derivatives and their hydrolysis in human liver and small intestine microsomes, and recombinant hCE-1 and hCE-2. Open and filled columns show hydrolase activity for S- and R-flurbiprofen derivatives, respectively. Protein concentrations: liver, 50 μg/ml; small intestine, 100 μg/ml; hCE-1, 50 μg/ml; hCE-2, 60 μg/ml; racemic concentration of ethylene glycol-flurbiprofen (EG-FP) and trimethylene glycol-flurbiprofen (TMG-FP): 250 μM; reaction time: liver and small intestine, 10 min; hCE-1, 20 min; hCE-2, 60 min; temperature: 37°C. Values represent the mean ± S.D. (n = 3). *, the difference between R- and S-isomers in the liver microsomes and recombinant hCE-1 was statistically significant (p < 0.05, n = 3).

Structure of propranolol (PL) derivatives.
Hydrolysis of Propranolol Derivatives.Figure 4 gives the structures of the propranolol derivatives used as model compounds with large alcohol groups. Compounds 1 to 6, 7 to 13, and 14 to 16 possess straight acyl chains, branched acyl chains, and cyclic acyl chains, respectively. The hydrolase activity of the human liver and small intestine microsomes and recombinant hCE-1 and hCE-2 are shown in Fig. 5. The human liver microsomes hydrolyzed R-isomers preferentially for all propranolol derivatives. The hydrolysis rates of compounds with straight acyl chains increased until butyryl propranolol (compound 3) and varelyl propranolol (compound 4) for R- and S-isomers, respectively. Although pivaroyl propranolol (compound 13) and cyclopropanoyl propranolol (compound 14) showed low hydrolysis rates, no relation between hydrolysis rate and structure was observed among branched and cyclic acyl derivatives. The recombinant hCE-1 showed a pattern of hydrolysis activity comparable to that of human liver microsomes for all propranolol derivatives.
The human small intestine and recombinant hCE-2 hydrolyzed both R- and S-form substrates at almost equal rates. The human small intestine and recombinant hCE-2 showed similar patterns for substrate specificity, including markedly lower activities for propranolol derivatives 9 and 11, as well as pivaroyl propranolol (13) and cyclopropanoyl propranolol (14). Interestingly, derivatives 9 and 11, with substituted methyl groups at the 3-position, were only slowly hydrolyzed, in contrast to the derivatives with substituted methyl groups at the 2-position (compounds 8 and 10), which were hydrolyzed as readily as their corresponding straight acyl derivatives (compounds 4 and 5, respectively). With the exception of compounds 9 and 11, R-isomers of propranolol derivatives were easily hydrolyzed by both hCE-1 and hCE-2 isozymes, whereas S-isomers were better substrates for hCE-2 than hCE-1.

Hydrolysis of propranolol derivatives in human liver and small intestine microsomes, and recombinant hCE-1 and hCE-2. Open and filled columns show hydrolase activity for S- and R-propranolol derivatives, respectively. Protein concentration: liver and small intestine, 10 μg/ml; recombinant, hCE-1, 30 μg/ml; hCE-2, 20 μg/ml; racemic concentration of substrate: 100 μM; reaction time: liver and small intestine, 5 min; hCE-1 and hCE-2, 15 min; temperature: 37°C. Values represent the mean ± S.D. (n = 3). There was a significant difference between R- and S-isomers for all substrates in the liver microsomes and recombinant hCE-1 (p < 0.05).
The major CES isozyme in human liver microsomes was identified using immunoinhibition experiments (Fig. 6). Racemic valeryl propranolol (4) was used as the substrate. Inhibition by anti-hCE-1 IgG (Yamada et al., 1994) was dose-dependent, and hydrolysis of R- and S-isomers was inhibited by 95% and 80%, respectively, at 500 μg/ml anti-hCE-1 IgG. The residual hydrolysis rates were nearly the same for R- and S-isomers, indicating that the residual activity was responsible for hCE-2. These data suggest that the major component of human liver hydrolase activity is hCE-1.
Hydrolysis of Benzoic Acid Derivatives. The hydrolysis of benzoic acid derivatives was determined in human small intestine and liver microsomes and the recombinant CES isozyme (Fig. 7). p- Amino-, o-amino-, and p-hydroxy-benzoic acid derivatives were used as substrates. Interestingly, the hydrolysis activity of recombinant hCE-1 and the liver microsomes decreased with increasing alcohol chain length of all types of benzoic acid derivatives. In contrast, recombinant hCE-2 and small intestine microsomes preferentially catalyzed those derivatives with longer alcohol chains. The responses toward benzoic acid derivatives showed that the hydrolase activity of the human liver and small intestine microsomes could be explained by hCE-1 and hCE-2, respectively.
To examine the different substrate requirements for hCE-1 and hCE-2, the kinetic parameters were determined using p-amino benzoic ethyl, propyl, and butyl esters. Km and Vmax values are listed in Table 2. It is interesting that the Km values for hydrolysis by recombinant hCE-1 varied little among the three compounds, but the Vmax value of the butyl ester was much smaller than that of the ethyl ester. Conversely, Vmax values for hydrolysis by recombinant hCE-2 were similar for all three compounds and Km values decreased with increasing alcohol chain length. These data suggest that the determining factor for the hydrolysis rate among the three p-amino benzoic acid esters is the Vmax value for hCE-1 and the Km value for hCE-2.
Kinetic parameters for hydrolysis of p-aminobenzoic acid ethyl, propyl, and butyl ester by hCE-1 and hCE-2
Km and Vmax were calculated by least squares method using the MULTI program (Yamaoka et al., 1981). Protein concentrations were: hCE-1, 45 μg /ml, and hCE-2, 25 μg /ml. Substrate concentrations were 10 to 500 μM, reaction time was 15 min, and temperature was 37°C. Values represent the mean ± S.D.
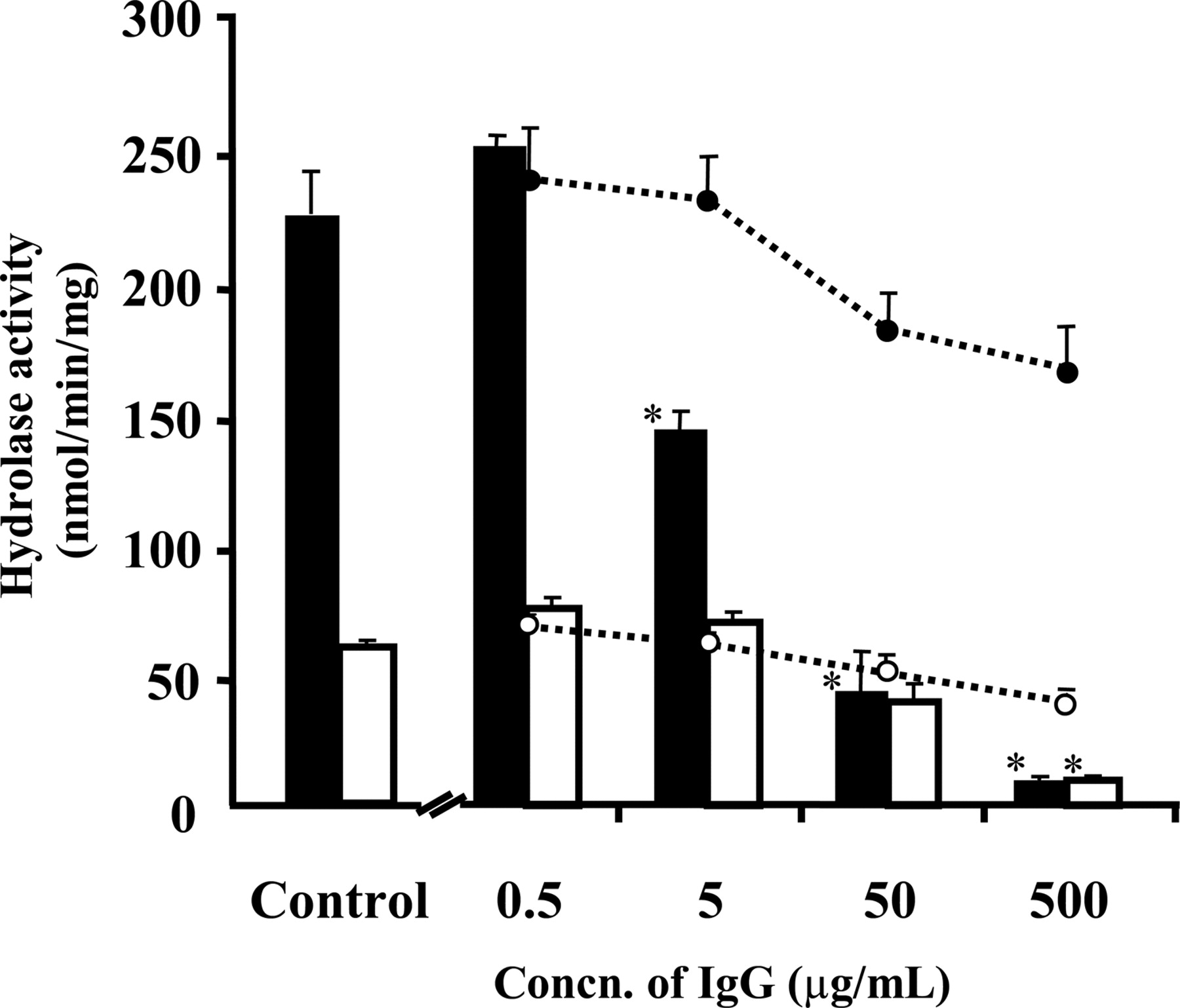
Effect of polyclonal anti-hCE-1 antibodies on hydrolase activity for racemic valeryl-propranolol in human liver microsomes. Columns and circles show hydrolase activity of human liver microsomes for valeryl-propranolol at 100 μM racemic concentration in the presence of anti hCE-1 rabbit IgG and rabbit IgG, respectively. Open columns and circles show the hydrolysis for S-valeryl-propranolol, and filled columns and circles indicate the hydrolysis for R-valeryl-propranolol. Values represent the mean ± S.D. (n = 3). * indicates statistically significant differences compared with hydrolase activity of control (p < 0.05).
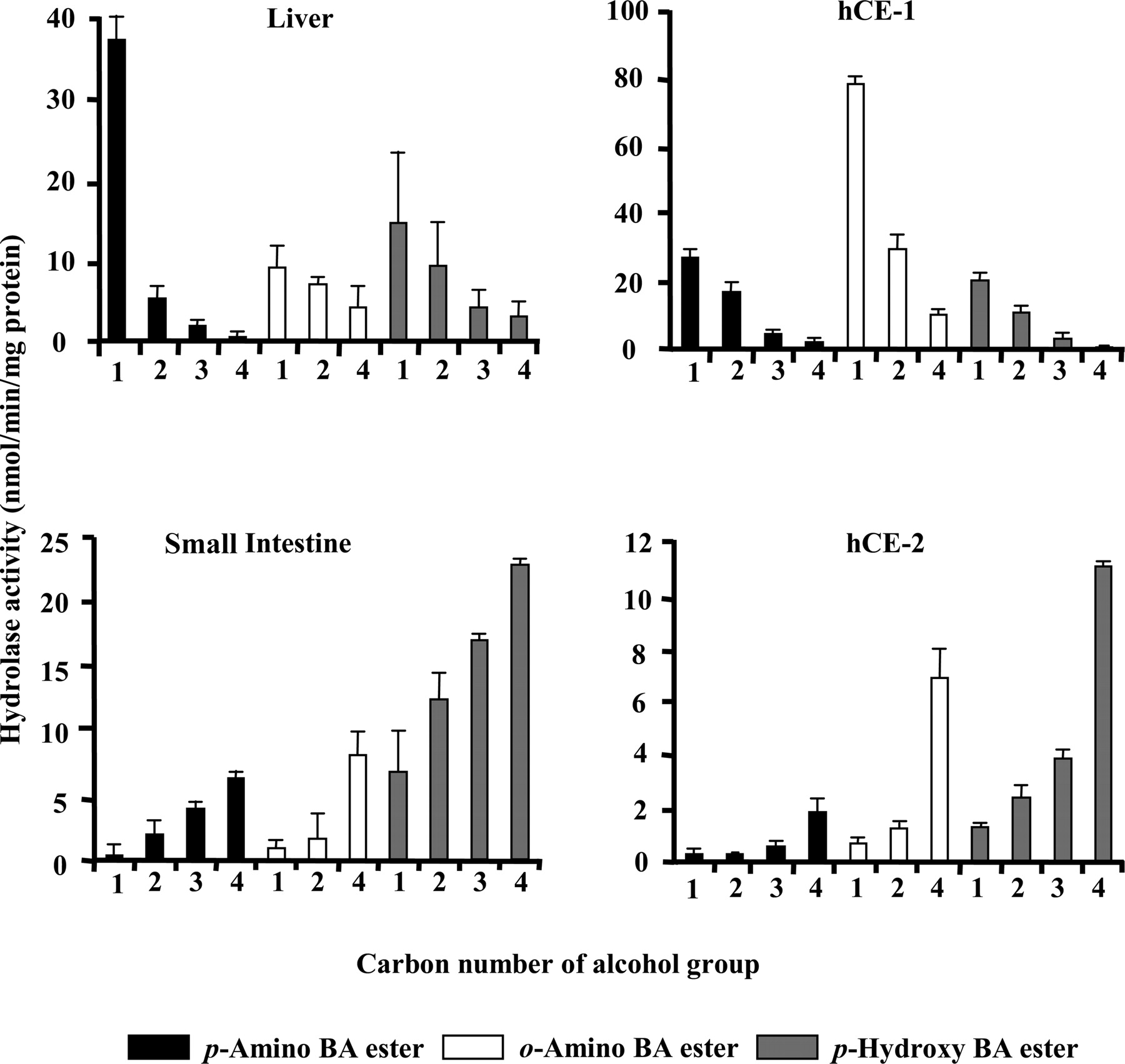
Hydrolysis of benzoic acid (BA) derivatives in human liver and small intestine microsomes, and recombinant hCE-1 and hCE-2. The numbers 1 to 4 indicate the carbon number of the alcohol group (1, methyl; 2, ethyl; 3, propyl; 4, butyl). Protein concentration: liver, 40 μg/ml; small intestine, 100 μg/ml; recombinant hCE-1, 40 μg/ml; hCE-2, 25 μg/ml; concentration of substrate: 500 μM; reaction time: liver and small intestine, 15 to 30 min; hCE-1 and hCE-2, 60 min; temperature: 37°C. Values represent the mean ± S.D. (n = 3).
Hydrolytic activity as a function of affinity for hCE-2 is a normal property of the enzyme reaction. However, hCE-1 showed a nearly identical Km value and increasing Vmax value with alcohol chain length. Since CES catalyzes ester hydrolysis in two steps, the Vmax value might depend on the binding velocity of the acyl group with the serine residue of CES and the releasing velocity of the acyl group from the acyl-enzyme intermediate upon attack of surrounding water. The acyl group for p-aminobenzoic acid derivatives is p-aminobenzoyl in all these substrates. The essential velocity for binding of the p-aminobenzoyl group with the serine residue of hCE-1 might be similar for the three ester compounds, given the invariance of the Km values and their similar structures. However, the velocity of the release of the p-aminobenzoyl group from the acyl-enzyme intermediate might be affected by alcohol released in the first step of the reaction. That is, interference may occur if the released alcohol can attack the acyl-enzyme intermediate. Therefore, p-aminobenzoic acid propyl ester was selected as a substrate, ethanol and butanol were added to the reaction mixture, and production of p-aminobenzoic acid and its ethyl or butyl ester was measured. The results are given in Table 3. Ethanol and butanol inhibited the production of p-aminobenzoic acid in reactions with both recombinant hCE-1 and hCE-2. The inhibition ratio was greater for hCE-1. The formation of ethyl ester on the addition of ethanol was nearly the same for hCE-1 and hCE-2. Interestingly, when the more hydrophobic alcohol, butanol, was added, p-aminobenzoic acid butyl ester was easily formed by hCE-1, but barely by hCE-2. These data suggest that hCE-1 catalyzes transesterification with hydrophobic rather than hydrophilic alcohols, and that hCE-2 possesses low transesterification ability.
Hydrolysis of p-amino benzoic acid (BA) propyl ester by hCE-1 and hCE-2 in the presence and absence of alcohol
Protein concentrations were: hCE-1, 45 μg/ml; hCE-2, 25 μg/ml. Substrate concentration was 500 μM; EtOH and BuOH concentrations were 0.5 to 25 mM. Reaction time was 60 min and temperature, 37°C. Values represent the mean ± S.D.
Discussion
The present study demonstrated that CES is the main hydrolyzing enzyme for ester compounds; suitable substrates are shown in Fig. 1. hCE-1 and hCE-2 were the predominant isoforms for hydrolysis in the human liver and small intestine, respectively. The predominance of hCE-2 activity in the small intestine was also confirmed by native PAGE, which showed only one band of hydrolase activity for 1-naphthylbutyrate (Fig. 2). In the liver, hydrolase activity was mainly attributed to hCE-1, although native PAGE revealed a relatively high hydrolase activity for hCE-2 in the liver microsomes (Fig. 2). As shown in Fig. 6, anti-hCE-1 antibody inhibited 80 to 95% of the hepatic hydrolysis of S- and R-valeryl-propranolol; the residual 5 to 20% of hydrolase activity was due to hCE-2, which showed nonenantioselective hydrolysis of valeryl-propranolol. This result closely agreed with a previous report showing the nearly complete inhibition of p-nitrophenylacetate hydrolase activity in human liver microsomes by anti-RH1 IgG, a cross-reactive antibody with hCE-1 (Hosokawa et al., 1995). These findings indicate the predominant contribution of hCE-1 to human hepatic hydrolase activity. Individual levels of both human liver CESs, hCE-1 and hCE-2, are highly variable. Xu et al. (2002) reported a 3-fold range variance for hCE-2 among 13 human liver microsomes, whereas Hosokawa et al. (1995) reported a more than 8-fold range variance of CES1 protein levels among 12 human liver microsomes. Therefore, in the present study, we used pooled human liver microsomes from 10 subjects whose expression levels of hCE-1 and hCE-2 showed interindividual differences. Our results nevertheless indicate that hCE-1 is the main component in human liver.
hCE-1 and hCE-2 exhibit 48% sequence identity, and differing substrate specificities have been proposed. That is, hCE-1 preferentially recognizes a substrate with large acyl and small alcohol moieties, whereas hCE-2 prefers substrates with smaller acyl and bulky alcohol moieties (Bosron and Hurley, 2002; Satoh et al., 2002). Our results regarding hydrolysis by hCE-2 concurred with this proposal; a bulky acyl moiety, such as flurbiprofen, was barely recognized by hCE-2, whereas propranolol derivatives with a small acyl moiety and bulky alcohol group were good substrates for hCE-2. These findings suggested the presence of sterically distorted regions, which affect the formation of an acyl-enzyme intermediate, in the active site of hCE-2. Interestingly, propranolol derivatives 9 and 11 (see Fig. 5) were scarcely hydrolyzed by hCE-2 or human small intestine microsomes. These compounds have a branched acyl moiety with a methyl group at the 3-position. In contrast, compounds 8 and 10 were easily hydrolyzed at almost the same rate as the corresponding straight acyl derivatives, although they possessed methyl groups at the 2-position. In general, the chemical hydrolysis of ester bonds is affected by substitution of the methyl group at the 2-position but not the 3-position, due to steric hindrance. The reduction in the hydrolysis rate resulting from substitution of the methyl group at the 3-position also suggests the presence of conformational interference in the active site of hCE-2.
In contrast to hCE-2, hCE-1 recognized substrates with either large or small acyl moieties. Flurbiprofen derivatives (a bulky acyl moiety) and R-propranolol derivatives (a small acyl moiety and bulky alcohol group) were recognized by hCE-1, although S-propranolol derivatives were poor substrates for hCE-1. Furthermore, acetyl derivatives such as p-nitrophenyl acetate and aspirin were hydrolyzed by both liver and small intestine microsomes (Fig. 1). The crystal structure of hCE-1 has been defined by Bencharit et al. (2003a,b). They reported that the substrate-binding site of hCE-1 was composed of a “small, rigid” pocket and a “large, flexible” pocket; the small, rigid pocket being selective, and the large, flexible pocket being promiscuous with regard to substrate specificity. These pockets allow hCE-1 to act on structurally distinct compounds containing either large or small acyl moieties. Therefore, R-propranolol derivatives might be easily recognized by hCE-1. In preliminary studies, we determined the Km and Vmax values for R- and S-butyryl-propranolol in human liver microsomes. The Km value (22.1 ± 0.8 μM) for the S-isomer was 10-fold smaller than that for the R-isomer (251 ± 18.8 μM), indicating the higher affinity of the S-enantiomer for hCE-1, despite the lower hydrolysis rate of the S-isomer. The Vmax value for S-butyryl-propranolol (95.7 ± 23.4 nmol/min/mg protein) was much smaller than that obtained for the R-enantiomer (1580 ± 4.0 nmol/min/mg protein). These preliminary findings suggest that both the R- and S-enantiomers are recognized by hCE-1, although their reactivity may be greatly affected by the conformational orientation upon incorporation into the active site pocket of hCE-1. It has been reported that S-cocaine and cis-cypermethrin analogs are poor substrates for hCE-1, in contrast to R-cocaine and trans-cypermethrin analogs (Brzezinski et al., 1997; Huang et al., 2005). The differences in hydrolysis rate between these enantiomers have been explained by steric clashes with the loop containing Gly142 and Gly143 in the rigid pocket (Bencharit et al., 2003b; Huang et al., 2005), where Gly residues form the oxianion hole (140–144) to stabilize the transition state of substrate via their amide nitrogen. Bencharit et al. (2003b) also determined the Z-site surface ligand-binding site for an inactive substrate. This Z-site is formed by interdigitation of the Ω1 and Ω2 loops adjacent to the active site of hCE-1. The enantioselective hydrolysis of propranolol derivatives is possibly explained by enantiomerically distinct active site orientations in hCE-1 due to structural clashes. Structural analysis will be required to establish the detailed nature of such enantioselectivity.
It was interesting that the substrate requirements for hydrolysis of various benzoate derivatives by hCE-1 and hCE-2 were quite different. Such substrate specificity might be explained by conformational fitting of substrates in the active sites. However, we propose that transesterification via hCE-1 may be due to different hydrolytic mechanisms in hCE-1 and hCE-2. CES-catalyzed hydrolysis proceeds in two steps. For benzoate derivatives, the first step is the formation of a covalent benzoyl-CES intermediate. At the same time, the released alcohol moiety is present in the active site gorge. Subsequently, the benzoyl-CES intermediate is attacked by histidine-activated water, and benzoic acid is released from the enzyme. Bencharit et al. (2002) reported the crystal structure of rabbit liver CES, in which 4-piperidino piperidine, a product of CPT-11 activation, was bound between the first N-acetylglucosamine of the Asn389 glycosylation site and the Trp550 side-chain of the C-terminal helix. There are two N-linked glycosylation sites in rabbit liver CES at Asn residues 79 and 389; Asn389 is modified via a long carbohydrate chain and plays a role as a novel exit pore for the release of small products from the active site of the enzyme. hCE-2 contains glycosylation sites at two positions (residues 103 and 267). Although there are no crystallographic data for hCE-2, one of these two glycosylation sites might function as an exit pore for the release of alcohol.
In contrast to hCE-2, hCE-1 maintains a glycosylation site at Asn79 but not at residue 389. Asn79 is modified by two N-acetylglucosamine groups and appears to be involved in the stabilization of the hCE-1 trimer by packing into the adjacent monomer (Bencharit et al., 2003a,b). hCE-1 is also capable of transesterifying cocaine in the presence of ethanol to cocaethylene (Brzezinski et al., 1994). During the two-step hydrolysis of cocaine, hCE-1 forms a covalent acyl-enzyme intermediate at the carboxylic methyl ester position of cocaine, which is then attacked by ethanol to create cocaethylene. Bencharit et al. (2003b) proposed the following mechanism based on their X-ray crystalline analysis. Ethanol enters the active site of hCE-1 through the side-door secondary pore adjacent to the large, flexible substrate-binding pocket. The entrance to the side-door secondary pore on the surface of hCE-1 is lined by structurally flexible residues including both β-strands (β14 and β15) and random coils. This flexibility may facilitate the passage of small molecules through the side-door. The alcohol produced may also be released from this side-door. The presence of the side-door secondary pore adjacent to the large, flexible substrate-binding pocket in hCE-1 might prolong the presence of the alcohol molecule so that the alcohol can attack the benzoyl-hCE-1 intermediate to reproduce the original substrate. Furthermore, the transition time of alcohol in hCE-1 might be prolonged with increasing hydrophobicity due to interaction of alcohol with the flexible site of hCE-1. This may be another explanation for the apparently slow hydrolytic rate of substrates with large alcohol moieties by hCE-1. Conversely, if hCE-2 can release an alcohol from the glycosylation site, its transesterification activity may be extremely low. This would agree with the observation of the low transesterification ability of hCE-2.
Although our proposal is a speculative mechanism of hydrolysis of hCE-1 and hCE-2, the fact that hCE-1 possesses transesterification activity suggests that, in addition to the binding structure of the substrate, this property should be considered in a complete analysis of the hydrolytic mechanism.
Footnotes
-
This work was supported in part by a Grant-in-Aid for Scientific Research (16590085) from the Japan Society for the Promotion of Science (T.I.)
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009381.
-
ABBREVIATIONS: CES, carboxylesterase; CPT-11, camptothecin-11 (irinotecan); hCE-1, human carboxylesterase-1; hCE-2, human carboxylesterase-2; HPLC, high-performance liquid chromatography; PAGE, polyacrylamide gel electrophoresis.
- Received January 19, 2006.
- Accepted July 11, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}