


Abstract
Carboxylesterases hydrolyze numerous endogenous and foreign compounds with diverse structures. Humans and rodents express multiple forms of carboxylesterases, which share a high degree of sequence identity (∼70%). Alignment analyses locate in carboxylesterases several functional subsites such the catalytic triad as seen in acetylcholinesterase. The aim of this study was to determine among human and rodent carboxylesterases the immunorelatedness, overlapping substrate specificity, differential sensitivity to serine enzyme inhibitors, tissue distribution, and tumor-related expression. Six antibodies against whole carboxylesterases or synthetic peptides were tested for their reactivity toward 11 human or rodent recombinant carboxylesterases. The antibodies against whole proteins generally exhibited a broader cross-reactivity than the anti-peptide antibodies. All carboxylesterases hydrolyzed para-nitrophenylacetate and para-nitrophenylbutyrate. However, the relative activity varied markedly from enzyme to enzyme (>20-fold), and some carboxylesterases showed a clear substrate preference. Carboxylesterases with the same functional subsites had a similar profile on substrate specificity and sensitivity toward phenylmethylsulfonyl fluoride (PMSF) and paraoxon, suggesting that these subsites play determinant roles in the recognition of substrates and inhibitors. Among three human carboxylesterases, HCE-1 hydrolyzed both substrates to a similar extent, whereas HCE-2 and HCE-3 showed an opposite substrate preference. All three enzymes were inhibited by PMSF and paraoxon, but they showed a marked difference in relative sensitivities. Based on immunoblotting analyses, HCE-1 was present in all tissues examined, whereas HCE-2 and HCE-3 were expressed in a tissue-restricted pattern. Colon carcinomas expressed slightly higher levels of HCE-1 and HCE-2 than the adjacent normal tissues, whereas the opposite was true with HCE-3.
Carboxylesterases structurally belong to a superfamily of α/β-fold proteins, which consist of alternate α-helix and β-sheets connected by loops with a varying length (Oakeshott et al., 1999). These enzymes hydrolyze chemicals containing such functional groups as a carboxylic acid ester, amide, and thioester (Parkinson, 1995; Satoh and Hosokawa, 1998). In the presence of an alcohol, carboxylesterases catalyze transesterification reaction, which accounts for the conversion of cocaine to ethylcocaine (Boyer and Petersen, 1992). Carboxylesterase activity is widely distributed in mammalian tissues, with the highest levels present in liver microsomes (Satoh and Hosokawa, 1998). High abundance of carboxylesterases in the liver is linked to certain cellular structural functions, particularly in directing protein targeting (Medda et al., 1987). For example, egasyn, a liver microsomal carboxylesterase, binds to β-glucuronidase via its active site, which retains this enzyme within the endoplasmic reticulum. In rabbits, microsomal carboxylesterases are shown to interact with and regulate the secretion of acute-phase response proteins, such as C-reactive protein (Macintyre et al., 1994). Given the fact that many therapeutic agents are esters or amides, carboxylesterase-mediated hydrolysis is given an important consideration in drug designs (Graffner-Nordberg et al., 1998; Buchwald and Bodor, 1999).
Multiple forms of carboxylesterases are identified in several mammalian species (Satoh and Hosokawa, 1998). Studies on cDNA cloning have identified multiple distinct gene products in rodents and humans, but the number is far less than that identified by electrophoresis (Mentlein et al., 1987; Satoh and Hosokawa, 1998). The cDNA-deduced amino acid sequences of these carboxylesterases have several notable features. They are synthesized as a larger precursor with an N-terminal signal peptide commonly seen in secretory proteins (Yan et al., 1994,1995; Satoh and Hosokawa, 1998). Carboxylesterases have four conserved cysteine residues and one or more putative N-glycosylation sites. A catalytic triad composing Ser, His, and Glu is located as that seen in acetylcholinesterase (Sussman et al., 1991). Some carboxylesterases have a C-terminal HXEL consensus sequence, through which the esterases are retained in the endoplasmic reticulum (Yan et al., 1995). Finally, mature carboxylesterases contain approximately 540 residues with a high degree of sequence identity (∼70%), even cross-species. The N-terminal half is highly conserved, whereas the C-terminal half is relatively diverse.
Carboxylesterases are shown to hydrolyze such compounds as choline esters, phospholipids, retinyl esters, and cholesteryl esters (Mentlein et al., 1984; Ghosh et al., 1995; Sun et al., 1997; Barr et al., 1998), suggesting that these esterases play an important physiological role. However, studies on carboxylesterases largely focus on detoxication of pesticides and metabolism of drugs and other xenobiotics (Satoh and Hosokawa, 1998). Carboxylesterases are structurally similar to acetylcholinesterase, a well established target of organophosphorus and carbamate insecticides during acute exposure (Sussman et al., 1991). Binding of carboxylesterases to these insecticides, therefore, is considered as a detoxication pathway. Tissue-specific expression of carboxylesterases is shown to determine the location and intensity of certain drugs. For example, serum carboxylesterase has a markedly higher rate at hydrolyzing procaine than its amide analog procainamide; therefore, procaine is used mainly as a local anesthetic, whereas procainamide is used for systematic indications such as cardiac arrhythmia (Parkinson, 1995). Carboxylesterase-based metabolism is used for activation of ester prodrugs. For example, irinotecan is rapidly converted by carboxylesterases to 7-ethyl-10-hydroxycamptothecin, a potent inhibitor of topoisomerase I (Saltz, 1997). Irinotecan is widely used for a wide range of malignancies. However, some patients respond to it poorly, presumably due to lower levels or polymorphistic variants of carboxylesterases (Canal et al., 1996; Saltz, 1997).
The aim of this study was to determine among human and rodent carboxylesterases the immunorelatedness, overlapping substrate specificity, differential sensitivity to serine enzyme inhibitors, tissue distribution, and tumor-related expression. Six antibodies against whole carboxylesterases or synthetic peptides were tested for their reactivity toward 11 human or rodent recombinant carboxylesterases. The antibodies against whole proteins generally exhibited a broader cross-reactivity than the anti-peptide antibodies. All carboxylesterases hydrolyzed para-nitrophenylacetate and para-nitrophenylbutyrate. However, the relative activity varied markedly from enzyme to enzyme (>20-fold), and some carboxylesterases showed a clear substrate preference. Among three human carboxylesterases, HCE-11 hydrolyzed both substrates to a similar extent, whereas HCE-2 and HCE-3 showed an opposite substrate preference. All three enzymes were inhibited by PMSF and paraoxon, but they showed a marked difference in relative sensitivities. HCE-1 was present in all tissues examined, whereas HCE-2 and HCE-3 were expressed in a tissue-restricted pattern. Colon carcinomas expressed slightly higher levels of HCE-1 and HCE-2 than the adjacent normal tissues, whereas the opposite was true with HCE-3.
Materials and Methods
Chemicals and Supplies.
Paraoxon, PMSF, para-nitrophenylacetate, andpara-nitrophenylbutyrate were from Sigma Chemical (St. Louis, MO). The goat anti-rabbit IgG conjugated with alkaline phosphatase or horseradish peroxidase and chemiluminescent substrate were from Pierce Chemical (Rockford, IL). Human tissue homogenates (protein medley) were from BD Biosciences Clontech (Palo, Alto, CA). Cell culture media, liver cDNA libraries, LipofectAMINE, and Plus Reagent were purchased from Invitrogen (Carlsbad, CA). Anti-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Unless otherwise indicated, all other reagents were purchased from Fisher Scientific (Pittsburgh, PA).
Molecular Cloning of Human, Rat, and Mouse Carboxylesterases.
Screening of cDNA libraries (human liver and placenta, rat liver, mouse liver) was conducted with a Gene-Trapp cDNA positive selection system (Invitrogen) as described previously (Hu and Yan, 1999). Briefly, double-stranded phagemid DNA was isolated from the cDNA library and converted to ssDNA by a sequential digestion with Gene II and Exo III. The ssDNA was denatured at 95°C for 1 min and chilled in ice for 1 min. Two oligonucleotides (TGTGACCATCTTTGGAGAGTC and TTTGGCGAGTCTGCGGGTGGC) were synthesized based on the sequence encoding a motif (GXSXG) common to all cloned mammalian carboxylesterases (Satoh and Hosokawa, 1998). The oligonucleotides (3 μg) were then biotinylated in a total volume of 25 μl with biotin-14-dCTP and terminal deoxynucleotidyl transferase. Hybridization between the ssDNAs (2.5 μg) from the library and the biotinylated oligonucleotides (20 ng) were conducted at 37°C for 1 h with the hybridization buffer provided with the kit. The hybridized ssDNAs were then captured by streptavidin-coated beads and repaired by a thermostable polymerase. The repaired ssDNAs were then transformed into HB101 bacteria. For sequencing, the phagemid DNA was isolated with a Spin Mini-prep kit (QIAGEN, Chatsworth, CA) and sequenced with vector/internal synthetic primers on both strands (Yan et al., 1995).
Transfection.
Human embryonic kidney cells (293T) were plated at a density of 60% in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. After reaching 85% confluence, cells were transfected by LipofectAMINE and Plus Reagent. A plasmid construct or the empty vector (4 μg/100-mm dish) was initially mixed with 20 μl of Plus Reagent diluted in 750 μl of serum-free medium for 15 min, and then mixed with 30 μl of LipofectAMINE diluted in 5 ml of serum-free medium for 15 min. The final transfection complexes were added to a monolayer of 293T cells. After a 3 h-incubation, the media were replaced by normal culture media and incubated for 48 h in a 37°C humidified incubator with 5.0% CO2. Cells were rinsed and harvested in 1.5 ml of phosphate-buffered saline. The cell suspension was sonicated by a Branson sonifier, and cell debris was removed by centrifugation at 12,000g for 10 min at 4°C. The supernatant was assayed for hydrolytic activity towardpara-nitrophenylacetate andpara-nitrophenylbutyrate.
Enzymatic Assays and Inhibition Studies.
The enzymatic activity was determined spectrophotometrically as described previously (Morgan et al., 1994). Unless otherwise indicated, the 1-ml sample cuvette contained 20 μg of lysates from transfected cells in 100 mM potassium phosphate buffer, pH 7.4, and 1 mM substrate at room temperature. Reactions were initiated by the addition of substrate (10 μl of 100 mM stock in acetonitrile), and hydrolytic rate was recorded from an increase in absorbance at 400 nm. The same experiments were conducted without cell lysates to correct for nonenzymatic hydrolysis. The extinction coefficient (E400) was determined to be 13 mM−1 cm−1(para-nitrophenylacetate) or 17 mM−1cm−1 (para-nitrophenylbutyrate). All assays were repeated at least three times. For inhibition studies, lysates (20 μg of protein in 1 ml of 100 mM potassium phosphate buffer) were incubated at room temperature with various concentrations of PMSF or paraoxon. After a 5-min incubation, the rate ofpara-nitrophenylacetate hydrolysis was spectrophotometrically measured as described above.
Homogenate Preparation from Human Colon Adenocarcinoma and Adjacent Normal Tissues.
Tissues were obtained from the Cooperative Human Tissue Network. Colon adenocarcinomas and the adjacent normal tissues (∼0.5 g) were collected from patients who underwent subtotal colon resection. The size of tumors was ∼5 cm in diameter, and the degree of differentiation of tumors was moderate or poor as determined by pathological examination. Tissues (∼0.5 g) were homogenized in buffer containing 20 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40, and a set of protease inhibitors (0.1 mM PMSF, 10 μg/ml pepstatin, 10 μg/ml leupeptin). The tissue homogenates were subjected to centrifugation at 35,000g for 60 min at 4°C. The supernatants were analyzed for the abundance of carboxylesterases by Western blots as described previously (Yan et al., 1995). Goat anti-rabbit IgG conjugated with alkaline phosphatase or horseradish peroxidase was used. The phosphatase activity was detected with colorimetric substrates nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate, whereas the peroxidase activity was detected with chemiluminescent substrate. Use of human tissues was approved by the University Institutional Review Committee.
Other Assays.
Immunization and antibody purification were described elsewhere (Zhu et al., 2000). Protein concentration was determined with a BCA kit (Pierce Chemical) as described by the manufacturer. Data are presented as mean ± S.D. of at least three separate experiments except where results of blots are shown, in which case a representative experiments is depicted in the figures.
Results
Immuno-cross Reactivity.
Mammalian species express multiple forms of carboxylesterases (Satoh and Hosokawa, 1998). These enzymes have a similar molecular mass (∼60 kDa) and share a remarkably high degree of sequence identity, particularly in the N-terminal half. As a result, some of the enzymes are indistinguishable on conventional Western blots. In the past several years, we have prepared antibodies against several major types of human or rodent carboxylesterases, either against whole carboxylesterases or peptides derived from some of these enzymes (Table1). These antibodies were tested for their cross-reactivity toward 11 human or rodent recombinant carboxylesterases. The enzymes included human HCE-1, HCE-2, HCE-3, and HCE-1 variant, rat hydrolase A (HA), B (HB), S (HS), and E, and mouse M-LK, M-E, and M-S. cDNAs encoding these enzymes were isolated by screening individual rat or mouse liver library as well as combined human liver and placental libraries (Hu and Yan, 1999; Yan et al., 1999). The isolated cDNAs were subjected to sequencing analyses, and identification of each cDNA was made based on the basic local alignment search tool search (≥98%). For clarity, the names used in this report and the corresponding names originally reported for each enzyme are summarized in Table 2. It should be emphasized that HCE-1 variant was originally identified to have two single amino acid deletions: alanine and glutamine at positions of −1 and 435, respectively (Kroetz et al., 1993). However, the cDNA encoding the variant in this study was found to have the alanine deletion only.
Antigens and antibody preparation
Carboxylesterases, species, and highly identical enzymes
Immunoreactivity was determined by Western blots. Cells (293T) were transiently transfected with the empty vector or a construct encoding a carboxylesterase, and cell lysates were prepared by sonication. Six antibodies (Table 1) were tested for their reactivity toward each recombinant carboxylesterase. As shown in Fig.1, all antibodies reacted strongly with respective recombinant carboxylesterases. Antibodies against whole carboxylesterases (e.g., anti-HA) exhibited a broader reactivity than anti-peptide antibodies (e.g., anti-HCE-3). The antibody against hydrolase S cross-reacted with all carboxylesterases to a comparable extent with an exception of HEC-2. All other antibodies generally displayed either a strong or a weak cross-reactivity (no medium). The anti-human carboxylesterase antibodies (anti-HCE-1, HCE-2, and HCE-3) showed little cross-reactivity toward each other with an exception of the anti-HCE-1 antibody, which reacted comparably with both HCE-1 and HCE-3. Finally, anti-HA, anti-HB, and anti-HCE-1 detected a minor product with a markedly higher electrophoretic mobility in some cell lysates. This minor product appeared to be a degraded product of recombinant carboxylesterases, particularly in the case when anti-HCE-1 antibody was used because this antibody was raised against a peptide derived from the C terminus (Table 1).

Immuno-cross-reactivity of human, rat, and mouse carboxylesterases.
Cells (293T) were transfected with an empty vector or a cDNA construct encoding a carboxylesterase. Cell lysates were prepared by sonication and centrifugation, as described under Materials and Methods. Lysates (4 μg) from transfected cells or pooled microsomes (1 μg) from rat or human liver were subjected to SDS-polyacrylamide gel electrophoresis. Samples were transferred electrophoretically to a Trans-Blot nitrocellulose membrane. The immunoblots were blocked in 5% nonfat dry milk and then incubated with the antibody (10 μg/ml) against HA, HB, HS, HCE-1, HCE-2, or HCE-3. The primary antibody was then located by alkaline phosphatase-conjugated goat anti-rabbit IgG. HCE-1* is identical to HCE-1 but has a longer 5′-untranslated region. The figure is representative of results obtained in three independent experiments. Carboxylesterases and their corresponding reported name(s) are compared based on the basic local alignment search tool search (Table 2). HE, hydrolase E.
To test whether the length of 5′-untranslated region influenced the expression of recombinant proteins, we included two HCE-1 clones with the HCE-1* having a longer 5′-untranslated region. As shown in Fig. 1, both constructs (HCE-1 and HCE-1*) yielded comparable intensities of immunostaining by all antibodies, suggesting that both constructs are transcribed/translated to a similar extent and the transient transfection experiments produce consistent results. Similarly, HCE-1v (an alanine deletion in the signal peptide) yielded a comparable immunostaining to HCE1, suggesting that this deletion exerts little effect on the overall expression of this variant (e.g., stability of mRNA and processing of the precursor). In addition, three individual transfection experiments were conducted with all constructs, and similar immunostaining intensities were consistently obtained (data not shown).
Hydrolysis of para-Nitrophenylacetate andpara-Nitrophenylbutyrate.
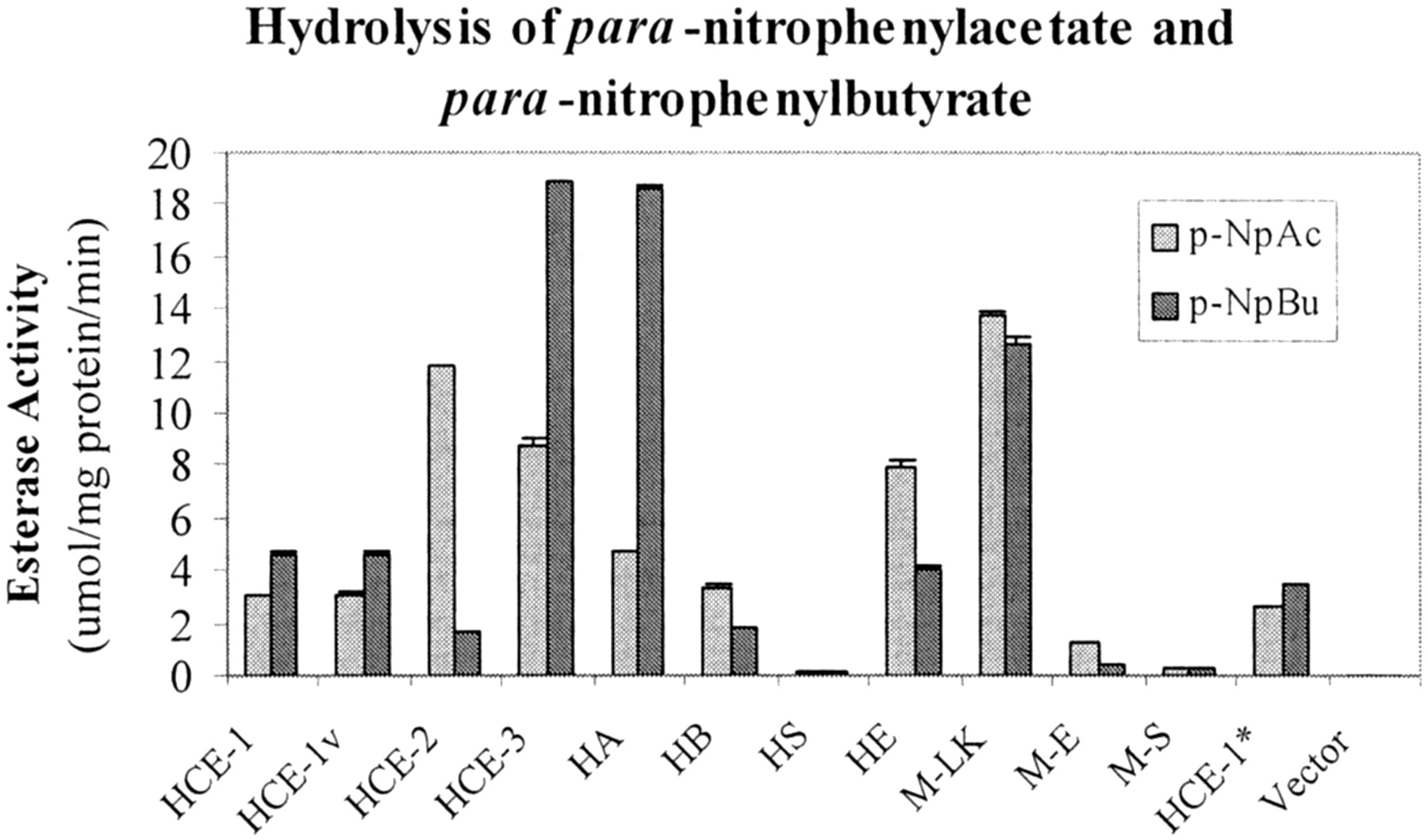
Next, we examined the hydrolytic activity of these carboxylesterases toward para-nitrophenylacetate andpara-nitrophenylbutyrate. These two esters were chosen for the following reasons. We previously demonstrated that human placental microsomes differentially hydrolyzed both substrates (Yan et al., 1999). More importantly, their structural relationship resembles that between acetyl- and butyrylcholine, two chemicals that are differentially hydrolyzed by acetyl- and butyrylcholinesterase (Chatonnet and Lockridge, 1989). As shown in Fig.2, transfection with the empty vector caused little hydrolytic activity toward both substrates. In contrast, transfection with plasmid constructs encoding carboxylesterases generally caused a marked increase in hydrolytic activity with two notable exceptions: rat hydrolase S and mouse M-S. The maximum activity (∼12 μmol/mg/min) toward para-nitrophenylacetate was observed in the lysates from cells transfected with HCE-2 and M-LK. In contrast, the maximum activity (18 μmol/mg/min) towardpara-nitrophenylbutyrate was observed in the lysates from cells transfected with HCE-3 and hydrolase A. Samples exhibiting similar immunostaining intensity had a marked difference in hydrolyzing the substrates assayed. Lysates from HCE-1 and HCE-3, for example, yielded a comparable immunostaining intensity by the anti-HCE-1 antibody; however, HCE-3 lysates exhibited a markedly higher activity than HCE-1 lysates (3- to 4-fold depending on the substrate used). In addition, some carboxylesterases hydrolyzed both substrates to a comparable extent, whereas others exhibited a clear substrate preference. For example, HCE-1 exhibited no apparent preference toward either substrate. In contrast, hydrolase A preferably hydrolyzedpara-nitrophenylbutyrate at a ratio of 4 overpara-nitrophenylacetate, whereas the opposite was true with carboxylesterases such as HCE-2 (a ratio of 6). These findings suggest that intrinsic catalytic properties of carboxylesterases are largely responsible for the observed difference on the overall hydrolytic activity among these enzymes although the difference in the expression levels is a likely contributing factor as well.

Hydrolytic activity of lysates from transfected cells toward para-nitrophenylacetate or para-nitrophenylbutyrate.
Lysates (20 μg) from cells transfected with an empty vector or a cDNA construct encoding a carboxylesterases were assayed for their activity to hydrolyze para-nitrophenylacetate orpara-nitrophenylbutyrate. The hydrolytic activity was spectrophotometrically determined at room temperature with 1.0 mM substrate. All assays were performed with individual samples (three transfection experiments) and repeated three times. Hydrolytic rates were expressed as the mean ± S.D. (micromoles per milligram of protein per minute).
Differential Inhibition by PMSF and Paraoxon.
Site-directed mutagenesis studies on cholinesterases demonstrate that mutants with an increase in hydrolyzing certain substrates also increase the reactivity toward a structurally related inhibitor (Vellom et al., 1993). Next, we examined whether the hydrolytic activity of carboxylesterase was correlated with their sensitivity to paraoxon or PMSF, two well-known serine enzyme inhibitors (Ordentlich et al., 1996; Das et al., 2000). As shown in Fig.3, both compounds effectively inhibited hydrolytic activity toward para-nitrophenylacetate in a concentration-dependent manner. Rat hydrolase S and mouse M-S were excluded from the inhibition studies because both had little activity toward this substrate (Fig. 2). Overall, PMSF is less potent than paraoxon and PMSF-mediated inhibition exhibited a greater variation than paraoxon-mediated inhibition from enzyme to enzyme (Fig. 3A). For example, PMSF at a concentration of 2 μM caused an inhibition of hydrolase A by as much as 95%, whereas the same concentration caused little change on M-LK-mediated hydrolysis. Some carboxylesterases exhibited a good correlation between hydrolytic activity and sensitivity to both inhibitors. The hydrolytic activity of rat hydrolase E toward para-nitrophenylacetate andpara-nitrophenylbutyrate (Fig. 2), for example, was higher than that of mouse M-E, and so was the sensitivity of hydrolase E to PMSF and paraoxon inhibition (Fig. 3). Among human carboxylesterases, HCE-3 was the most sensitive to PMSF, whereas HCE-1 and HCE-2 were moderately inhibited. In contrast, HCE-2 exhibited a marked resistance to paraoxon inhibition, whereas HCE-1 and HCE-3 were comparably inhibited (Fig. 3).

Differential inhibition of hydrolytic activity of carboxylesterases by PMSF and paraoxon.
Lysates (20 μg/ml) from transfected cells were incubated at room temperature with various concentrations of PMSF or paraoxon for 5 min before the addition of 1 mM para-nitrophenylacetate. Rates in the presence of an inhibitor were recorded as a percentage of the corresponding rates in the absence of this inhibitor (100%). All assays were performed in triplicate.
Tissue Distribution of HCE-1, HCE-2, and HCE-3.
Hydrolytic activity is present ubiquitously in mammalian tissues; however, to what extent carboxylesterases contribute to this activity remains unclear (Satoh and Hosokawa, 1998). We have demonstrated that antipeptide antibodies are highly specific among human carboxylesterases (Fig. 1). Therefore, we used these antibodies to determine tissue distribution of HCE-1, HCE-2, and HCE-3. Overall, the anti-HCE-1 antibody detected cross-reactive proteins among all tissues tested (Fig. 4). The immunostaining intensity was comparable with exceptions of the liver, placenta, spleen, and brain. The liver had the highest immunostaining intensity, whereas the placenta, spleen, and brain showed the weakest intensity. In contrast, cross-reactive proteins with HCE-2 or HCE-3 were more tissue-restricted. High levels of HCE-2 cross-reactive proteins were detected in the liver, kidney, and ileum, whereas high levels of HCE-3 cross-reactive proteins were detected in the adrenal and brain. Some organs such as the kidney, adrenal, and ileum expressed cross-reactive proteins detected by all three antibodies. The antibody against HCE-1 cross-reacts with HCE-3, whereas the antibody against HCE-2 or HCE-3 is highly specific to respective enzyme (Fig. 1), suggesting that these organs express at least HCE-2 and HCE-3 (anti-HCE-1 cross-reacts with HCE-3). Interestingly the liver expressed no HCE-3 although this organ contains the highest levels of many xenobiotic-metabolizing enzymes.

Tissue distribution of HCE-1, HCE-2, and HCE-3.
Microsomes (5 μg) from the liver, kidney, and placenta or homogenates (10 μg) from other organs were subjected to SDS-polyacrylamide gel electrophoresis. Samples were transferred electrophoretically to a Trans-Blot nitrocellulose membrane. The immunoblots were detected with the antibody (10 μg/ml) against HCE-1, HCE-2, or HCE-3. The primary antibody was then located by alkaline phosphatase- or horseradish peroxidase-conjugated goat anti-rabbit IgG. The alkaline phosphatase activity was detected by colorimetric substrate (HCE-1 and HCE-2), whereas peroxidase activity was detected by chemiluminescent substrate (HCE-3). As controls, lysates from respective transfected cells were analyzed simultaneously.
Tumor-Related Expression of HCE-1, HCE-2, and HCE-3.
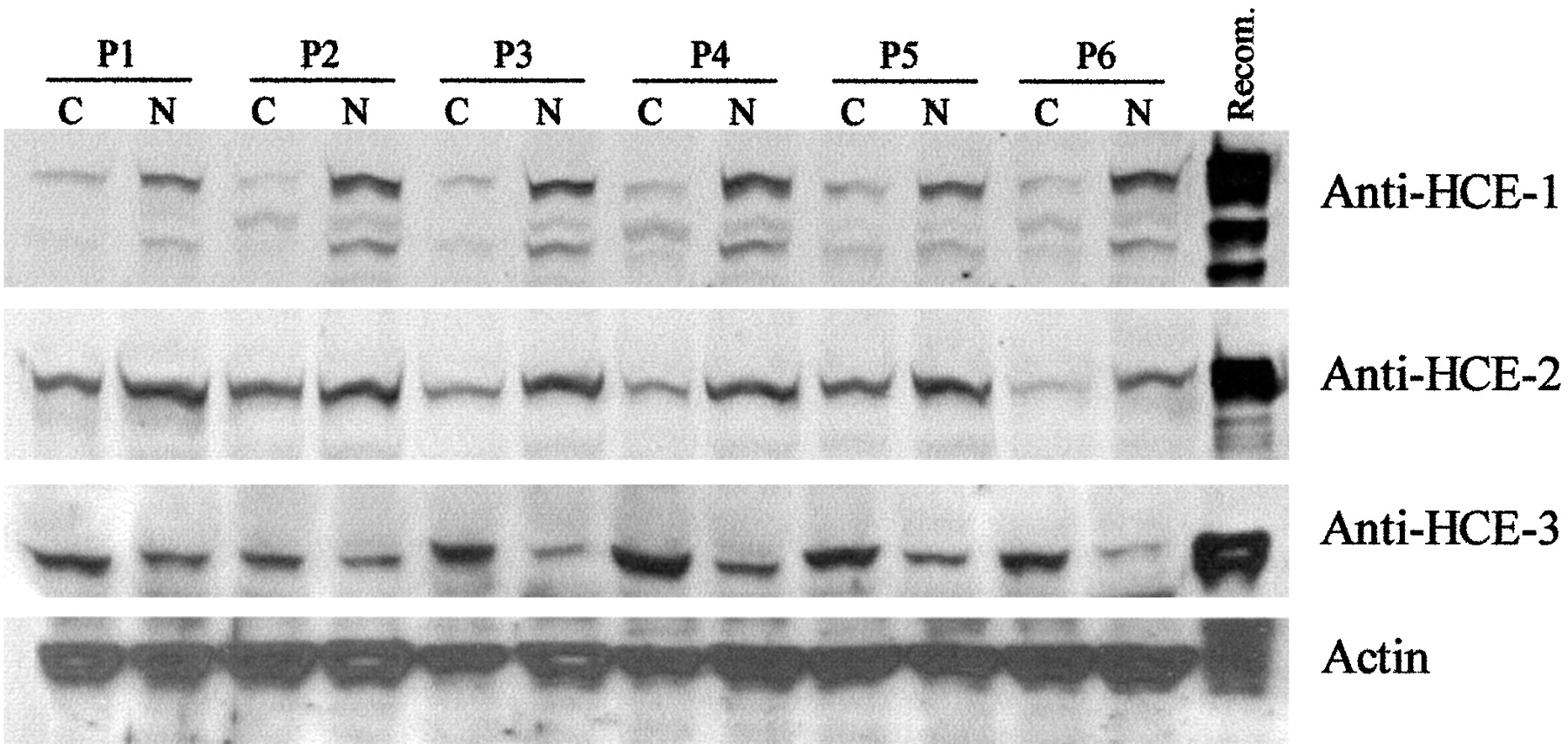
Carboxylesterases activate ester or amide prodrugs, and HCE-2 not HCE-1 has been shown to effectively activate irinotecan, a widely used chemotherapeutic agent for a variety of malignancies, including colon carcinoma (Ewesuedo and Ratain, 1997; Saltz, 1997; Stucky-Marshall, 1999). Next, we examined whether human carboxylesterases are differentially expressed in colon cancer and its adjacent normal tissue. Paired samples from patients who underwent subtotal colon resection were collected, and tissue homogenates were analyzed for the abundance of HCE-1, HCE-2, and HCE3. As shown in Fig.5, all three proteins were present in colon carcinomas and the adjacent normal homogenates. However, the relative abundance varied between the tumor and normal tissues. Without exceptions, levels of both HCE-1 and HCE-2 were slightly higher in the adjacent normal tissues than those in the carcinomas. In contrast, levels of HCE-3 were slightly higher in the carcinomas than those in the adjacent normal tissues (Fig. 5). HCE-3 was previously reported to be abundantly present in brain blood vessels (Yamada et al., 1994; Mori et al., 1999), and tumor-related increase on the expression of this carboxylesterase suggests that this enzyme plays a role in angiogenesis, although its precise action remains to be determined.

Expression of HCE-1, HCE-2, and HCE-3 in colon adenocarcinoma and the adjacent normal tissues.
Tissue homogenates (10 μg) from colon adenocarcinomas and the adjacent normal tissues or lysates (1 μg) from, respectively transfected cells were subjected to SDS-polyacrylamide gel electrophoresis and transferred electrophoretically to a Trans-Blot nitrocellulose membrane. The immunoblots were detected with an antibody (10 μg/ml) against HCE-1, HCE-2, or HCE-3. As a control, the blots were also detected by an antibody against actin (representative blot on actin abundance shown in the figure). P, patient; C, cancer; N, normal.
Discussion
Mammalian carboxylesterases structurally belong to a superfamily of α/β-fold proteins, which consist of alternate α-helix and β-sheets connected by loops with a varying length. These enzymes hydrolyze endogenous and foreign chemicals containing such functional groups as a carboxylic acid ester, amide, and thioester. Studies on molecular cloning demonstrate that these enzymes share a high degree of sequence identity. In this report, we have tested 11 human and rodent carboxylesterases on their immunorelatedness, substrate specificity, sensitivity to inhibitors, tissue distribution, and tumor-related expression. Antibodies against whole carboxylesterases generally exhibit a broader cross-reactivity than anti-peptide antibodies. All recombinant carboxylesterases hydrolyze bothpara-nitrophenylacetate andpara-nitrophenylbutyrate with some carboxylesterases having a substrate preference. The hydrolytic activity is inhibited by PMSF and paraoxon, but the sensitivity to each inhibitor differs markedly from enzyme to enzyme. Human HCE-1 is widely distributed among 14 normal tissues, whereas HCE-2 and HCE-3 are more tissue-restricted. In addition, human carboxylesterases are differentially expressed between colon carcinoma and the adjacent normal tissues. HCE-1 and HCE-2 are slightly more abundant in the adjacent normal tissues, whereas the opposite is true with HCE-3.
The substrate preference shown by some carboxylesterases betweenpara-nitrophenylacetate andpara-nitrophenylbutyrate resembles that by acetylcholinesterase and butyrylcholinesterase on acetylcholine and butyrylcholine (Chatonnet and Lockridge, 1989; Vellom et al., 1993). Acetylcholinesterase rapidly hydrolyzes acetylcholine but exhibits a much diminished activity toward butyrylcholine. In contrast, butyrylcholinesterase shows far less selectivity on the acyl group and effectively hydrolyzes both choline esters. X-ray structural analyses and site-directed mutagenesis studies reveal that phenylalanines 288 and 290 in the acyl pocket (numbered according to Torpedo californica acetylcholinesterase; Sussman et al., 1991) are responsible for such a differential hydrolysis of the choline esters (Harel et al., 1992; Ordentlich et al., 1993; Vellom et al., 1993). Replacement of one (Phe-288) or both phenylalanines with butyrylcholinesterase leucine and valine markedly increases hydrolytic activity toward butyrylcholine. Carboxylesterases and cholinesterases have several features in common: α/β-folding in overall structure, the Ser-His-Glu triad in catalysis, and a moderate identity in primary sequence (∼30%). However, sequence alignment analyses of carboxylesterases locate at residues 288 and 290 neither the phenylalanines as seen in acetylcholinesterase nor leucine and valine as seen in butyrylcholinesterase. Instead, carboxylesterases contain a well conserved serine at residue 288 and a less conserved proline at residue 290. The highly conserved serine suggests that preferable hydrolysis by carboxylesterases betweenpara-nitrophenylacetate andpara-nitrophenylbutyrate is specified by the overall sequence of the acyl pocket but not by residues 288 and 290 alone. In support of this possibility, HCE-3 and hydrolase A have an identical sequence in this stretch (16 amino acids) and both preferably hydrolyzepara-nitrophenylbutyrate (Robbi et al., 1990; Mori et al., 1999). In contrast, HCE-2 and hydrolase E preferably hydrolyzepara-nitrophenylacetate. HCE-2 has a 12-residue deletion in the acyl pocket, and hydrolase E mismatches HCE-3 and hydrolase A by 40% in this region (Robbi et al., 1990; Pindel et al., 1997).
Rat hydrolase S and mouse M-S show little activity toward either substrate, although abundant expression in the respective transfected cells is detected by Western blots (Fig. 1). Both hydrolase S and M-S, like HCE-2, have a deletion in the acyl pocket (Pindel et al., 1997). However, such a deletion may not be completely responsible for the lack of hydrolytic activity. Instead, the expressed hydrolase S and M-S in the lysates may lack the conformation necessary to confer catalysis. Several lines of evidence support this possibility. First, we have previously demonstrated with immunoprecipitation experiments that serum hydrolase S rapidly hydrolyzes para-nitrophenylacetate (Yan et al., 1995), providing direct evidence that mature hydrolase S is capable of hydrolyzing this substrate. Second, serum hydrolase S has a much higher molecular weigh (∼72 kDa) than its calculated molecular mass (∼58 kDa) due to extensive glycosylation, and such a post-translational modification has been found to play a major role in folding carboxylesterases into a catalytically active enzyme (B. Yan and D. Yang, 1995; unpublished data). In this study, the major product (>90%) of both hydrolase S and M-S in the lysates has a molecular mass of ∼60 kDa, suggesting that they are incompletely glycosylated. It should be emphasized that hydrolase S and M-S represent carboxylesterases that are most extensivelyN-linked glycosylated (five putative sites); therefore, they are easily subjected to being incompletely glycosylated, particularly in an overexpression system such as transfected cells. In addition, mature hydrolase S and E-S are secreted into the serum due to the lack of the C-terminal retention tetrapeptide (HXEL) (Yan. et al., 1995), and the inability of their microsomal precursors to hydrolyze either substrate suggests that they contribute insignificantly to the overall intracellular hydrolysis.
In addition to the acyl pocket, studies on cholinesterase have revealed that several other functional subsites play roles in substrate recognition and inhibitor reactivity. Sequence alignment analyses reveal that carboxylesterases have similar functional subsites, including the catalytic triad, hydrophobic subsite, acyl pocket, peripheral anionic subsite, H-bond network, and oxyanion hole (Ordentlich et al., 1996, 1998). In this report, we demonstrate that HCE-3 and hydrolase A, identical on all subsites with only one substitution in the H-bond network (Glu→Asp-443), have the same substrate preference and are comparably inhibited by both paraoxon and PMSF (Figs. 2 and 3), providing direct evidence on the importance of these subsites in catalysis and inhibition of carboxylesterases. Substrate recognition and inhibitor reactivity likely use similar functional subsites but hydrolytic activity is not always correlated with the sensitivity to all inhibitors. For example, M-LK rapidly hydrolyzes both para-nitrophenylacetate andpara-nitrophenylbutyrate. However, this enzyme is highly sensitive to paraoxon but only moderately inhibited by PMSF (Fig. 3), suggesting that the same functional architecture reacts differently with inhibitors or substrates. Studies on insecticide resistance reveal in acetylcholinesterase, nine residues that are responsible for altered sensitivity to commonly used insecticides (Newcomb et al., 1997; Walsh et al., 2001). Surprisingly, only two of these residues are conserved between carboxylesterases and acetylcholinesterase, suggesting that these two types of enzymes, although highly similar on their overall structure, react differentially with a vast array of insecticides. Given the fact that mammalian species express multiple forms of carboxylesterases, which are regulated in species- and chemical-dependent manners (Satoh and Hosokawa, 1998; Zhu et al., 2000), carboxylesterases are likely contributing significantly to species and individual variation on organophosphate toxicity.
Immuno-cross-reactivity, on the other hand, appears to be determined by the overall sequence identity. For example, hydrolase B has the highest sequence identity with hydrolase A (70.1%), hydrolase E (71.8%), M-E (72.6%), and M-LK (82.0%); therefore, the anti-HB antibody cross-reacts strongly with these enzymes except hydrolase A. The anti-HB antibody was immunoabsorbed against purified hydrolase A; therefore, antibody against epitopes shared by hydrolase A and B was removed (Morgan et al., 1994). Hydrolase S has a comparable sequence identity (66.4–82.0%) with all carboxylesterases except HCE-2 (46.1%); therefore, the anti-HS antibody cross-reacts to a similar extent with all carboxyl-esterases but HCE-2 (Fig. 1). It is interesting to note that hydrolase S and HCE-2 share a much higher sequence identity (65.6%) in the N-terminal half than the overall sequence identity (46.1%); however, the anti-HS antibody shows no cross-reactivity toward HCE-2 (Fig. 1), suggesting that the C-terminal half of carboxylesterases contains epitopes with stronger immunogenicity. Based on the immuno-cross-reactivity detected by the anti-peptide antibodies, it appears that the C-terminal tetrapeptide is sufficient to invoke production of antibody. For example, the anti-HCE-2 antibody was raised against a peptide ending with HTEL, this antibody cross-reacts moderately with carboxylesterases having this C-terminal tetrapeptide (hydrolase E and M-E). In addition, the anti-HCE-1 antibody was raised against a peptide ending with HIEL, this antibody cross-reacts even with carboxylesterases having HVEL (e.g., HCE-3) but not HTEL (e.g., HEC-2). Apparently, substitution of the isoleucine with a polar residue threonine but not a nonpolar residue valine significantly affects immunoreactivity with the HCE-1 antibody.
Tissue-dependent and tumor-related expression of human carboxylesterases has both physiological and pharmacological significance. HCE-2, for example, is abundantly present in the liver, intestine, and kidney, internal organs that constantly encounter foreign chemicals (Fig. 4). Therefore, HCE-2 likely plays important roles in xenobiotic metabolism. In contrast, HCE-3 is abundantly expressed in the adrenal and brain, suggesting that this enzyme is largely involved in the metabolism of endogenous compounds (Fig. 4). In support of this notion, HCE-3 preferably hydrolyzespara-nitrophenylbutyrate, whereas HCE-2 preferably hydrolyzes para-nitrophenylacetate (Fig. 2). Endogenous compounds such as phospholipids and cholesteryl esters are generally more lipophilic (Sun et al., 1997; Barr et al., 1998). Local activation of prodrugs by carboxylesterases likely has profound clinical consequence. Irinotecan is effectively used for the treatment of a variety of cancers, including refractory colon adenocarcinomas in adults (Ewesuedo and Ratain, 1997; Saltz, 1997; Stucky-Marshall, 1999). However, side effects include granulocytopenia, gastrointestinal toxicity, and renal failure (Merrouche et al., 1997; Persons et al., 1998). Irinotecan is rapidly converted by HCE-2 to its cytotoxic metabolite, 7-ethyl-10-hydroxy-camptothecin. In this report, we demonstrate that HCE-2 is abundantly expressed in the intestine and kidney. Activation of irinotecan by HCE-2 in these tissues is likely responsible for its therapeutic action as well as toxicity.
Footnotes
-
This work was partially supported by National Institute of Environmental Health Sciences Grant ES07965 and a New Investigator Award from the American Association of Colleges of Pharmacy.
- Abbreviations used are::
- HCE
- human carboxylesterase
- PMSF
- phenylmethylsulfonyl fluoride
- ssDNA
- single-stranded DNA
- HA hydrolase A
- HB, hydrolase B
- HS
- hydrolase S
- Received December 10, 2001.
- Accepted February 1, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}