


Abstract
The CYP2C subfamily of cytochrome P450 monooxygenases is responsible for the metabolism of approximately 20% of therapeutic drugs and many endogenous compounds in humans. These enzymes can be induced by prior treatment with drugs, resulting in changes in drug efficacy. Induction of human CYP2C enzymes by xenobiotics occurs at the transcriptional level and is reported to involve the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR). In the present study, we report that murine CYP2C37 mRNA is induced by phenobarbital and phenytoin. In contrast, the mouse PXR agonist 5-pregnen-3β-ol-20-one-16α-carbonitrile did not induce CYP2C37 mRNA, suggesting that PXR does not regulate this gene. The induction of CYP2C37 mRNA by phenobarbital and phenytoin is essentially abolished in CAR-null mice; thus, induction of Cyp2c37 by these xenobiotics is CAR-dependent. A functional CAR response element (CAR-RE) was identified at –2791 base pairs from the translation start site of the Cyp2c37 gene. Mutation of this CAR-RE abolished mouse CAR transactivation of a Cyp2c37 –2.9-kilobase pair luciferase reporter construct in HepG2 cells.
The human CYP2C subfamily of cytochrome P450 (P450) monooxygenases is responsible for the metabolism of approximately 20% of all clinically prescribed drugs, other xenobiotics, and many endogenous compounds. Clinically prescribed substances known to be metabolized by the human CYP2C enzymes include phenytoin (anti-convulsant), warfarin (anticoagulant), tolbutamide (antidiabetic), torsemide (diuretics), and numerous nonsteroidal anti-inflammatory drugs (Goldstein and de Morais, 1994; Miners and Birkett, 1998). Recent work has shown that prior exposure to certain clinical drugs and herbal medicines can induce the expression of CYP2C8, CYP2C9, and CYP2C19 (Gerbal-Chaloin et al., 2001; Raucy et al., 2002; Chen et al., 2004; Ferguson et al., 2005). Drug-induced expression of P450 enzymes leads to increased metabolic activity potentially altering the effectiveness of the inducing agent or coadministered drugs, resulting in adverse drug reactions.
P450 expression is frequently regulated at the transcriptional level and is mediated by receptors such as aryl hydrocarbon receptor, constitutive androstane receptor (CAR), pregnane X receptor (PXR), and peroxisome proliferator-activated receptor. Once activated, these receptors bind response elements located within the 5′-flanking regions of target genes, thus inducing gene transcription (Nebert and Jones, 1989; Aldridge et al., 1995; Goodwin et al., 1999, 2002a; Gerbal-Chaloin et al., 2002; Chen et al., 2003, 2004; Wang et al., 2004a; Ferguson et al., 2005). Induction of the CYP2B and CYP3A enzymes by clinically prescribed drugs such as phenobarbital and rifampicin has recently been attributed to CAR and PXR, respectively (Honkakoski et al., 1998; Sueyoshi et al., 1999; Wang et al., 2003; Chen et al., 2004; Ferguson et al., 2005). These xenobiotic signaling pathways are extensively reviewed in Pascussi et al. (2004), Honkakoski and Negishi (2000), and Sueyoshi and Negishi (2001).
The mouse is used increasingly as a model for human disease and is an excellent system to investigate the drug-induced transcriptional regulation of P450 genes due to the availability of receptor-null mice. At present, 15 murine Cyp2c genes have been identified (Nelson et al., 2004; Wang et al., 2004b); however, only the Cyp2c29 and Cyp2c44 genes have been examined for drug induction (DeLozier et al., 2004; Jackson et al., 2004). Previous studies in our laboratory identified a phenytoin-responsive module (PHREM) located within the 5′-flanking sequence of the murine Cyp2c29 gene and demonstrated that the PHREM was necessary for induction by phenobarbital and phenytoin (Jackson et al., 2004). In contrast, studies in our laboratory showed that Cyp2c44 is not inducible by either the CAR activator phenobarbital or the PXR agonist pregnenolone 16α-carbonitrile (PCN) (DeLozier et al., 2004). An alignment of the 5′-flanking sequence of all currently known murine Cyp2c genes indicated that the PHREM that is present in Cyp2c29 was unique to this gene. Nevertheless, pilot studies indicated that Cyp2c37 was inducible by phenobarbital. Although the PHREM of Cyp2c29 was not conserved within the 5′-flanking sequence of Cyp2c37, several putative CAR response elements (CAR-REs) were identified by computational analysis. Using mutant and deletion Cyp2c37 luciferase promoter constructs, we identified a functional CAR-RE located ∼2.8 kb from the translation start site. In vivo studies demonstrate that both phenobarbital and phenytoin induce CYP2C37 mRNA. In contrast, the mPXR agonist PCN did not induce CYP2C37 mRNA, suggesting that PXR does not regulate Cyp2c37. Induction of CYP2C37 mRNA by phenobarbital and phenytoin was essentially abolished in CAR-null mice; thus, phenobarbital and phenytoin induction of Cyp2c37 is CAR-dependent, similar to Cyp2c29.
Materials and Methods
Materials and Reagents. Phenobarbital (sodium salt), phenytoin (5,5-diphenylhydantoin, sodium salt), PCN, and 1, 4-bis-[2-(3, 5-dichloropyridyloxy)]benzene (TCPOBOP) were purchased from Sigma-Aldrich (St. Louis, MO). DMSO and all other common reagents not listed were purchased from Sigma-Aldrich or other common vendors. Cell culture media, fetal bovine serum, 100× penicillin-streptomycin-glutamine solution and trypsin/EDTA were purchased from Invitrogen (Carlsbad, CA). All oligonucleotides were purchased from Sigma-Genosys (The Woodlands, TX) at 50 nmol scale and desalted. HepG2 cells were purchased from The American Type Culture Collection (Manassas, VA).
Animals. C3H/HeNCrlBR(C3H) mice were purchased from Charles River Laboratory (Wilmington, MA). A CAR-null mouse (Ueda et al., 2002) was first crossbred with C3H to generate CAR-heterozygous offspring. Subsequently, CAR-heterozygous offspring were repeatedly backcrossed with C3H mice until the genetic background became over 95% C3H. The obtained heterozygous mice were bred to produce the wild-type and CAR-null C3H mice. PXR-null 129S1/Sv*129 × 1/SvJ*C57BL/6 and congenic wild-type mice were obtained from Jeff Staudinger (University of Kansas, Lawrence, KS; Staudinger et al., 2001b) and maintained at the National Institute of Environmental Health Sciences (NIEHS). Mice were fed with a standard solid diet and tap water ad libitum for 5 days. Eight to 15-week-old male mice received corn oil (vehicle), phenobarbital (80 mg/kg), or phenytoin (80 mg/kg) once daily via gavage at a volume of 10 ml/kg for 4 consecutive days. Mice treated with PCN (80 mg/kg) were orally dosed once daily for 3 consecutive days using corn oil as vehicle. Animals were sacrificed 24 h after the last dose. The livers were removed for total RNA isolation and microsome isolation. The NIEHS Animal Care and Use Committee approved all animal procedures.
Total RNA Isolation and Quantitative RT-PCR. Total RNA was extracted using an ABI 6100 Nucleic Acid PrepStation. All chemicals for the ABI 6100 were purchased from Applied Biosystems (Foster City, CA). Total RNA from individual mice was isolated and stored at –80°C. Before reverse transcription, equal amounts of RNA from each individual RNA sample were pooled within each experimental group consisting of three to five mice according to treatment and genotype. Quantitative RT-PCR analysis was performed using a two-step process. An initial reaction with MuLV Reverse Transcriptase (Applied Biosystems) was followed by a subsequent quantitative PCR using 2× SYBR Green Master Mix (Applied Biosystems). Reverse transcription was performed with 100 ng of total RNA combined with 1× PCR buffer II, 0.4 μl (8 units) of RNase inhibitor (Applied Biosystems), 5.5 mM MgCl2, 0.5 mM dATP, dCTP, dTTP, and dGTP (each), 2.5 μM random hexamers (Applied Biosystems), and 0.5 μl (25 units) of MuLV Reverse Transcriptase in a final volume of 20 μl. Reverse transcription reactions were incubated in a PCR System 9700 Thermocycler (Applied Biosystems) using the following cycling parameters: 25°C for 10 min, 42°C for 60 min, 95°C for 5 min (inactivation), and 4°C hold. The subsequent quantitative PCR was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems). PCRs contained 0.5 μl of cDNA template, 1× SYBR Green buffer Master Mix, and 2.5 pmol of forward and reverse primer in a final volume of 10 μl. Gene-specific primer set sequences are as follows: Cyp2b10 (forward, 5′-ACCCCACGTTCCTCTTCA3-′; reverse, 5′-CAGCAGGCGCAAGAACTGA-3′); Cyp2c29 (forward, 5′-GTATTTGGGCTCAAAGCCTACTGTCA-3′; reverse, 5′-CAGGGTCATGAGTGTAAATCGTCTCA-3′), Cyp2c37 (forward, 5′-GATGGCAATCAACCATTGC-3′; reverse, 5′-GCCGATCACATGCTCAATT-3′), Cyp3a11 (forward, 5′-GTCAAACGCCTCTCCTTGCTG-3′; reverse 5′-GGCTTGCCTTTCTTTGCCTTC-3′), and β-actin (forward, 5′-CCTAGAAGCATTTGCGGTGCACGATG-3; reverse, 5′-TCATGAAGTGTGACGTTGACATCCGT-3′). PCR cycling parameters were as follows: 50°C for 2-min hold, 95°C for 10-min hold, 94°C for 30 s (denaturation), 60°C for 30 s (annealing), and 72°C for 30 s (extension). Denaturation, annealing, and extension temperatures and times were repeated for 42 cycles. PCR products were analyzed using gel electrophoresis, dissociation curve analysis, and dye terminator DNA sequencing (Applied Biosystems) to determine single product formation and gene specificity. Standard curves (log of template dilution versus Ct value) for each gene-specific primer set were used to determine relative mRNA content for each target gene. Each gene-specific PCR was performed in triplicate within each pooled experimental group. The triplicate values obtained from each gene-specific PCR were used to determine a relative starting template amount mean and standard error for each of the experimental groups.
Electrophoretic Mobility Shift Assay (EMSA). EMSA was performed on a 5% polyacrylamide gel using 0.5× Tris borate-EDTA running buffer. Oligonucleotides were labeled with [α-32P]dCTP and probe was purified by Microspin G-25 columns (Amersham Biosciences) to remove unincorporated deoxynucleoside-5′-triphosphates. Oligonucleotide sequences used in EMSA were as follows: CYP2C9 –1839 CAR-RE (forward, 5′-CTAGACCAAACTCTTCTGACCTCT-3′; reverse, 5′-CTAGAGAGGTCAGAAGAGTTTGGT-3′); Cyp2c37 –1890 CAR-RE (forward, 5′-CTAGAGTTCTCTCCTGGATGAATTTGGGT-3′; reverse, 5′-CTAGACCCAAATTCATCCAGGAGAGAACT-3′); Cyp2c37 –2065 CAR-RE (forward, 5′-CTAGGTTACTGTGCTGGGTGAACTGTGTT-3′; reverse, 5′-CTAGAACACAGTTCACCCAGCACAGTAAC-3′); Cyp2c37 –2791 CAR-RE (forward, 5′-CTAGAAAAGCAAACTTTTCTGAACTCCATG; reverse, 5′-CTAGCATGGAGTTCAGAAAAGTTTGCTTTT-3′); Cyp2c37 –2820 CAR-RE (forward, 5′-CTAGAGCCCGTATCACAAAGTTCAACAAG-3′; reverse, 5′-CTAGCTTGTTGAACTTTGTGATACGGGCT-3′); Cyp2c37 –3119 CAR-RE (forward, 5′-CTAGATTAGTGAAATCAAAATGTGATGTATGAAATTCAAG-3′; reverse, 5′-CTAGCTTGAATTTCATACATCACATTTTGATTTCACTAAT-3′); Cyp2c37 –3205 CAR-RE (forward, 5′-CTAGCAAATAGAACAACATAAACTGAGAC; reverse, 5′-CTAGGTCTCAGTTTATGTTGTTCTATTTG-3′). Labeled probe (∼100,000 cpm per reaction) was applied to each binding reaction in 2 μl of 5× binding buffer, 0.5 μg/μl poly(dI-dC), and 1 μl of each in vitro transcribed/translated protein (mCAR and hRXR) in a final volume of 10 μl. The 5× binding buffer was composed of 20% glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM dithiothreitol, 250 mM NaCl, and 50 mM Tris-HCl (pH 7.5). After addition of probe, the reactions were performed at room temperature for 20 min before being loaded on a polyacrylamide gel and electrophoresed. Gels were then dried and exposed to film for 16 to 24 h at –80°C.
Expression Vectors and Cloning of theCyp2c375′-Flanking Region. The expression construct pCR3-mCAR was described previously (Sueyoshi et al., 1999), and pGEMT-hRXR was a kind gift by Ronald Evans (Salk Institute for Biological Studies, San Diego, CA). PCR was performed to isolate a 1.97-kb fragment of the Cyp2c37 5′-flanking sequence using the following primers: forward, 5′-TGCTGTTAGCTGGTCTTGTTCTCTC-3′; reverse, 5′-CCATGGAGATTCTTCTTACTGACACA-3′. The reverse primer introduced an NcoI restriction site at the 3′ end of the PCR product. The PCR fragment was generated using mouse genomic DNA from Promega (Madison, WI). This fragment was then subcloned into pCR2.1 Topo TA vector (Invitrogen) following the manufacturer's protocol. The endonucleases BglII and NcoI were used to remove a 1.8-kb fragment of the Cyp2c37 5′-flanking region from the pCR2.1 Topo TA subclone. This 1.8-kb fragment was then ligated into a BglII- and NcoI-digested pGL3Basic luciferase reporter vector (Promega) producing the Cyp2c37 –1.8-kb luciferase reporter. The Cyp2c37 5′-flanking sequence from –1842 bp to –2887 bp was amplified from murine genomic DNA (Promega) by PCR using forward primer 5′-ACGCGTGGGAAATATAGGGCAATGTGCATC-3′ and reverse primer 5′-GAAGATCTATAGCACAGGGTCAGCTC-3′. The forward primer introduced a Mlu I site in the 5′ end of the PCR product for Cyp2c37 5′-flanking sequence. This ∼1-kb fragment was then subcloned into pCR2.1 Topo TA vector (Invitrogen), then digested by BglII to excise an ∼876-bp fragment using interior BglII sites, and finally ligated into a BglII-digested Cyp2c37 –1.8-kb luciferase reporter to create the Cyp2c37 –2.7-kb luciferase reporter. The subclone for the –1842-bp to –2887-bp Cyp2c37 5′-flanking sequence was digested by StuI and Mlu I to remove an ∼273-bp fragment (–2887 bp to –2614 bp). Subsequent ligation of this fragment into a StuI- and a Mlu I-digested Cyp2c37 –2.7-kb luciferase reporter created the Cyp2c37 –2.9-kb luciferase reporter. To create the Cyp2c37 –3.6-kb luciferase reporter, murine genomic DNA was again used as a template to amplify the Cyp2c37 5′-flanking sequence from –2540 bp to –3598 bp using a forward primer, 5′-CGCGTTTGTAGAGTTAGAAAACACAATTTGC-3′ (which also introduced a Mlu I site) and the reverse primer 5′-CCCAGTTTCTCATGCTCAAATGGAA-3′. The PCR product –2540 bp to –3598 bp of the Cyp2c37 5′-flanking sequence was subcloned into pCR2.1 Topo TA vector (Invitrogen) and digested with Mlu I and StuI to excise an ∼1050-bp fragment of Cyp2c37 5′-flanking sequence. This fragment was then ligated into a StuI- and Mlu I-digested Cyp2c37 –2.9-kb luciferase reporter, producing the Cyp2c37 –3.6-kb luciferase reporter. Mutations of putative binding sites in Cyp2c37 luciferase reporters were produced by site-directed mutagenesis using QuikChange (Stratagene, La Jolla, CA) on the Cyp2c37 –2.9-kb luciferase reporter background. Mutant oligonucleotides used in the site-directed mutagenesis reactions are as follows: –2791Bmut forward, 5′-ATAAAAGCAAACTTTTCCCCCCTCCATGCAATAAAAACAGTG-3′; –2791Bmut reverse, 5′-CACTGTTTTTATTGCATGGAGCCCCGAAAAGTTTGCTTTTAT-3′; –2791Amut forward, 5′-CAAGTTATAAAAGCCCCCCTTTCCCCCCTCCATGC-3′; –2791Amut reverse, 5′-GCATGGAGGGGGGAAAGGGGGGCTTTTATAACTTG-3′ (mutations underlined). Dye terminator DNA sequencing (Applied Biosystems) was used to verify all sequences.
Transcriptional Activation Assays. HepG2 cells were maintained in Eagle's minimum essential media (Invitrogen) supplemented with 10% fetal bovine serum and 1× penicillin-streptomycin-glutamine solution (Invitrogen). Cells were plated into 24-well plates at a density of ∼100,000 cells/well. Transfections included 100 ng of nuclear receptor (pCR3-mCAR), 100 ng of each luciferase promoter construct, and 1 ng of pRL-tk transfection control. Twenty-four hours after transfection, cells were treated with drugs or vehicle and incubated for an additional 24 h. Cells were subsequently lysed with 100 μl of 1× passive lysis buffer (Promega) for 30 min at room temperature with gentle rocking, and dual luciferase assays (Promega) were then performed on cell lysates according to the manufacturer's procedures.

Drug response of hepatic CYP2C37 mRNA in C3H mice. Mice were treated via gavage with corn oil, phenobarbital (80 mg/kg), or phenytoin (80 mg/kg) for 4 consecutive days and sacrificed 24 h after the last dose for isolation of hepatic total RNA. Quantitative RT-PCR was performed to determine hepatic CYP2C37 mRNA content in response to phenobarbital and phenytoin. Target gene was normalized to a reference gene, β-actin. Values expressed above represent the relative abundance of gene-specific mRNA ± S.E. p values were determined using Dunnett's method comparing treated to corn oil control. **, p ≤ 0.01; ***, p ≤ 0.001.
Statistics. Results from luciferase assays and quantitative RT-PCR are expressed as mean ± standard error of triplicate determinations. Single statistical comparisons were made using Dunnett's method; however, the Tukey-Kramer HSD test was used for multiple statistical comparisons when appropriate. The statistical tests were performed using JMP version 5.1 software (SAS Institute, Inc., Cary, NC) and the criterion of significance was set at p ≤ 0.05.
Results
Induction of Hepatic CYP2C37 mRNA in Wild-Type Mice. C3H wild-type mice were initially treated with corn oil (10 ml/kg), phenobarbital (80 mg/kg), or phenytoin (80 mg/kg) via gavage for 4 consecutive days. Twenty-four hours after the last dose was administered, RNA was isolated from the liver and quantitative RT-PCR was performed. CYP2C37 mRNA was induced ∼10-fold by phenobarbital and ∼20-fold by phenytoin (Fig. 1).
Element Identification and Binding Analysis. An alignment of ∼5 kb of all currently known murine Cyp2c 5′-flanking regions was conducted using Vector NTI Advance 9.0 (Invitrogen) to investigate the conservation of the previously identified PHREM within this murine gene subfamily. The alignment indicated that the PHREM is not conserved within the corresponding region of the Cyp2c37 gene, nor is it conserved in the other Cyp2c genes; thus, it is unique to Cyp2c29 (Fig. 2A). These findings suggested that induction of Cyp2c37 by phenytoin and phenobarbital is mediated by an alternative responsive region.
SeqLab GCG (Accelrys, San Diego, CA) was used to search 10 kb of the Cyp2c37 5′-flanking region for CAR/PXR response elements, defined as imperfect direct repeats of AGGTCA spaced by three to five nucleotides (DR-n). Several putative response elements were found upstream of the Cyp2c37 translation start site, including DR-3 motifs at –3205 bp, –3102 bp, –2820 bp, –2065 bp, and –1890 bp; one DR-5 motif at –3113 bp; and one DR-4 motif at –2791 bp (Fig. 2B). Luciferase reporters containing various lengths of the Cyp2c37 5′-flanking region were constructed and cotransfected with mCAR into HepG2 cells. The transfected cells were treated with mCAR activity modulators TCPOBOP and androstenol to facilitate the identification of CAR responsive regions. Cyp2c37 –1.8 kb and –2.7 kb luciferase reporter constructs were not activated by the presence of mCAR regardless of whether ligands were added (Fig. 3). However, the Cyp2c37 –2.9 kb and –3.6 kb luciferase reporters exhibited constitutive mCAR activation of ∼3-fold by mCAR that was not significantly induced further by TCPOBOP. The constitutive transactivation of the –2.9-kb and the –3.6-kb Cyp2c37 luciferase reporters was repressed by the addition of 10 μM androstenol (Forman et al., 1998) (Fig. 3). This effect could be partially reversed by the addition of 250 nM TCPOBOP, a mCAR-specific activator (Tzameli et al., 2000). These data indicated the presence of a CAR responsive region between –2.7 kb and –3.6 kb within Cyp2c37 5′-flanking sequence.
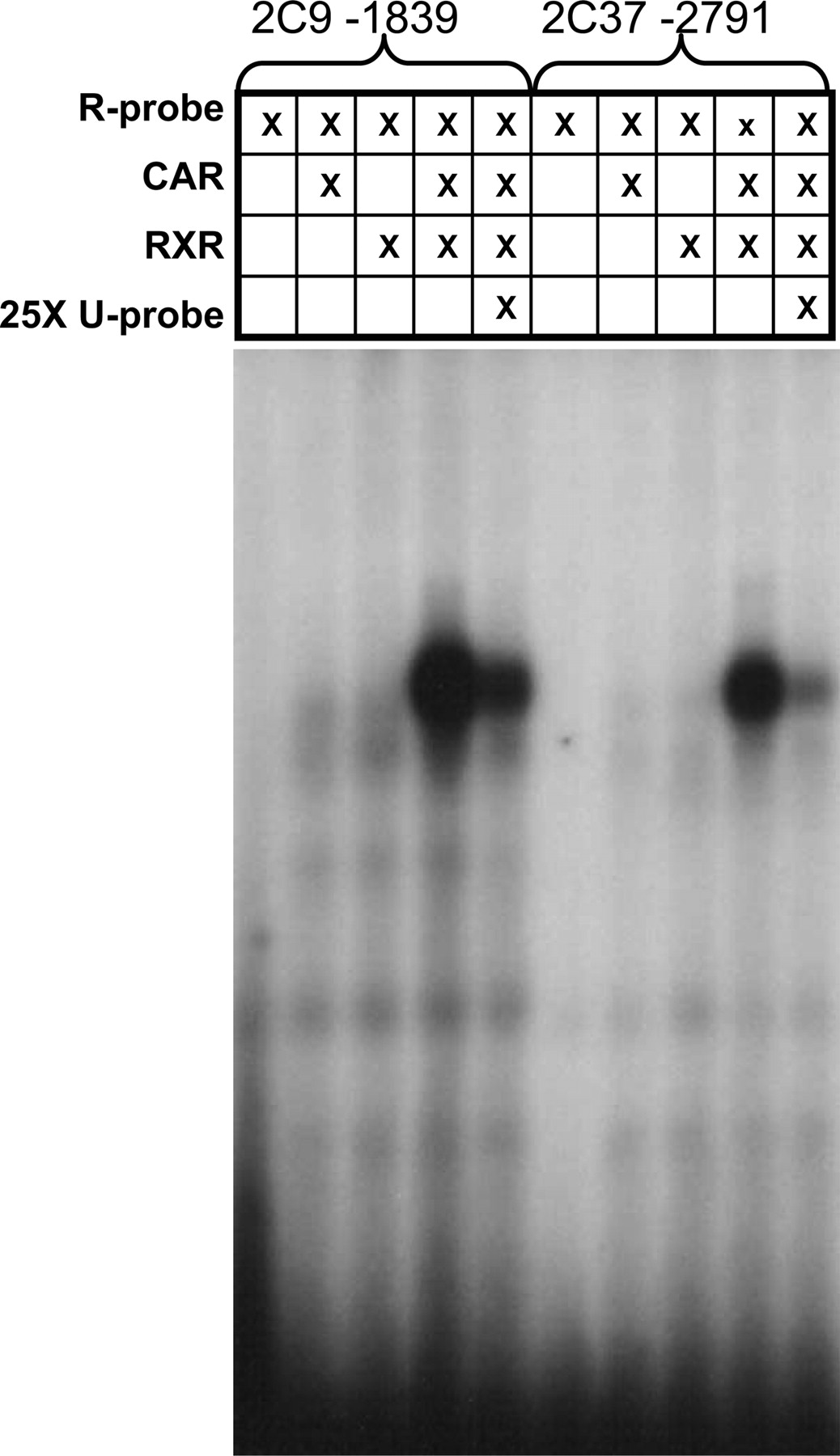
EMSAs were performed to identify binding of mCAR to any of the previously identified putative response elements within this region. EMSA results of radiolabeled oligonucleotides indicated that only the putative DR-4 response element, located at –2791 bp from the start of the Cyp2c37 translation, bound mCAR (Fig. 4). This interaction was reduced with 25-fold molar excess of nonradiolabeled oligonucleotides. The –2791 element is similar to a CAR-RE previously identified within the human CYP2C9 5′-flanking sequence at –1839 bp (Gerbal-Chaloin et al., 2002).

Alignment of CYP2C 5′-flanking regions and summary illustration of Cyp2c37 and Cyp2c29 5′-flanking regions. A, alignment of Cyp2c29 PHREM and surrounding sequence with corresponding 5′-flanking sequence of all presently known murine CYP2C genes. B, a schematic representation of the Cyp2c37 and Cyp2c29 5′-flanking region providing a summary of putative and functional transcription factor binding sites.
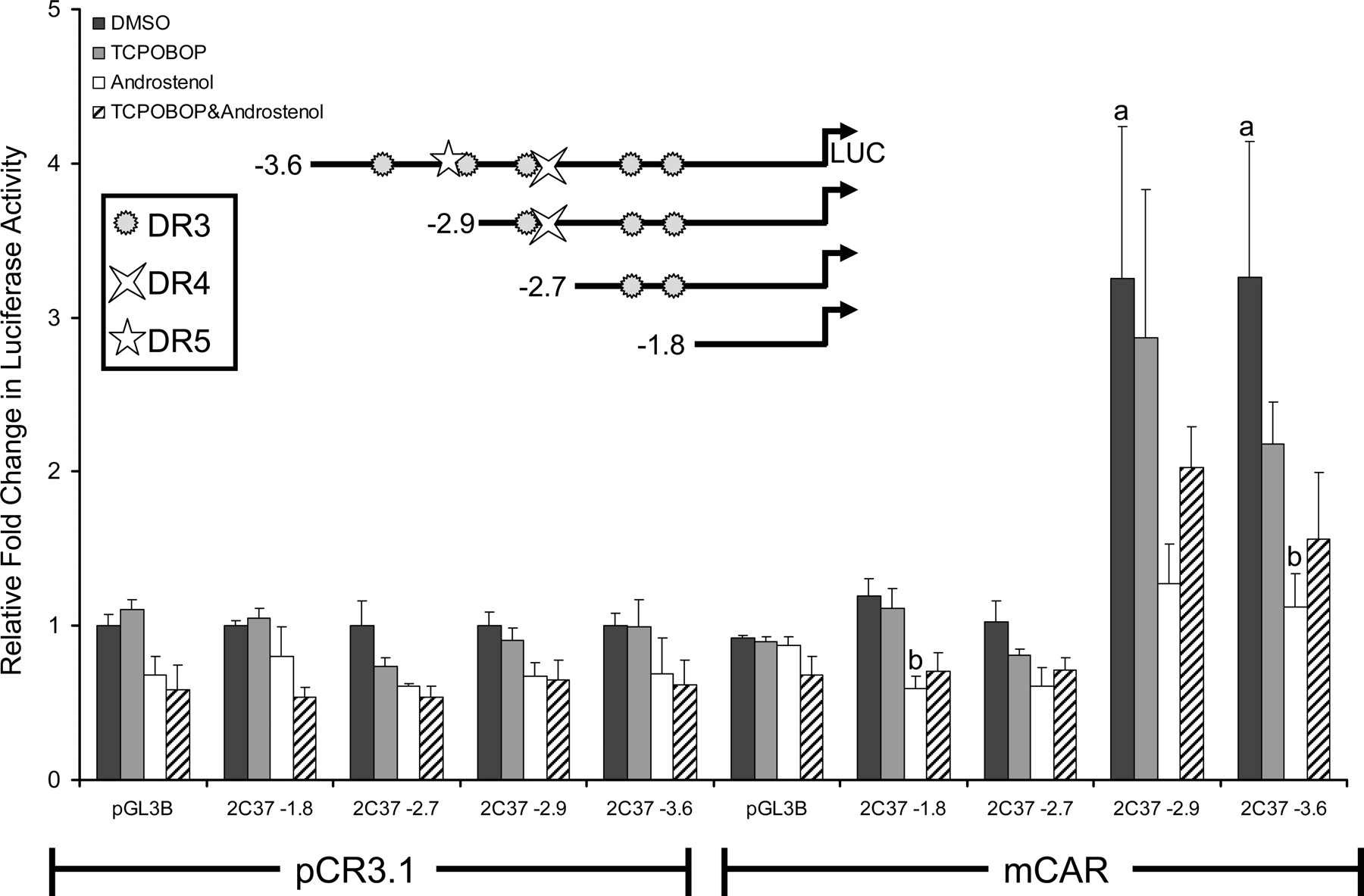
Transcriptional activation analysis of the Cyp2c37 5′-flanking region by mCAR. HepG2 cells were cotransfected with pRL-Tk (internal transfection control), expression vectors mCAR (pCR3) or empty (pCR3.1), and pGL3 Basic luciferase vectors containing varying lengths of the Cyp2c37 5′-flanking region to evaluate mCAR effects on gene reporter activity. Androstenol (10 μM) and TCPOBOP (250 nM) were used to modulate mCAR activity. These data represent the results of three independent transfections. p values were determined using the Tukey-Kramer HSD test. a, p ≤ 0.05; mCAR significantly increased luciferase activity compared with no receptor DMSO control group. b, p ≤ 0.05; androstenol significantly decreased luciferase activity compared with mCAR DMSO treatment group.

Analysis of mCAR binding to –2791 putative CAR-RE using EMSA. Radiolabeled –2791 putative CAR-RE (100,000 cpm 32P) oligonucleotides were incubated with in vitro transcribed and translated mCAR, hRXR, or mCAR/hRXR proteins. In parallel experiments, incubation was performed in the presence of a 25-fold molar excess of nonradiolabeled probe. EMSA data demonstrated that the Cyp2c37 putative –2791 CAR-RE binds mCAR/RXR as indicated by the presence of a band shift. This interaction could be specifically competed out using nonradiolabeled Cyp2c37 –2791 CAR-RE oligonucleotides. The positive control, CYP2C9 –1839 CAR-RE, showed similar results.
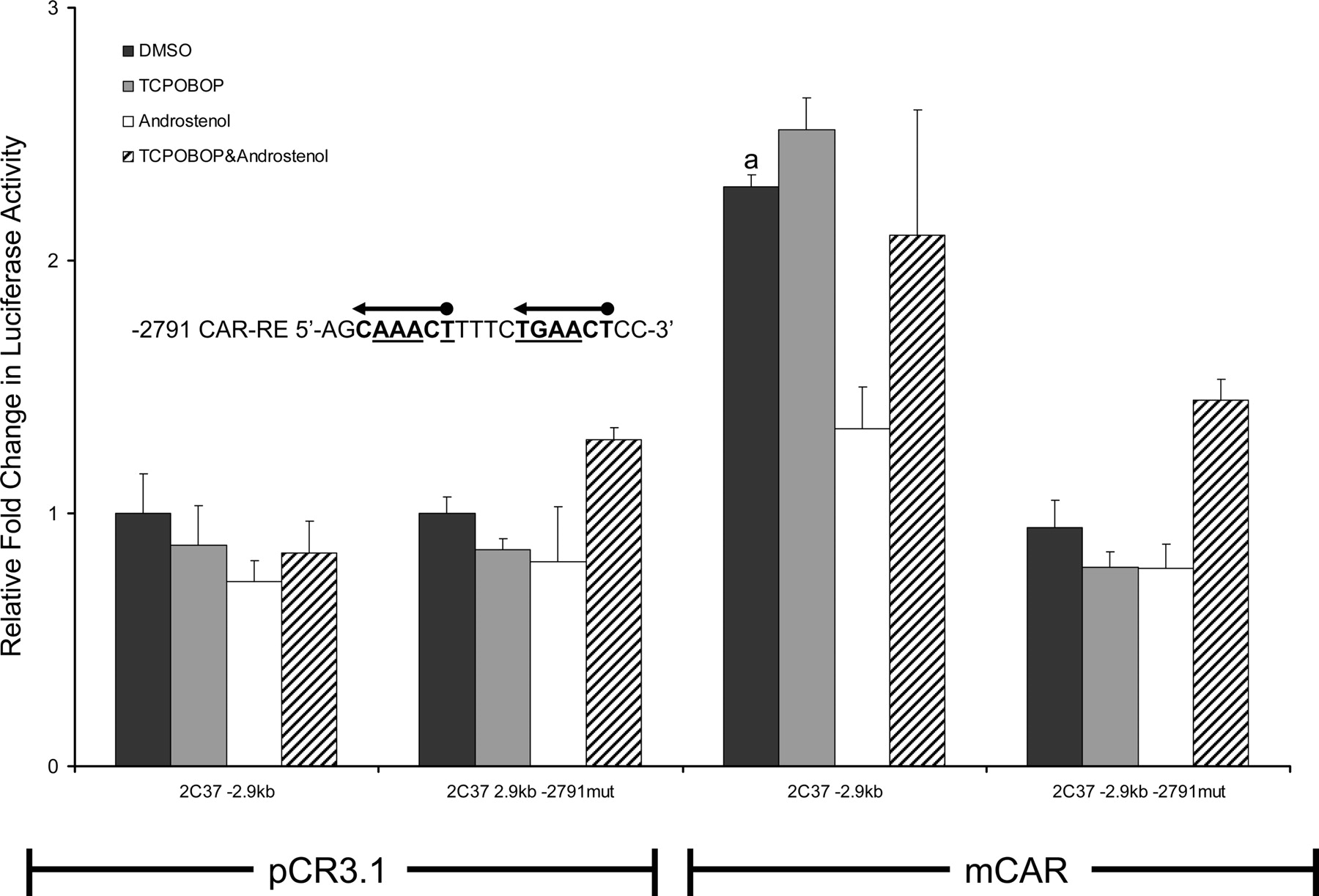
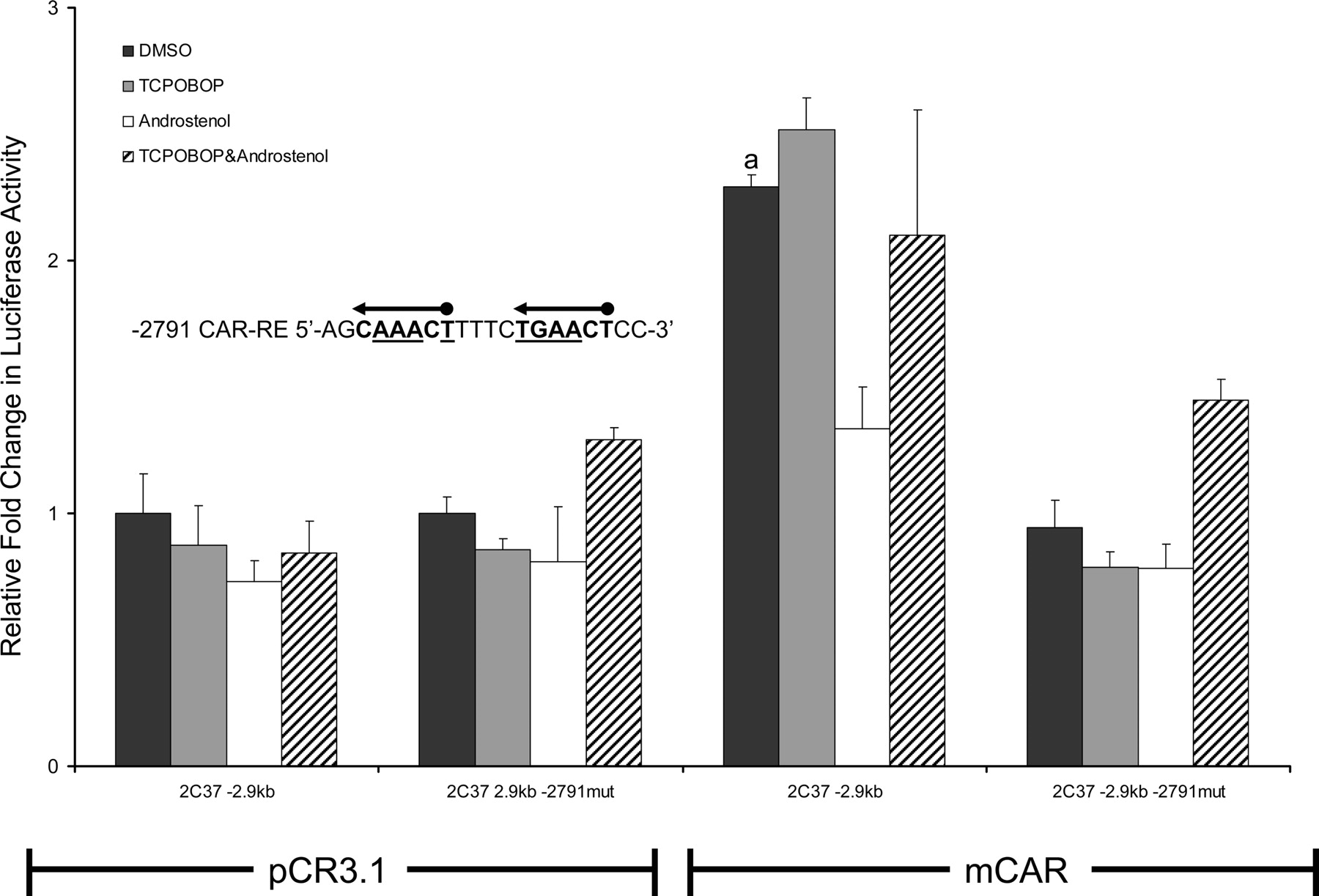
Functional analysis of the Cyp2c37 –2791 CAR-RE mutant. HepG2 cells were cotransfected with pRL-Tk (internal transfection control), expression vectors mCAR (pCR3) or empty (pCR3.1), and Cyp2c37 wild-type or –2.9 kb Cyp2c37 mutant luciferase reporter to evaluate mCAR effects on gene reporter activity. Underlined nucleotides in Cyp2c37 –2791 putative CAR-RE sequence were mutated to deoxycytidines using site-directed mutagenesis (Stratagene). These data represent the results of three independent transfections. Mutation of –2791 imperfect DR-4 CAR-RE abolished mCAR transactivation. This reporter was also not responsive to androstenol (10 μM) repression. p values were determined using the Tukey-Kramer HSD test. a, p ≤ 0.05; mCAR significantly increased luciferase activity compared with no receptor DMSO control group. b, p ≤ 0.05; androstenol significantly decreased luciferase activity compared with mCAR DMSO treatment group.
Cyp2c37 –2791 CAR-RE Functional Analysis. Mutagenesis was performed to determine whether the Cyp2c37 –2791 CAR-RE site is necessary for mCAR constitutive transactivation of the Cyp2c37 –2.9-kb luciferase reporter. Mutation of the Cyp2c37 –2791 CAR-RE completely abolished mCAR transactivation of the Cyp2c37 –2.9-kb luciferase reporter (Fig. 5).
Evaluation ofCyp2c37Induction in CAR-Null and PXR-Null Mice. CAR-null (Fig. 6) and PXR-null mice (Fig. 7) were used to examine whether induction of hepatic CYP2C37 mRNA by phenytoin is mediated by CAR or PXR in vivo. Hepatic CYP2B10 mRNA and CYP3A11 mRNA were used as positive controls to evaluate xenobiotic activation by CAR and PXR, respectively. In CAR-null mice (Fig. 6), induction of CYP2B10 mRNA by phenytoin and phenobarbital was completely abolished as expected. The induction of CYP2C29 and CYP2C37 mRNA by phenobarbital was dramatically reduced (∼98% and ∼87%, respectively) in CAR-null mice compared with their congenic controls. Similarly, induction of CYP2C29 and CYP2C37 mRNA by phenytoin in CAR-null mice was reduced by ∼99% and ∼92%, respectively. However, the induction of CYP3A11 mRNA by phenytoin was only moderately decreased (∼66%) in CAR-null mice.
We next examined induction in PXR-null mice in parallel with the congenic wild-type mice (Fig. 7). CYP3A11 mRNA was induced ∼6-fold by PCN in wild-type mice, and this induction was completely abolished in PXR-null mice. In contrast, induction of CYP3A11 mRNA by phenytoin was reduced ∼45% in PXR-null mice. Induction of CYP2B10, CYP2C29, and CYP2C37 mRNA, respectively, by phenytoin was attenuated but not abolished in PXR-null mice (Fig. 7). Taken together, these data suggest that induction of CYP2C29, CYP2C37, and CYP2B10 mRNA by phenytoin and phenobarbital is mediated predominantly by CAR. This hypothesis is further supported by the observation that PCN, a known mPXR agonist, does not induce CYP2C29, CYP2C37, or CYP2B10 mRNA. In contrast, the induction of CYP3A11 mRNA by phenytoin appears to be mediated by CAR and PXR as suggested by the fact that induction is not abolished in either CAR-null or PXR-null mice.

Evaluation of Cyp2c37 induction by phenytoin in CAR-null mice. Mice were treated as described above. Quantitative RT-PCR was performed to evaluate target gene mRNA content in response to phenobarbital and phenytoin. Target genes were normalized to a reference gene, β-actin. CYP2B10, CYP2C29, and CYP2C37 mRNA was induced by phenobarbital and phenytoin in congenic wild-type mice, but induction was abolished in CAR-null mice. CYP3A11 mRNA was also induced by phenobarbital and phenytoin in wild-type mice; however, induction of CYP3A11 mRNA by phenytoin was not removed in CAR-null mice. Values expressed above represent the relative abundance of gene-specific mRNA ± S.E. p values were determined using the Tukey-Kramer HSD test. a, p < 0.05, significantly higher than corn oil controls. b, p < 0.05, significantly lower than wild-type treated with phenobarbital. c, p < 0.05, significantly lower than wild-type treated with phenytoin.

Evaluation of Cyp2c37 induction by phenytoin and PCN in PXR-null mice. Mice were treated with phenytoin as described above. Mice treated with PCN (80 mg/kg) were treated orally for 3 consecutive days. Quantitative RT-PCR was performed to evaluate target gene mRNA content in response to PCN and phenytoin. Target genes were normalized to a reference gene, β-actin. CYP2B10, CYP2C29, and CYP2C37 mRNA induction by phenytoin was reduced, not abolished in PXR-null mice. CYP3A11 mRNA was induced by PCN in congenic wild-type mice, but was eliminated in PXR-null mice. In contrast, induction of CYP3A11 mRNA by phenytoin was reduced, not abolished in PXR-null mice. Values expressed above represent the relative abundance of gene-specific mRNA ± S.E. p values were determined using the Tukey-Kramer HSD test. a, p < 0.05; significantly higher than corn oil controls. b, p < 0.05; significantly lower than wild-type treated with PCN. c, p < 0.05, significantly lower than wild-type treated with phenytoin.
Discussion
Several studies have examined the drug-induced transcriptional regulation of the human CYP2C subfamily of P450 enzymes by CAR and PXR; however, relatively little is known of the regulation of their murine counterparts. Currently, a total of 15 murine Cyp2c genes have been identified within a cluster on chromosome 19 (Nelson et al., 2004; Wang et al., 2004b). Within this large P450 gene subfamily, only the Cyp2c29 gene has been shown to be drug-inducible thus far (Jackson et al., 2004). Herein, we show that hepatic CYP2C37 mRNA is induced by phenobarbital and phenytoin, becoming the second known drug-inducible murine Cyp2c gene. We demonstrate using CAR-null mice that the induction of CYP2C37 mRNA is primarily CAR-dependent. In addition, we identify a functional DR-4 CAR-RE at –2791 bp from the translation start site of the Cyp2c37 gene.
Alignments of the 5′-flanking regions of the murine Cyp2c genes indicate that particular subsets of Cyp2c members including Cyp2c29, Cyp2c39, and Cyp2c38 share high sequence homology; however, as a group, the 5′-flanking regions are not highly conserved. The alignments show that the PHREM is exclusive to Cyp2c29 and is not conserved in any of the other corresponding 5′-flanking regions. These findings suggested that induction of hepatic CYP2C37 mRNA is mediated by an alternative drug responsive region. A number of putative CAR binding sites were identified; however, only the putative DR-4 CAR-RE (–2791) bound mCAR, and mutagenesis of this site indicated that it is necessary for mCAR transactivation. These results are consistent with our previous studies examining transcriptional regulation of the Cyp2c29 gene by CAR (Jackson et al., 2004). In addition, the alignment of the 5′-flanking regions of the murine Cyp2c genes indicated that the newly identified –2791 CAR-RE was unique to Cyp2c37 and this region was not conserved among the other Cyp2c members (Fig. 8A). Although neither responsive region was conserved, the lack of conservation does not prove that the other subfamily members are noninducible after exposure to phenobarbital or phenytoin.

Alignment of CYP2C 5′-flanking regions and summary illustration of common mCAR and mPXR activators. A, alignment of Cyp2c37 –2791 CAR-RE and surrounding sequence with corresponding 5′-flanking sequence of all presently known murine CYP2C genes. B, illustration showing the shared activators of the xenobiotic sensing nuclear receptors PXR and CAR and their respective genes that are induced.
Using quantitative RT-PCR, we determined that CYP2C37 mRNA is induced by phenytoin and phenobarbital in wild-type mice, but in CAR-null mice the induction of CYP2C37 mRNA was essentially eliminated. These data indicate that phenobarbital and phenytoin induction is primarily CAR-dependent. Phenytoin induction of CYP2C37 and CYP2C29 mRNA was reduced moderately in PXR-null mice; however, a significant amount of induction remained. Although it is possible that that PXR is directly involved in the induction of Cyp2c37 and Cyp2c29 by phenytoin, its effects could be indirect. First, induction of Cyp2c37 and Cyp2c29 by phenytoin was essentially abolished in CAR-null mice, suggesting minimal contribution of PXR. Second, the mPXR-specific agonist PCN did not induce these genes in wild-type mice. A study by Maglich et al. (2002) demonstrated that expression of mCAR is induced by PCN in wild-type mice, but not in PXR-null mice. These results suggest that CAR expression is regulated by PXR in mice; therefore, CAR expression may be lower in PXR-null mice. A reduction in the expression of CAR or other nuclear receptors could indirectly attenuate the induction of CYP2C37 and CYP2C29 mRNA by phenytoin in PXR-null mice. Thus, PXR could be involved indirectly in regulating murine CYP2C gene regulation. In humans, both CAR and PXR appear to regulate the drug-induced expression of CYP2C8 and CYP2C9 (Chen et al., 2004; Ferguson et al., 2005; Al-Dosari et al., 2006) by compounds like the CAR agonist 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-3,4-dichlorobenzyl)oxime (CITCO) and the PXR agonist rifampicin. In contrast, the present studies suggest that CAR is the primary regulator of murine Cyp2c37 and Cyp2c29 drug induction, suggesting a species difference in CYP2C regulation.
Although initially monitored to determine PXR activation by PCN, CYP3A11 mRNA was observed to be induced by phenytoin in wild-type mice. Induction of CYP3A11 mRNA by phenytoin was reduced, but not abolished in either CAR-null or PXR-null mice. In contrast, PCN induction of CYP3A11 mRNA was abolished in PXR-null mice, consistent with PXR-mediated induction (Xie et al., 2000; Staudinger et al., 2001a,b; Goodwin et al., 2002b). Thus, phenytoin appears to act as a mPXR and mCAR activator similar to dieldrin and clotrimazole (Fig. 8B) (Zhang et al., 2004).
In conclusion, we have demonstrated that CYP2C37 mRNA is induced by phenobarbital and phenytoin. We have shown that the induction of hepatic CYP2C37 mRNA by phenobarbital and phenytoin is CAR-dependent. We identified a functional DR-4 CAR-RE located at –2791 bp from the translation start site of Cyp2c37, which we propose mediates CAR-dependent drug induction of the Cyp2c37 gene. The mPXR agonist PCN does not induce murine Cyp2c37 or Cyp2c29. Thus, our studies also suggest that CAR is the predominate regulator of these murine Cyp2c genes (Fig. 8B). Furthermore, our studies indicate that phenytoin probably activates both mCAR and mPXR in the induction of the Cyp3a11 gene.
Acknowledgments
We especially thank Dr. Jeff Staudinger (University of Kansas, Department of Pharmacology and Toxicology) forthe gift of the PXR-null mice to Dr. Masahiko Negishi [National Institute of Environmental Health Sciences (NIEHS), Laboratory of Reproductive and Developmental Toxicology]. We acknowledge Rick Moore (NIEHS, Laboratory of Reproductive and Developmental Toxicology) for excellence in maintaining and managing our CAR-null and PXR-null breeding programs. In addition, we recognize Dr. Grace Kissling (NIEHS, Biostatistics Branch) for expertise in statistical analysis. We also thank Louise Harris (NIEHS, Animal Facility) for expertise in animal dosing.
Footnotes
-
This research in entirety is a portion of the dissertation research in progress by Jonathan P. Jackson and was supported by the Intramural Research Program of the National Institutes of Health and National Institute of Environmental Health Sciences. This work was previously presented at Experimental Biology 2006 meeting, April 1–5, 2006, San Francisco, CA.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012005.
-
ABBREVIATIONS: P450, cytochrome P450; hRXR, human retinoid X receptor; PXR, pregnane X receptor; DR-n, direct repeat spaced by n nucleotides; PHREM, phenytoin responsive module; CAR, constitutive androstane receptor; CAR-RE, CAR responsive element; PCN, 5-pregnen-3β-ol-20-one-16α-carbonitrile; TCPOBOP, 1,4-bis-[2-(3, 5-dichloropyridyloxy)]benzene; DMSO, dimethyl sulfoxide; C3H, C3H/HeNCrlBR; EMSA, electrophoretic mobility shift assay; bp, base pair(s); mCAR, murine CAR; kb, kilobase(s); RT-PCR, reverse transcription-polymerase chain reaction; HSD, honestly significant difference.
-
↵1 Current affiliation: CellzDirect, Inc., Austin, TX.
- Received July 14, 2006.
- Accepted August 23, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}