


Abstract
CYP2C9 is distinguished by a preference for substrates bearing a negative charge at physiological pH. Previous studies have suggested that CYP2C9 residues R97 and K72 may play roles in determining preference for anionic substrates by interaction at the active site or in the access channel. The aim of the present study was to assess the role of these two residues in determining substrate selectivity. R97 and K72 were substituted with negative, uncharged polar and hydrophobic residues using a degenerate polymerase chain reaction-directed strategy. Wild-type and mutant enzymes were expressed in bicistronic format with human cytochrome P450 reductase in Escherichia coli. Mutation of R97 led to a loss of holoenzyme expression for R97A, R97V, R97L, R97T, and R97E mutants. Low levels of hemoprotein were detected for R97Q, R97K, R97I, and R97P mutants. Significant apoenzyme was observed, suggesting that heme insertion or protein stability was compromised in R97 mutants. These observations are consistent with a structural role for R97 in addition to any role in substrate binding. By contrast, all K72 mutants examined (K72E, K72Q, K72V, and K72L) could be expressed as hemoprotein at levels comparable to wild-type. Type I binding spectra were obtained with wild-type and K72 mutants using diclofenac and ibuprofen. Mutation of K72 had little or no effect on the interaction with these substrates, arguing against a critical role in determining substrate specificity. Thus, neither residue appears to play a role in determining substrate specificity, but a structural role for R97 can be proposed consistent with recently published crystallographic data for CYP2C9 and CYP2C5.
The cytochrome P450 (P4501) group of enzymes are the predominant catalysts of phase I xenobiotic metabolism in humans and other mammals, catalyzing a variety of monooxygenation reactions including, among others, aliphatic and aromatic hydroxylation, epoxidation, N- and O-dealkylation, and N-and S-oxidation. In humans, approximately 15 enzymes from the CYP1–3 families are responsible for the metabolic clearance of most lipophilic chemicals. Among this group, forms from the same subfamily show discrete but often overlapping substrate specificities.
CYP2C9, one of the most important forms in overall drug metabolism, is distinguished from other human CYP2C forms by a preference for substrates bearing a negative charge at physiological pH (Smith and Jones, 1992; Mancy et al., 1995); however, neutral or positively charged substrates may also be substrates. Various models have been developed to explain the preference for anionic substrates. Mansuy and colleagues originally proposed a pharmacophore model based on examination of tienilic acid derivatives and other CYP2C9 substrates (Mancy et al., 1995). In this model, a positively charged residue bordering the substrate binding site was proposed to interact electrostatically with the negative center on the substrate.
Homology modeling of the CYP2C9 protein based on crystal structures available for other P450 enzymes has also been used to generate hypotheses regarding the active site topology of CYP2C9. In particular, R97 and R105 were nominated as key cationic residues in the CYP2C9 active site (Payne et al., 1999; Rao et al., 2000; Ridderstrom et al., 2000). In site-directed mutagenesis experiments (Ridderstrom et al., 2000), substitution of R97 with alanine increased the Km for diclofenac 4′-hydroxylation, whereas mutation of R105 was without effect. However, all human CYP2C forms and many other P450s have arginine at position 97; therefore, this cannot be the sole determinant of the selectivity of CYP2C9.
An alternative hypothesis (Lewis et al., 1998), based upon a different homology model, proposed that K72, suggested to be positioned within the substrate access channel, may be responsible for substrate selection. This residue is analogous to R47 in CYP102 (P450 BM3), where it interacts with substrate via a hydrogen-bonded water network (Haines et al., 2001).
The purpose of the present study was to assess the role of these two residues, R97 and K72, in binding of substrates. Specifically, it was hypothesized that if either residue was critical for binding negatively charged substrates, a concerted charge reversal on the residue in question (by site-directed mutagenesis) and simultaneously on prototypical P450 2C9 substrates (by chemical modification of the substrates) should lead to a retention of the binding interaction.
Materials and Methods
Chemicals. Ibuprofen, naproxen, phenytoin, and diclofenac were obtained from Sigma-Aldrich (St. Louis, MO). The known amide and amine analogs of ibuprofen (Fig. 1) were prepared according to the literature protocols (Brooks and Rodriques, 1990). The bicistronic expression construct for CYP2C9 (pCW′/2C9/hNPR) was obtained as previously described (Cuttle et al., 2000).
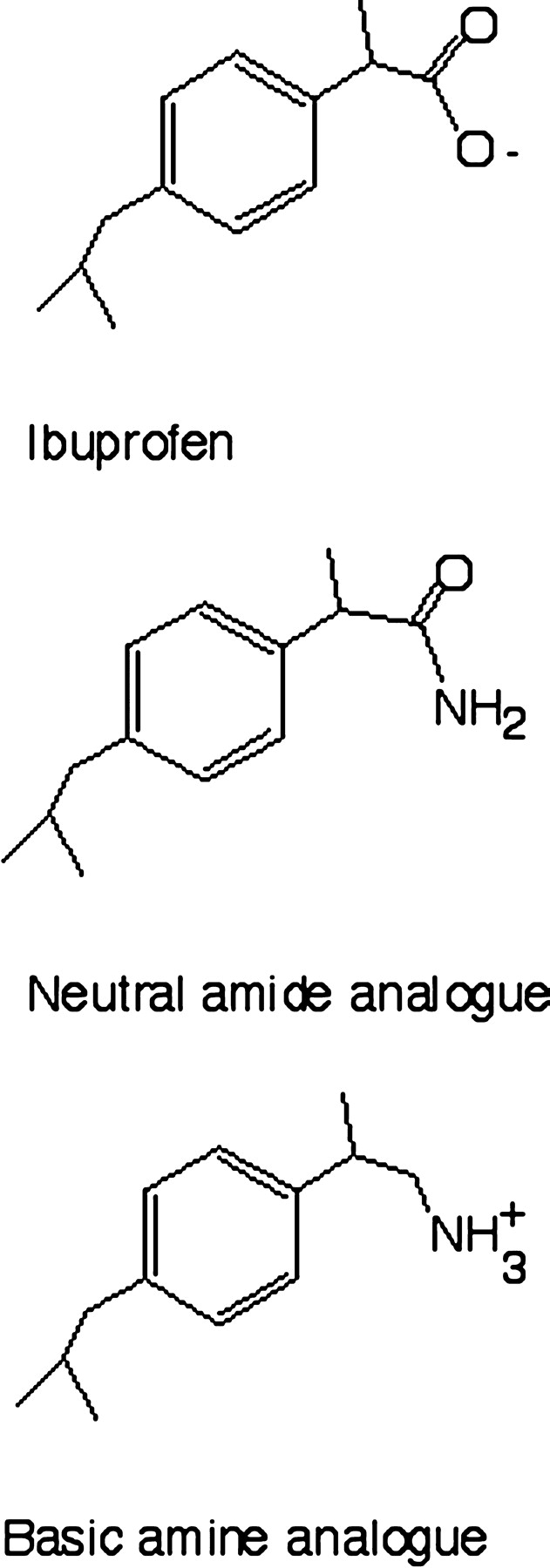
Ibuprofen and analogs made in this study
Mutagenesis. Site-directed mutants were created by PCR-directed mutagenesis using degenerate primers in a “mega-primer” approach. Briefly two primary PCR amplifications were undertaken using a degenerate mutagenic primer and a flanking primer. In the second step, the products of this initial amplification were used to generate a primer dimer. The resulting full-length fragment was either subcloned directly into the vector or amplified further by a final PCR step using flanking primers and then subcloned. Sequences of the mutagenic and flanking primers are listed in Table 1. Possible substitutions at R97 were as follows: E (GAA), A (GCA), T (GTA), Q (CAA), P (CCA), L (CTA), K (AAA), T (ACA), I (ATA). Substitutions possible at K72 were: H (CAT), Q (CAG), L (CTT, CTG), N (AAT), K (AAG), I (ATT), M (ATG), D (GAT), E (GAG), V (GTT, GTG).
Primers used in this study
For mutation of R97, the primary amplifications were performed with 0.25 μM concentrations of the respective primers, 5 to 75 ng of pCW′/2C9/hNPR plasmid DNA, and 2.5 U of Taq polymerase (Invitrogen, Carlsbad, CA) with all other reagents and concentrations as recommended by the manufacturer. The following thermal cycling conditions were used: 10 cycles of 94°C for 30 s, 65°C for 30 s, and 72°C for 1 min; 25 cycles of 94°C for 30 s, 65°C, for 30 s, and 72°C for 1 min plus a 10-s extension per cycle; and a 5-min polishing extension at 72°C. In the second amplification, 30% of the product of each of the initial amplifications was used in place of primer and template. In the final amplification, 1% or 0.1% of the product of the second amplification was used as template with the flanking primers. Cycling conditions for the second and final amplifications were the same as for the initial amplification. The resulting product was cut with NdeI and AvaII and cloned into the cognate sites of pCW′/2C9/hNPR.
For mutation of K72, the same overall procedure was used with the following exceptions. Pfu polymerase (Stratagene, La Jolla, CA) was used and cycling conditions were identical in both the primary and secondary amplifications: 10 cycles of 94°C for 30 s, 61°C for 30 s, and 75°C for 1.5 min; 20 cycles of 94°C for 30 s, 61°C for 30 s, and 75°C for 1.5 min plus a 10-s extension per cycle; and a 5-min polishing extension at 75°C. The amount of each primary PCR product used in the secondary amplification was increased to 60% and the product of the second amplification was cut with NdeI and BamHI and cloned directly into the cognate sites of pCW′/2C9/hNPR. All mutants were sequenced throughout the region subjected to PCR amplification to determine which mutation had been incorporated and to ensure that no unwanted mutations were introduced.
Expression of Recombinant P450s in Bacteria. Expression was undertaken as previously described (Notley et al., 2002; Gillam et al., 1993) in E. coli strain DH5aF′IQ (Invitrogen) in the presence of coexpressed chaperones GroES and GroEL.
Difference Spectroscopy. Hemoprotein concentrations were determined by measuring Fe(II)·CO versus Fe(II) difference spectra as previously described (Guengerich, 1994). Type I spectra were recorded with 2.5 to 1000 μM ibuprofen and diclofenac and 0.2 μM P450 using a Cary 100 Conc UV-visible spectrophotometer. Spectral binding data were analyzed by nonlinear regression using the program DNRP53 (Duggleby, 1984) to obtain values for ΔAmax and Ks. The equations for 1) a single saturable site and 2) a saturable (high affinity) site and a second nonsaturable (low affinity) site were used to fit data. The F-test of variance was used to determine which model best fit the data.
Homology Modeling. An initial model of CYP2C9 was constructed based on the original published crystal structure of modified CYP2C5 (Williams et al., 2000) using the Homology, Biopolymer, and Discover 3 modules of the Insight II suite of software (v. 2000.1; Molecular Simulations Inc./Accelerys, Sydney, Australia) operating on an O2 Silicon Graphics workstation. Coordinates were assigned for designated structurally conserved regions; then, the F-G loop (which is not specified in the original P450 2C5 structure) and residues 278–280 (absent in CYP2C5) were built. The coordinates for the heme group were taken directly from the P450 2C5 structure. After all loops and gaps were spliced, the resulting structure was subjected to energy minimization using Discover_3 and the consistent valence force field. Hydrogen atoms were included in the model and a pH of 7.4 was specified. Forcefield parameters for the heme were specified separately, and the heme and the Cys435 residue were kept fixed throughout all the minimizations. Minimization was initially performed using the steepest descent method (1000 steps), followed by a series of minimization stages (1000 steps each), using the conjugate gradient method, until a local energy minimum was obtained. For assessment of the effect of the R97A and R97P mutations, the recently published CYP2C9 crystal structure (Williams et al., 2003) was used as a template, and R97 was replaced with alanine or proline using the Biopolymer module of Insight II. The modified structure was then subjected to the same minimization as above, followed by a dynamics step of 2000 fs at 298K and another three minimization stages. The native CYP2C9 structure was subjected to the same process in parallel as a control.
Other Methods. NADPH-P450 reductase concentrations were quantified as described previously (Guengerich, 1994) using a specific activity of 3200 nmol of cytochrome c reduced min-1 nmol hNPR-1 (Yasukochi and Masters, 1976). Protein concentrations were determined using a modification of the method of Lowry et al. (1951; Guengerich, 1994).
Results
Expression of Mutant and Wild-Type Recombinant P450 2C9. Nine R97 mutants and four K72 mutants were obtained using the degenerate strategy. Levels of recombinant P450s and hNPR obtained with coexpression of chaperones are shown in Table 2. No R97 mutants were successfully expressed in holoenzyme form in the absence of chaperones (data not shown). However, apoprotein was expressed for all mutants at levels generally lower than for wild-type CYP2C9 as revealed by immunoblotting with a polyclonal antibody raised to recombinant CYP2C19 (Cuttle et al., 2000). Low levels of hemoprotein were detected in R97Q, R97K, R97I, and R97P mutants with chaperone coexpression (Fig. 2; Table 2). By contrast, all K72 mutants were expressed as intact holoenzyme at levels comparable to wild-type (Table 2). Expression of hNPR was within the expected range for all preparations (Table 2).
Expression of P450 2C9 mutants
P450 2C9 mutants were coexpressed in bicistronic format with hNPR in E. coli cotransformed with the expression vector pGro7 encoding the bacterial chaperones GroES and GroEL. Values represent the mean of two to four separate expression trials.

Fe(II)·CO versus Fe(II) spectra for R97 mutants and wild-type P450 2C9.
Bacteria were disrupted by sonication and membranes were isolated by differential centrifugation. Bacterial membranes were diluted in 100 mM potassium phosphate buffer, pH 7.4, containing 20% (w/v) glycerol and 0.2% (v/v) Emulgen 913, and difference spectra were recorded according to the method of Guengerich (1994).
Substrate Binding by Mutant and Wild-Type Recombinant P450 2C9. Type I binding spectra were obtained for wild-type and mutant CYP2C9 forms expressed in bacterial membranes with the prototypical P450 2C9 substrates ibuprofen and diclofenac. Table 3 lists the ΔAmax and Ks values derived from nonlinear regression analysis of the spectral data. Mutation of K72 had no marked or consistent effect on the Ks characterizing the interaction of CYP2C9 with these substrates. The most significant change was seen with the K72L mutation, which decreased Ks for ibuprofen by 3-fold but increased the Ks for diclofenac 2-fold. Maximum amplitudes of type I spectra were reduced for all mutants and both substrates compared with wild-type. Binding spectra were also recorded for naproxen, phenytoin, and the amide and amine analogs of ibuprofen; however, no classical spectral perturbation could be observed with these compounds.
Type I binding interaction with ibuprofen and diclofenac
Type I spectra were acquired using a P450 concentration of 0.2 μM and with ligand concentrations of 2.5 to 1000 μM. Estimates of binding parameters were derived by nonlinear regression analysis of n = 28–56 data points.
Homology modeling of CYP2C9. In the initial homology model of CYP2C9 derived here, which was based on the initial CYP2C5 structure, R97 was located in the active site of the model as predicted from other earlier models (Payne et al., 1999; Ridderstrom et al., 2000) and specifically toward the beginning of the B-C loop; however, the side chain was directed away from the substrate binding cavity after repeated energy minimization of the structure.
The published CYP2C9 crystal structure (Williams et al., 2003; PDB accession code 1OG2) into which the R97A mutation had been introduced was subjected to a molecular dynamics simulation at 298K. During the simulation, the region of the B-C loop containing the A97 appeared to undergo a general shift away from the heme such that the Cα-Cβ bond of A97 appeared to move through an angle of ∼60 degrees toward F114 and I99. In the wild-type control, R97 remained directed toward the heme propionate groups when the structure of the native enzyme was subjected to the same simulation (Fig. 3). When a longer dynamics simulation was run on the R97A mutant at 298K, the structure became unstable after 1600 to 5000 fs, whereas the wild-type and R97P structures were stable for at least 20,000 fs.

Molecular dynamics simulation of the effect of the R97A mutation.
Modeled structures of the wild-type CYP2C9 (A, C) and R97A mutant (B, D) before (light gray) and after (dark gray) molecular dynamics simulation and energy minimization. For clarity, only residues 95–100 and the heme prosthetic group are shown in close-up (A, B). In panels B and D, the whole molecule is shown as a backbone trace to illustrate overall movement of the models during the simulation, with residue 97 depicted in ball and stick representation. The published CYP2C9 crystal structure (Williams et al., 2003; PDB accession code: 1OG2) into which had been introduced the R97A mutation was subjected to a molecular dynamics simulation at 298K. During the simulation, the region of the B-C loop containing the A97 mutation appeared to undergo a general shift away from the heme such that the Cα-Cβ bond of A97 appeared to move through an angle of ∼60 degrees toward F114 and I99, indicated by the arrow (B). When the structure of the native enzyme was subjected to the same simulation in the wild-type control (A), although there was considerable lateral movement of the B-C loop and the rest of the protein (as also seen in the R97A mutant), R97 remained directed toward the heme propionate groups.
Discussion
In the present study, mutation of R97 compromised expression of CYP2C9 holoenzyme, yet apoprotein expression was still observed. Expression of an hNPR holoprotein was also within the expected range. This observation suggested that mutation of R97 destabilized the tertiary structure of the enzyme so as to compromise heme incorporation or retention. Ridderstrom et al. (2000) previously reported the successful expression of an R97A mutant in yeast, enabling its kinetic characterization with respect to diclofenac metabolism. It is unclear why this mutant appears to produce intact hemoprotein in yeast but not in bacteria. Differences in protein folding and/or heme insertion pathways between the two hosts may be critical. Similar discrepancies have been seen between yeast and mammalian systems (Oscarson et al., 1999), suggesting that yeast may be a more permissive expression system for poorly stable mutants. Differences in methods of enzyme work-up between systems may also affect the stability of the mutant enzyme. Alternatively, the mutant protein may be better stabilized by the membrane environment of yeast or by yeast chaperones during folding. We attempted to enhance expression here by coexpression of bacterial chaperones, a strategy found to enable the bacterial expression of otherwise intractable P450 forms (E. M. J. Gillam, L. M. Notley, D. J. Mitchell, I. H. Hanna, and F. P. Guengerich, unpublished observations). This led to a low level of hemoprotein being detected for R97K, R97Q, R97I, and R97P mutants; however, hemoprotein was still undetectable for the other variants. While this report was in preparation, Dickman et al. (2003) reported that mutation of R97 similarly led to instability of the expressed protein in the baculovirus system.
These studies highlight the fact that very different effects of mutations can be detected using different expression systems and suggest that whereas the yeast system is more tolerant of structural modifications, bacterially expressed enzymes are more sensitive to perturbations in structure. These results find a precedent in recent data obtained for CYP2D6. D301 of CYP2D6 was believed to be responsible for interaction with substrate based on site-directed mutagenesis data using enzyme expressed in yeast (Ellis et al., 1995). However, D301 mutants appear to be unstable or poorly expressed in the bacterial system (Hanna et al., 2001), and recent data suggest that D301 may be involved in maintaining the structural integrity of the enzyme by stabilizing the B-C loop (Guengerich et al., 2002; Kirton et al., 2002; Paine et al., 2002). A role for D301 in direct interactions with some substrates cannot be excluded; however, an alteration in overall protein structure may indirectly perturb substrate interactions. Similarly, we cannot exclude a role for R97 of CYP2C9 in interactions with certain substrates, but our observations are consistent with a structural role for R97 in CYP2C9, rather than, or in addition to, any role in substrate binding.
A structural role for R97 was not proposed based on earlier homology models, nor was it indicated by our initial homology model based on the original CYP2C5 structure (Williams et al., 2000). However, during preparation of this report, two critical studies were published that support a structural role for R97. First, Johnson and colleagues (Wester et al., 2003) reported the crystal structure of a substrate-bound form of CYP2C5. A key difference between this and the earlier structure was in the assignment of electron density around R97. Residue R97 was seen to be directed toward the heme propionate groups in the new structure, suggestive of a stabilizing ion-pairing interaction. Thus, the data from the present study suggesting a structural role for R97 are entirely consistent with this interaction. This slight discrepancy between the two crystal structures was sufficient to bias the initial homology model derived here, illustrating the critical dependence of homology models on the template structural data. Very recently, the R97-heme interaction has been confirmed for CYP2C9 by publication of the crystal structure showing an interaction between the guanidino group of R97 and the two heme propionate groups (Williams et al., 2003).
As noted above, a low level of hemoprotein expression was detected for R97K, R97Q, R97I, and R97P mutants. It is intriguing that two of the mutants (R97K and R97Q) that could be expressed, albeit at trace levels, retained some character of the arginine at position 97. The R97K mutant retains a positive charge in this site, suggesting that lysine may partially substitute for arginine. Such a substitution of R97 with lysine does occur in CYP2C22 (Emi et al., 1990; Nagata et al., 1990); however, the typical G-E-E-F-S-G-R-G motif found in most other CYP2C forms in this region is altered dramatically in CYP2C22, to G-E-E-F-S-D-K-M, perhaps to accommodate the lysine substitution without loss of overall structural integrity. The G-X-G motifs at either end of the B-C loop have been proposed to allow opening and closing of this part of the structure upon substrate binding (Wester et al., 2003). Thus, in CYP2C22, we would predict a loss of flexibility in this region.
The R97Q mutant would retain a residue of comparable size and shape with some hydrogen bonding potential; this may partially fulfill the role played by the arginine. When Gln was modeled into the CYP2C9 structure, favorable distances were seen for hydrogen bonding between the amide protons and the heme propionate carboxyl oxygens.
To investigate further the effect of mutation of R97, the R97A mutation was modeled into the published crystal structure of CYP2C9 (Williams et al., 2003; PDB accession code: 1OG2). After a limited molecular dynamics simulation and energy minimization, the region of the B-C loop containing the alanine side chain appeared to have shifted away from the heme and up toward I99. We can speculate that the Ala substitution both removes the anchoring electrostatic interaction with the heme and also allows greater flexibility in the region in general. Leu, Val, and Thr in this position would also be expected to be able to rotate away from the heme; however, Gln and Ile would be predicted to be constrained by steric interactions with F114, N116, and I99. Proline, being sterically constrained by the cyclic structure, may stabilize this region of the B-C loop in a manner compatible with heme binding. Indeed, both the wild-type and R97P mutant appeared stable during an extended molecular dynamics simulation under conditions where the R97A mutant became energetically unstable.
In contrast to R97 mutants, K72 mutants were expressed easily as holoenzyme, suggesting that the residue at this position had no deleterious effect on folding or heme insertion. K72 is not conserved in either CYP2C19 or CYP2C8 but is present in CYP2C18. K72 is analogous to R47 in P450 BM3, a residue shown to reside in the substrate access channel and to interact indirectly with substrate via a hydrogen-bonded water network (Haines et al., 2001). In homology models based on either of the CYP2C5 structures or CYP102, as well as in the CYP2C9 crystal structure, the side chain of K72 is similarly positioned at the turn of the beta1/1–1/2 sheet and predicted to point toward the space between the B-C loop and F-G loop/F′ helix, the region proposed to be one possible access channel in CYP102 (Ravichandran et al., 1993). Thus, K72 was proposed to play an analogous role in selection of substrates at the CYP2C9 active site from a homology model based on CYP102 (Lewis et al., 1998) and, again, very recently from the CYP2C9 structure (Williams et al., 2003). However, the results obtained in the present study fail to support a critical role for K72 in determining substrate specificity. Ks values for ibuprofen and diclofenac were relatively constant over the mutants compared with wild-type; the slight changes seen would be more consistent with a subtle conformational change elicited by the K72 mutations influencing binding to other critical residues. The general reduction in amplitude of spectra in mutants compared with wild-type CYP2C9 may again reflect slight conformational changes in the mutants leading to a reduced ability of ligands to induce the type I spectral change. We cannot exclude the possibility that substrates interact with K72 indirectly via a network of hydrogen-bonded water molecules in the same way as for R47 in CYP102, and the changes in binding data seen reflect subtle alterations in this network.
In light of these results, it is not surprising that complementary inversion of the charge on residue 72 and that on the substrate failed to lead to a type I spectral interaction being seen with either of the ibuprofen analogs. Although steric or other factors related to the analogs or residues introduced might also lead to failure to see such an interaction, the simplest explanation is that K72 has no significant role in substrate selection. This conclusion is consistent with the prediction that substrates may enter via a channel parallel to the I helix on the other side of the B-C loop rather than near the K72 (Wester et al., 2003), or that a conformational change on binding or reduction occurs which alters the overall conformation of substrate relative to enzyme (Williams et al., 2003). Importantly, our data reflect a reversible interaction with the resting form of the enzyme rather than metabolism, and we cannot exclude the possibility that mutation of K72 may affect turnover in the absence of a difference in initial binding. However, the simple hypothesis that K72 is the key determinant in substrate selection by CYP2C9 is not supported by our study.
Acknowledgments
We thank Drs. F. P. Guengerich, E. F. Johnson, L. Dickman, and A. R. Rettie for communicating results of papers in preparation and N. Rosic and L. M. Notley for assistance with some of the bacterial expression studies.
Footnotes
-
↵1 Abbreviations used are: P450, cytochrome P450; PCR, polymerase chain reaction; hNPR, human NADPH-cytochrome P450 reductase; ΔAmax, the maximal amplitude of the type I binding spectrum; PDB, Protein Data Bank.
-
This study was supported in part by an Australian Research Council Small Grants Scheme.
- Received September 17, 2003.
- Accepted January 13, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}