


Abstract
Drug-drug interactions may cause serious adverse events in the clinical setting, and the cytochromes P450 are the enzyme system most often implicated in these interactions. Cytochrome P450 2C is the second most abundant subfamily of cytochrome P450 enzymes and is responsible for metabolism of almost 20% of currently marketed drugs. The most abundant isoform of this subfamily is CYP2C9, which is the major clearance pathway for the low therapeutic index drugs warfarin and phenytoin. Considering the importance of CYP2C9 to drug-drug interactions, the in vitro-in vivo extrapolation of drug-drug interactions for CYP2C9 may be confounded by the presence of polymorphic variants and the possibility of multiple binding regions within the CYP2C9 active site, leading to the potential for genotype- and substrate-dependent inhibition. To address the issues of genotype-dependent enzyme inhibition as well as probe substrate correlations, the inhibitory potency (Ki) of 28 effector molecules was assessed with five commonly used probes of CYP2C9 in both the CYP2C9.1 and CYP2C9.3 proteins. The inhibition of CYP2C9.1 and CYP2C9.3 by the battery of inhibitors with five substrate probes demonstrated differential inhibition potency not only between the two genotypes but also across substrate probes. Furthermore, the substrate probes fell into three distinct classes depending on genotype, suggesting that multiple probes may be needed to fully assess inhibition of CYP2C9 in vitro. Thus, both genotype and choice of probe substrate must be considered when attempting to predict potential CYP2C9 drug-drug interactions from in vitro data.
Drug-drug interactions (DDIs) are one of the primary causes of serious adverse events occurring in clinical practice (Dambro and Kallgren, 1988). The most commonly observed DDIs result from inhibition of target-drug metabolism by a coadministered drug. The cytochrome P450 enzymes are the primary family of oxidative drug-metabolizing enzymes and, as such, are implicated in a substantial number of DDIs. Cytochrome P450 2C is the second most abundant subfamily of P450 enzymes and is responsible for metabolism of almost 20% of the drugs currently available in the market (Rendic and Di Carlo, 1997). CYP2C9 is an important member of the subfamily, serving as the primary metabolic pathway of the narrow therapeutic index drugs warfarin and phenytoin as well as numerous other therapeutic entities (Rettie and Jones, 2005). Interactions with warfarin or phenytoin metabolism are of substantial clinical concern and can result in serious adverse events.
With the increased mechanistic knowledge of P450 enzyme function and the role of P450s in drug metabolism, a more systematic approach has been taken by investigators and the pharmaceutical industry for predicting drug-drug interactions. The U.S. Food and Drug Administration as well as the Pharmaceutical Research Manufacturer's Association has defined guidelines for preclinical in vitro and in vivo studies for the prediction of DDIs (Bjornsson et al., 2003; http://www.fda.gov/cder/guidance/clin3.pdf). Oftentimes, for in vitro experiments, one probe substrate is used for a single P450 enzyme to determine the drug interaction potential of a new chemical entity during the discovery screening phase. These results are then extrapolated to interaction potential with other compounds metabolized by the same P450. Unfortunately, this approach is not always successful. One factor that may play a role in unsatisfactory extrapolations is the phenomenon of multiple binding regions within the enzyme active site. This phenomenon of multiple binding regions has been described for many of the P450s, but most frequently with CYP3A4 (Korzekwa et al., 1998; Shou et al., 1999; Schrag and Wienkers, 2001). With this in mind, Kenworthy et al. (1999) studied the correlation of inhibitory potential of 34 drugs with 10 commonly used in vitro probe substrates for CYP3A4. Their findings suggested three groupings of probe substrates and, thus, possibly three binding regions within the active site of CYP3A4. The authors suggested that to correctly predict the inhibition potential of CYP3A4 inhibitors, researchers should use three separate probe substrates for CYP3A4. As with CYP3A4, evidence suggests the presence of more than one binding region in the CYP2C9 active site (Williams et al., 2003), including the presence of atypical kinetic profiles (Korzekwa et al., 1998), heteroactivation (Hutzler et al., 2001), and NMR data (Hummel et al., 2004). Therefore, the possibility exists that multiple probe substrates of CYP2C9 might be needed to predict potential DDIs with CYP2C9.
Further confounding the issue of predicting in vivo DDIs from in vitro data is the occurrence of polymorphic variant alleles of CYP2C9. Twenty-four variant alleles of CYP2C9 have been reported to date, with all variants displaying reduced substrate turnover compared with wild-type enzyme (Lee et al., 2002). The most clinically significant variant allele that occurs with substantial frequency is the CYP2C9*3 allele, which results in a protein with markedly reduced substrate turnover compared with the wild-type enzyme (Higashi et al., 2002). However, it is unknown whether the same enzyme features of variant enzyme proteins that result in decreased catalysis of substrate also alter the inhibitory potential of competing compounds. To address this issue of genotype-dependent enzyme inhibition as well as probe substrate correlations, the inhibitory potency (Ki) of 28 effector molecules was assessed with five commonly used probes of CYP2C9 using both the CYP2C9.1 and CYP2C9.3 proteins. Correlations of inhibition of probe substrate activity were compared against each other within the same enzyme as well as between the two enzyme variants.
Materials and Methods
Chemicals. Diclofenac, (S)-warfarin, phenytoin, tolbutamide, tenoxicam, and p-hydroxyphenytoin were purchased from Sigma-Aldrich (St. Louis, MO). The metabolites 4′-hydroxy diclofenac, 4-hydroxy tolbutamide, and 7-hydroxy warfarin were purchased from BD Gentest (Woburn, MA). (S)-Flurbiprofen and 4′-hydroxyflurbiprofen were obtained from the Pfizer compound library. CYP2C9.1 and CYP2C9.3 Supersome enzymes were purchased from BD Gentest. All other materials were purchased from commercial sources and were of the highest purity available.
Ki Determination. The incubation times and protein concentrations used were within the linear range, with respect to time and protein, of each assay. Incubations were carried out using five common probe substrates of CYP2C9 [diclofenac, (S)-flurbiprofen, (S)-warfarin, phenytoin and tolbutamide]. Twenty-eight known inhibitors exhibiting a wide range of inhibition potencies were selected for study. Stock solutions of all the inhibitors were made in dimethyl sulfoxide and then diluted 100 times with acetonitrile before addition to the incubation mixtures. Three concentrations of each substrate (0.5Km, Km, and 2Km) and four concentrations of each inhibitor (100-fold range) were used for determination of Ki in a 96-well plate format. In brief, each reaction was carried out in duplicate, and 1 pmol of CYP2C9.1 enzyme (2 pmol when phenytoin was the substrate) and 2 pmol of CYP2C9.3 (4 pmol in the case of phenytoin) was used per incubation. Each incubation reaction mixture contained enzyme, substrate, and inhibitor suspended in phosphate buffer (50 mM, pH 7.4) and was preincubated for 3 min in an incubator-shaker at 37°C. The reactions were initiated by the addition of NADPH (1 mM final concentration). Organic solvent concentrations did not exceed 2% v/v. Solvent concentrations were the same for all experiments and turnover rates did not differ significantly from minimal solvent controls. The reaction was terminated with 50 μlof acetonitrile containing 1 μM tenoxicam (internal standard) except for diclofenac, in which 100 μl was used. Length of the incubations for diclofenac, (S)-flurbiprofen, and tolbutamide was 10 min for CYP2C9.1 and 20 min for CYP2C9.3. For (S)-warfarin and phenytoin, the incubations were carried out for 20 and 40 min for CYP2C9.1 and CYP2C9.3, respectively.
It is of note that, to assure validity of the results and to allow comparison of inhibition profiles from different sets of experiments, a number of precautions were taken. To avoid batch-to-batch variability in enzyme, all samples of each variant were taken from the same batch provided by the manufacturer. The experiments were planned to minimize the amount of enzyme in each incubation to reduce the potential impact of nonspecific binding of both substrate and inhibitor, and incubation times were limited to 20 min or less to avoid substrate or inhibitor depletion.
Liquid Chromatography/Tandem Mass Spectral Analysis. The liquid chromatography/mass spectrometry system consisted of an API 4000 triple quadrupole mass spectrometer with an atmospheric pressure electrospray ionization source (MDS SCIEX, Concord, ON, Canada), and two LC-10ADvp pumps with an SCL-10ADvp controller (Shimadzu, Columbia, MD). A Thermo Electron Aquasil-C18 column (2.1 × 20 mm, 3.0 μm; Thermo Electron Corporation, Waltham, MA) was used for separation with initial conditions of 10% mobile phase B, followed by a gradient of 10% B to 90% B over 1 min (solvent A = 0.1% formic acid, solvent B = 100% acetonitrile), followed by an immediate return to initial conditions that were maintained for 1 min with a flow rate of 0.5 ml/min before the next injection.
The compounds were detected in negative ion mode. The deprotonated molecular ions were formed using an ion spray voltage of –3500 V, curtain gas of 10 V, collision gas of 8 eV, and source temperature of 600°C for all compounds. Product ions were formed at collision energies of –16 eV (4′-hydroxydiclofenac, m/z 309.8→265.8), –12 eV (4-hydroxyflurbiprofen, m/z 258.9→214.9), –28 eV (7-hydroxywarfarin, m/z 322.9→176.6), –26 eV (4-hydroxytolbutamide, m/z 285.0→185.9), –18 eV (p-hydroxyphenytoin, m/z 266.8→224.0), and –14 eV (tenoxicam, m/z 335.9→271.8).
Data Analysis. The Ki of each inhibitor was calculated via nonlinear regression of the data to a competitive inhibition equation (eq. 1, using GraphPad Prism 4; GraphPad Software, San Diego, CA), except for amiodarone (all probe substrates and variants), (S)-ibuprofen [(S)-warfarin substrate probe, both variants], and quinine [(S)-flurbiprofen substrate probe, both variants], which were fit using a partial competitive inhibition equation (eq. 2). The goodness of fit was determined by visual inspection of the data with the Dixon plot and r2 values. Simple linear regression was used to determine the correlation between the Ki values of pairs of substrates using GraphPad Prism 4. 

Clustering Analysis. Statistical and clustering analyses of the inhibition potency data were performed using Spotfire DecisionSite 8.1 (Spotfire, Inc., Somerville, MA). An unweighted pair group method with arithmetic mean clustering algorithm was used to determine similarity between the inhibition data sets and to form successively larger clusters using a Euclidean distance similarity measure. Data were entered as inhibition potency (Ki) values. Compounds that exhibited activation or Ki values above 100 μM were entered as a Ki of 100 μM. For instances in which the modifier was also a substrate, a variant-specific average value of Ki for that modifier exhibited by the other four substrate probes was calculated and used.
Estimation of Potential in Vivo Inhibition. To estimate the potential in vivo effects of inhibition of CYP2C9.1 and CYP2C9.3 with each of the substrate probes, data were computed using an equation for competitive inhibition (eq. 1) to estimate velocities in the presence and absence of inhibitor. Values for Km and Vmax in the absence of inhibitor were determined experimentally; for each substrate, the estimated Ki was used and literature-derived values of Cmax (total plasma concentrations) for inhibitor and substrate were inserted into the equation to determine the predicted velocity of the reaction.
Results
For each of the substrate probes [(S)-flurbiprofen, (S)-warfarin, tolbutamide, phenytoin, and diclofenac], inhibition profiles and the resulting inhibition constant (Ki) were determined with a set of 28 inhibitors in both the CYP2C9.1 and CYP2C9.3 enzymes (Tables 1 and 2, respectively). Potency of inhibition across the 28 inhibitors spanned several orders of magnitude for each substrate. A number of interesting trends were noted upon examination of the data.
Ki values (μM) obtained with five probe substrates, i.e., (S)-flurbiprofen, (S)-warfarin, phenytoin, tolbutamide, and diclofenac, using CYP2C9.1
Global standard error for data fitting was less than 15% and r2 > 0.9 for each effector.
Ki values (μM) obtained with five probe substrates, i.e, (S)-flurbiprofen, (S)-warfarin, phenytoin, tolbutamide, and diclofenac, using CYP2C9.3
Global standard error for data fitting was less than 15% and r2 > 0.9 for each effector.

CYP2C9.1 linear correlation graphs of the log inhibition data. A, diclofenac versus (S)-flurbiprofen; B, phenytoin versus (S)-flurbiprofen; C, tolbutamide versus (S)-flurbiprofen; D, (S)-warfarin versus (S)-flurbiprofen; E, diclofenac versus phenytoin; F, tolbutamide versus phenytoin; G, diclofenac versus tolbutamide; H, diclofenac versus (S)-warfarin; I, tolbutamide versus (S)-warfarin; J, phenytoin versus (S)-warfarin.
Comparing within only the CYP2C9.1 enzyme results, using a recently proposed system of binning inhibition potency [Ki < 1 μM (high concern), Ki 1–10 μM (moderate concern), Ki > 10 μM (low concern)] (Obach et al., 2006), it is noted that 21 of the 28 inhibitors exhibited a substantially lower Ki value (increased inhibition potency) against (S)-warfarin hydroxylation than against any of the other probe substrates. In fact, the estimated Ki value was <1 μM for 16 of the 28 inhibitors of (S)-warfarin hydroxylation. In contrast, only eight, six, nine, and nine of the inhibitors exhibited Ki values <1 μM against (S)-flurbiprofen hydroxylation, phenytoin hydroxylation, tolbutamide hydroxylation, and diclofenac hydroxylation, respectively. An additional eight compounds exhibited Ki values between 1 and 10 μM toward (S)-warfarin hydroxylation, resulting in 24 (of 28) compounds exhibiting Ki values less than 10 μM toward this reaction. For the other four probe substrates, the majority of inhibitors fell within this 1 to 10 μM Ki range. There was some switching of “bins” (i.e., Ki < 1 μM versus Ki 1–10 μM versus Ki > 10 μM) of inhibitors across the five substrates. Notably, Vivid Green inhibited the metabolism of (S)-warfarin, phenytoin, tolbutamide, and diclofenac relatively potently (Ki = 0.5–1.7 μM) but was a much weaker inhibitor of (S)-flurbiprofen metabolism (Ki = 8.2 μM). In contrast, quinine was a relatively potent inhibitor of (S)-flurbiprofen metabolism (Ki = 1.1 μM) but was a very poor inhibitor of the metabolism of the other four probe substrates (Ki = 20 to >100 μM). Indomethacin was a very potent (Ki = 0.7 μM) inhibitor of (S)-warfarin hydroxylation but a relatively weak (Ki > 10 μM) inhibitor of all other probe substrates. Finally, (S)-ibuprofen was a poor (Ki > 40 μM) inhibitor of (S)-warfarin hydroxylation but a relatively potent (Ki ∼4 μM) inhibitor of the other four probe substrates. Closer analysis of Table 1 indicates that for a number of inhibitors, the Ki values varied 10-fold across substrates.
Another method used to determine whether Ki values assessed with CYP2C9 are substrate-dependent was to calculate the number of inhibitors that showed a greater than 3-fold difference in inhibition potency compared with a standard. Due to its prevalent use in industry as a CYP2C9 substrate probe, diclofenac was chosen as the standard. Eighteen inhibitors of (S)-warfarin hydroxylation exhibited greater than a 3-fold difference in inhibition potency (Ki values) compared with inhibition of diclofenac hydroxylation in CYP2C9.1. Except for amiodarone and quercetin, the inhibitory potency was always greater toward (S)-warfarin hydroxylation than diclofenac hydroxylation. It is interesting that for five compounds (mibefradil, indomethacin, benzbromarone, ketoconazole, and dapsone), the Ki value was at least 10-fold lower toward (S)-warfarin hydroxylation compared with diclofenac hydroxylation.
When inhibition of (S)-flurbiprofen and diclofenac metabolism was compared, 12 inhibitors of (S)-flurbiprofen hydroxylation exhibited greater than 3-fold differences in Ki values compared with diclofenac as a substrate probe with CYP2C9.1. In five of these cases (omeprazole, quinine, quercetin, Vivid Green, and dapsone), the difference was at least 10-fold. In contrast to the results with (S)-warfarin, in four of the five cases (omeprazole, quercetin, Vivid Green, and dapsone), the Ki values were 10-fold lower toward diclofenac hydroxylation than (S)-flurbiprofen metabolism.
With respect to the inhibition of phenytoin hydroxylation by CYP2C9.1 compared with inhibition of diclofenac hydroxylation, fewer differences were noted in Ki values. Only four inhibitors differed by more than 3-fold between phenytoin and diclofenac hydroxylation inhibition (benzbromarone, nicardipine, omeprazole, and piroxicam). Only one inhibitor differed in potency by 10-fold (nicardipine). In every case, the inhibition constant was lower (greater inhibition) with respect to diclofenac hydroxylation compared with the inhibition of phenytoin. Finally, for CYP2C9.1, in only one instance did the inhibition of tolbutamide hydroxylation differ by more than 3-fold compared with the inhibition of diclofenac hydroxylation, and this was with the compound piroxicam.
These same 28 inhibitors were analyzed for their ability to inhibit metabolism of the same five probe substrates in the CYP2C9.3 enzyme. Relatively few changes in inhibition potency were noted with respect to (S)-warfarin hydroxylation when compared with the Ki values obtained in CYP2C9.1 enzyme. Only two inhibitors differed by more than 3-fold with respect to Ki (indomethacin and benzbromarone). The largest number of compounds for which Ki estimates differed by more than 3-fold between the two enzymes occurred with the substrate probe flurbiprofen. For 16 inhibitors, the Ki values differed by more than 3-fold between the CYP2C9.1 and CYP2C9.3 enzymes. For three compounds (benzbromarone, piroxicam, and dapsone), the inhibition constant varied more than 40-fold. Even more intriguing is that in the cases of quercitin and Vivid Green, greater inhibition was noted in the CYP2C9.3 enzyme. With respect to inhibition of phenytoin hydroxylation, only two compounds (benzbromarone and nicardipine) exhibited Ki values that differed between the variants, but the differences were substantial (benzbromarone was ∼10-fold less potent and nicardipine was ∼10-fold more potent). For inhibition of tolbutamide hydroxylation, only three compounds (diclofenac, (S)-ibuprofen, and sulfamethizole) were less potent inhibitors (∼3-fold less potent) in the CYP2C9.3 enzyme. Finally, five compounds differed in inhibition potency more than 3-fold with respect to their inhibition of diclofenac hydroxylation (benzbromarone, tolbutamide, (S)-warfarin, (S)-ibuprofen, and gemfibrozil) between the CYP2C9 variants and, in all cases, became less potent inhibitors in CYP2C9.3.
Log-transformed inhibition data were analyzed for correlation between the substrate probes for CYP2C9.1 (Table 3; Fig. 1), between the substrate probes for CYP2C9.3 (Table 4; Fig. 2), and for each substrate probe comparing inhibition profiles between the CYP2C9.1 and CYP2C9.3 variants (Table 5). The inhibition profiles of diclofenac, phenytoin, and tolbutamide were highly correlated for CYP2C9.1 (r2 > 0.92), whereas (S)-flurbiprofen and (S)-warfarin showed lower correlations with each other (r2 = 0.69) and with the three aforementioned probes (r2 < 0.76). The inhibition profiles of diclofenac, phenytoin, and tolbutamide were also highly correlated for CYP2C9.3 (r2 > 0.88). The probes (S)-flurbiprofen and (S)-warfarin showed lower correlations with each other (r2 = 0.69) and with diclofenac, phenytoin, and tolbutamide (r2 ≅ 0.69-0.84). In comparing differences in inhibition profiles for each probe between the CYP2C9.1 and CYP2C9.3 variants, diclofenac, tolbutamide, and (S)-warfarin were highly correlated (r2 > 0.88), phenytoin exhibited a lower correlation (r2 = 0.73), and (S)-flurbiprofen exhibited the lowest correlation (r2 = 0.55).
r2 values obtained from the linear regression of the Ki values for two probes using CYP2C9.1
r2 values obtained from the linear regression of the Ki values for two probes using CYP2C9.3
r2 values obtained from the linear regression of the Ki values for probes using CYP2C9.1 and CYP2C9.3
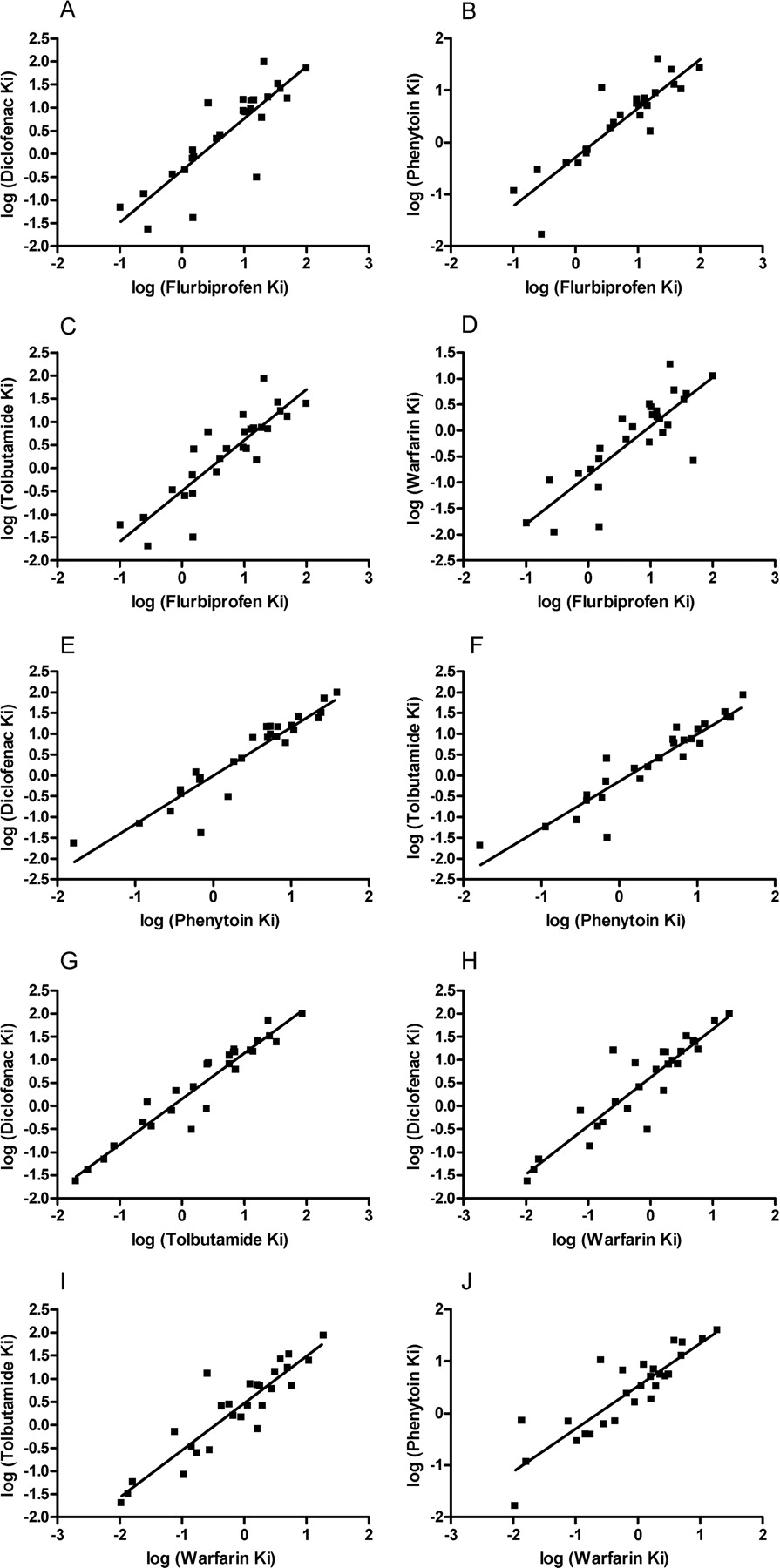
CYP2C9.3 linear correlation graphs of the log inhibition data. A, diclofenac versus (S)-flurbiprofen; B, phenytoin versus (S)-flurbiprofen; C, tolbutamide versus (S)-flurbiprofen; D, (S)-warfarin versus (S)-flurbiprofen; E, diclofenac versus phenytoin; F, tolbutamide versus phenytoin; G, diclofenac versus tolbutamide; H, diclofenac versus (S)-warfarin; I, tolbutamide versus (S)-warfarin; J, phenytoin versus (S)-warfarin.
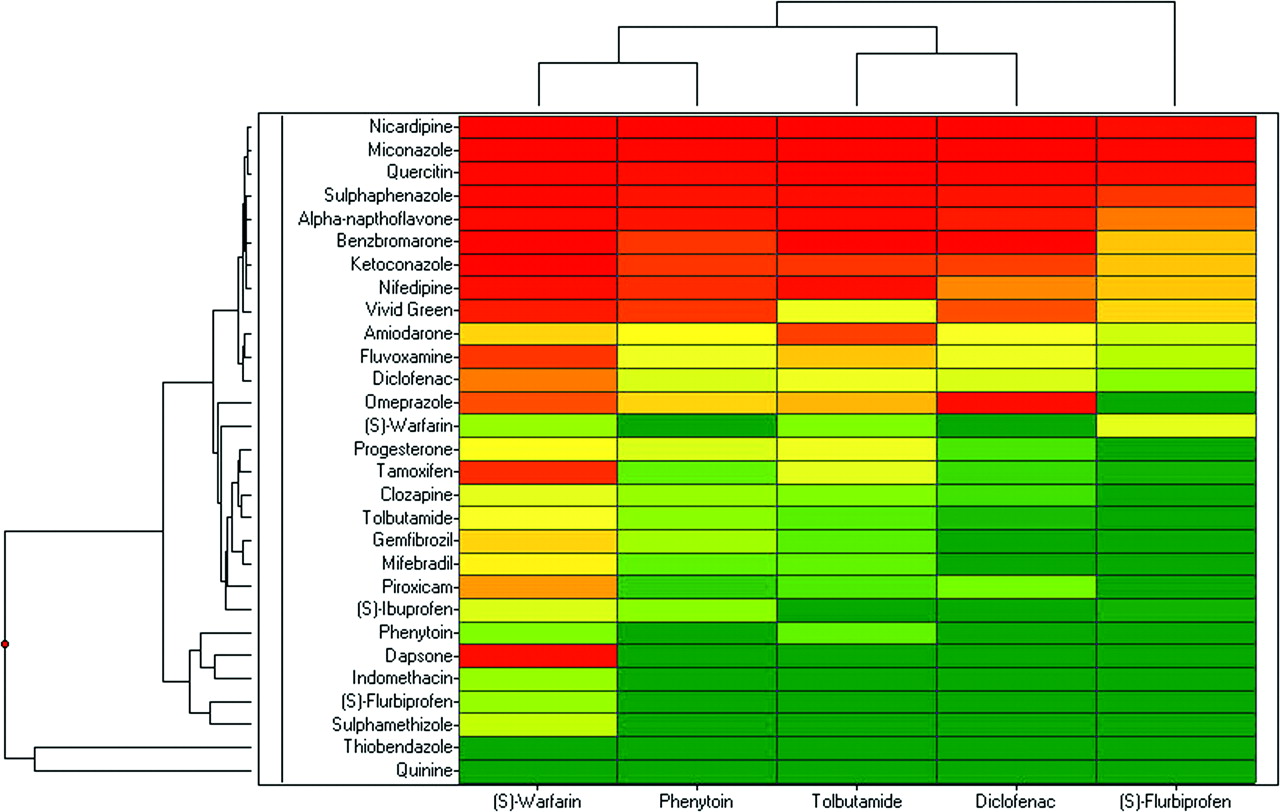
Hierarchical clustering analysis was performed on the nontransformed inhibition potency data using an unweighted pair group method with arithmetic mean clustering algorithm and a Euclidean distance similarity measure. Results from the clustering analysis for the CYP2C9.1 and CYP2C9.3 data were visualized as a dendrogram (Figs. 3 and 4, respectively) in which the horizontal axis of the dendrogram represents the Euclidean linkage distance between substrate clusters. For the panel of modifiers, the vertical axis of the dendrogram represents the Euclidean linkage distance between effector clusters. The clustering analysis for CYP2C9.1 probe substrate inhibition data suggested three distinct groupings of probe substrate similarities: diclofenac, tolbutamide, and phenytoin (Euclidean linkage distance 13.5); (S)-warfarin (Euclidean linkage distance 27.2); and (S)-flurbiprofen (Euclidean linkage distance 76.6). These results correlated well with the linear regression analysis of probe substrate correlation mentioned above. However, the clustering analysis for the CYP2C9.3 inhibition data for probe substrate similarity differed from the groupings obtained by linear regression analysis of inhibition data (and from that observed with CYP2C9.1), although three distinct groupings were still obtained: (S)-warfarin and (S)-phenytoin (Euclidean linkage distance 42.9), diclofenac and tolbutamide (Euclidean linkage distance 51.7), and (S)-flurbiprofen (Euclidean linkage distance 79.1). Inhibition potency results were also visualized as a heat map (Figs. 3 and 4). Heat map coloration indicates relative closeness of inhibition potency data to maximal (<1 μM, red), moderate (1–10 μM, yellow), or minimal (>10 μM, green) inhibition. Again, confirming the Ki rankings discussed previously, compounds exhibited lower Ki values toward (S)-warfarin as a probe substrate compared with the other probes, in general.

CYP2C9.1 inhibitor hierarchical clustering dendrogram and heat map (red <1 μM; 1 μM < yellow <10 μM; green >10 μM)
The potential in vivo significance of differential inhibition of probe substrate metabolism in the CYP2C9.1 and CYP2C9.3 variants is depicted in Table 6. It can be noted that the estimated percentage of remaining activity, in general, was greater in the CYP2C9*3 enzyme, suggesting a reduced inhibition of metabolism in the CYP2C9*3 genotype. However, in some cases, such as with nicardipine inhibition of phenytoin metabolism, a substantially greater inhibition is predicted to occur with the CYP2C9.3 genotype. Not unexpectedly, these differences were both substrate- and inhibitor-dependent.
Potential in vivo significance of differential inhibition of probe substrate metabolism in CYP2C9.1 and CYP2C9.3 variants
Discussion
Prediction of in vivo enzyme inhibition (and potential drug-drug interactions) from in vitro data remains an important area of investigation among drug metabolism researchers. Although several studies have been conducted to assess correlations of in vitro predictions of drug-drug inhibition interactions and their potential extrapolation to in vivo data, none has assessed the potential impact of genetic variants on these predictions. Furthermore, correlation analysis of inhibition potency for multiple substrate probes had previously been assessed in the CYP3A4 enzyme (Kenworthy et al., 1999), but not for CYP2C9. The P450 isoform CYP2C9 shares many characteristics with CYP3A4, such as substrate-dependent atypical kinetics profiles, heteroactivation, and large active site volume, which all suggest the possibility for multiple binding regions within the same active site. In addition, CYP2C9 is a polymorphically expressed enzyme, and the CYP2C9.3 variant, in particular, exhibits substantially reduced substrate turnover, which may further confound predictions of drug-drug interaction potential.
For the CYP2C9.1 enzyme, both correlation analysis and Euclidean linkage analysis indicated that the substrate probes diclofenac, phenytoin, and tolbutamide were highly correlated, suggesting that they could be used interchangeably as substrate probes for CYP2C9.1. However, the substrate probes (S)-flurbiprofen and (S)-warfarin were not correlated with each other, or with the other three probes, suggesting that they may represent different binding regions (or orientations) within the active site and exhibit different susceptibilities to inhibition. Together, these data suggest multiple binding modes or regions in the CYP2C9.1 active site. Thus, it appears that as with CYP3A4 (Kenworthy et al., 1999), the use of multiple probes might be required to accurately assess the potential for drug-drug inhibition interactions. One combination of probes that could be considered would be the commonly used probe diclofenac in combination with (S)-warfarin as a probe for inhibition studies. This would allow obtainment of information regarding the two CYP2C9 substrates [phenytoin and (S)-warfarin] that possess the lowest therapeutic index and thus are most worrisome in terms of potential drug-drug interactions. If one were to choose a single probe, as has more recently been suggested with CYP3A4 (Galetin et al., 2005), then (S)-warfarin would seem the most logical choice since the Ki values of the inhibitors toward this substrate are the lowest, and the drug is commonly prescribed and requires close monitoring. One must, however, be cognizant that (S)-warfarin is turned over very slowly, and thus, excess consumption of inhibitor must be monitored, potential for shunting to other metabolites is possible in human liver microsomal preparations, and nonspecific binding of inhibitor to the microsomes must be checked since higher protein concentrations may be required, due, again, to the slow turnover.

CYP2C9.3 inhibitor hierarchical clustering dendrogram and heat map (red <1 μM; 1 μM < yellow <10 μM; green >10 μM)]
It is of interest that, for the CYP2C9.3 variant, correlation analysis suggested the same grouping of probe substrates as observed with CYP2C9.1, but Euclidean linkage analysis suggested that (S)-warfarin and phenytoin were correlated, tolbutamide and diclofenac were correlated, and (S)-flurbiprofen was not correlated with any of the other substrate probes. Thus, the difference as compared with CYP2C9.1 is that phenytoin now tracks with (S)-warfarin in the CYP2C9.3 enzyme. The lack of flexibility of the probes (S)-warfarin and phenytoin may make them more sensitive to active site alterations than the more flexible probes. Despite the relatively conservative change in the CYP2C9.3 enzyme (I359L), some effect on binding orientation is potentially occurring in addition to substantially reduced metabolic rate.
With respect to the inhibitors, hierarchical clustering resulted in similar groupings between the CYP2C9.1 and CYP2C9.3 variants based upon variant-specific average values for each modifier. Eight of the 10 most potent inhibitors were shared between the variants. For CYP2C9.1, four inhibitors exhibited Euclidean linkage distances furthest from the rest of the cluster (indomethacin, dapsone, thiobendazole, and quinine; Euclidean linkage distance >48). Seven inhibitors for the CYP2C9.3 variant (phenytoin, dapsone, indomethacin, flurbiprofen, sulfamethizole, thiobendazole, and quinine; Euclidean linkage distance >41) exhibited Euclidean linkage distances that were furthest from the rest of the cluster. Both the least potent and most potent inhibitors were shared by CYP2C9.1 and CYP2C9.3.
Recently proposed guidelines indicate that compounds with Ki values <1 μM are of high concern for drug-drug interactions, compounds with Ki values between 1 and 10 μM are of moderate concern, and compounds with Ki values greater than 10 μM tend to be of lesser concern (Obach et al., 2006). Using the substrate diclofenac as the comparator (because it is the most commonly used CYP2C9 substrate probe), when the inhibition of each compound was compared for the other substrates, inhibitors of (S)-warfarin metabolism showed the greatest number of changes in classification in both CYP2C9.1 and CYP2C9.3 enzymes (i.e., were now classified as being in a category of greater concern). With (S)-warfarin as substrate probe, 12 inhibitors of CYP2C9.1 and 10 inhibitors of CYP2C9.3 became classified as being of more concern, and 8 of the 12 in CYP2C9.1 and 4 of the inhibitors of CYP2C9.3 moved from the moderate to the high concern classification, as compared with when diclofenac was used as the probe. Again, comparing to results with diclofenac, changes in classification of inhibitors were mixed for the other three probe substrates [phenytoin, tolbutamide, and (S)-flurbiprofen] as some inhibitors became classified as being of more concern and some of less concern, depending on the probe substrate. Thus, only with the probe (S)-warfarin were the inhibitors almost universally classified as being of greater concern. With the other probe substrates, it was more difficult to predict the relative inhibitory potency of an inhibitor comparing to diclofenac.
The inhibition profiles for (S)-flurbiprofen and (S)-warfarin differ from those of the other substrate probes in several ways. (S)-Flurbiprofen exhibits substantial variability in the degree of its inhibition by the studied compounds when compared across genotypes, particularly when compared with a standard such as diclofenac. For the inhibition of (S)-flurbiprofen, the inhibition potency of a compound may be significantly increased or reduced depending upon the particular substrate-inhibitor combination when compared between the two CYP2C9 variants. This is further exemplified by the low correlation of the inhibition profile of (S)-flurbiprofen between the two CYP2C9 variants. The active site residues Arg-108 and Phe-114 have been reported to be important in the binding, orientation, and metabolism of nonsteroidal anti-inflammatory drugs such as flurbiprofen and diclofenac in the CYP2C9 active site, forming an anionic binding site and a pi-stacking region, respectively (Davies et al., 2004; Dickmann et al., 2004; Wester et al., 2004). Although the recently published crystal structure of CYP2C9 with (S)-flurbiprofen bound as a ligand indicates interaction with the Arg-108 and Phe-114 residues (Wester et al., 2004), the rigid biphenyl structural core of (S)-flurbiprofen may not allow orientation to the same binding modes or full access to a hydrophobic pocket that are exhibited by less conformationally restrained probes such as diclofenac and tolbutamide. Thus, use of (S)-flurbiprofen as a substrate probe may allow analysis of a different set of interactions than would be assessed using the probes that correlate well together such as phenytoin, tolbutamide, and diclofenac.
The substrate probe (S)-warfarin also exhibits a unique inhibition profile. Not only is the inhibition profile different from that of any of the other probes, but the potency of inhibitors is generally increased when (S)-warfarin is used as a substrate probe. It should be noted that (S)-warfarin may exist in several distinct tautomer conformations in solution (Heimark and Trager, 1984; He et al., 1999). Although previous studies indicate that the ring-closed form of (S)-warfarin is the preferred conformation for metabolism, perhaps other tautomers contribute to nonproductive binding of (S)-warfarin. Differential or allosteric binding modes and nonproductive binding may in part explain the differences in inhibition potency and profile observed with (S)-warfarin (Seifert et al., 2006).
In summary, inhibition of CYP2C9.1 and CYP2C9.3 by a battery of inhibitors with five substrate probes demonstrated differential inhibition potency not only between the two genotypes but also across substrate probes. Furthermore, the substrate probes fell into three distinct classes depending on genotype, suggesting that multiple probes may be needed to fully assess inhibition of CYP2C9 in vitro. A combination of inhibition studies using diclofenac and (S)-warfarin may provide the broadest “chemical space” with respect to CYP2C9 inhibition, but use of (S)-warfarin as a single probe may also provide acceptable results, with recognition of the aforementioned potential caveats.
Acknowledgments
We thank Dr. Charles Locuson for valuable discussions during the preparation of the manuscript.
Footnotes
-
This study was supported by Pharmacokinetics, Dynamics and Metabolism at Pfizer and by National Institutes of Health Grants GM063215 and GM069753 (to T.S.T.).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.010926.
-
ABBREVIATIONS: DDI, drug-drug interaction; P450, cytochrome P450.
- Received May 1, 2006.
- Accepted September 8, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}