


Abstract
Identification and characterization of the pregnane X receptor (PXR) as a key regulator of cytochrome P450 3A (CYP3A) gene expression has led to an increased understanding of the molecular basis of many drug-drug interactions. Mice lacking PXR (PXR-KO) were used in the present study to delineate the role of PXR in regulating hepatomegaly and regulating the activity of CYP3A, organic anion transporting polypeptide-2 (Oatp2), andCyp7a1 (cholesterol 7α-hydroxylase) gene products in vivo. Pregnenolone-16α-carbonitrile (PCN) produced hepatomegaly in the wild-type mice but not in the PXR-KO mice. PCN increased both the number of proliferating cell nuclear antigen immuno-positive nuclei and apparent cell size in the wild-type mice but not in the PXR-KO mice. To determine the role PXR plays in regulating CYP3A activity, 6β-hydroxylation of testosterone and the duration of the loss of righting reflex following administration of the muscle-relaxant zoxazolamine were measured. PCN increased the level of testosterone 6β-hydroxylation and decreased the duration of the loss of righting-reflex time following zoxazolamine administration in wild-type mice, but did not effect either of these parameters in PXR-KO mice. PCN increased the hepatic uptake of [3H]digoxin, an Oatp2 substrate, in wild-type mice but not in the PXR-KO mice. Similarly, PCN decreased bile acid excretion in wild-type mice but not in the PXR-KO mice. Taken together, these data demonstrate a pivotal role for PXR in the regulation of drug-induced hepatomegaly and in the metabolism (CYP3A), transport (Oatp2), biosynthesis (Cyp7a1), and excretion of xenobiotics and bile acids in vivo.
The pregnane X receptor (PXR1; NR1I2) is the molecular target of many xenobiotics including the synthetic steroid pregnenolone-16α-carbonitrile (PCN) and mediates the xenobiotic-inducible transcription of CYP3A family members across species (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998; Jones et al., 2000; Xie et al., 2000a). Recent research has led to the idea that in addition to serving as a broad specificity “xenosensor”, PXR function is also activated by lithocholic acid, thereby regulating the expression ofCyp3a11, organic anion transporting polypeptide (Oatp2), and Cyp7a1 genes in mice in a coordinate manner (Staudinger et al., 2001).
For many years it has been known that pretreatment of rodents with certain steroids produces protection from various forms of toxicants. Subsequently, it was shown that these “catatoxic” or protective steroids exert their effect by accelerating the metabolism of the toxicant (Selye, 1971). A screen designed to identify additional protective compounds revealed that PCN is one of the most potent catatoxic chemicals (Selye, 1970). It was later shown that the protective nature of PCN probably involves the stimulation of transcription and activity of the CYP3A subfamily of the cytochrome P450 family of monooxygenases in rodents (Newman and Guzelian, 1983; Schuetz et al., 1984), which are highly expressed in liver and intestine. CYP3A family members encode broad specificity heme-containing monooxygenases that catalyze the oxidation of a wide variety of structurally dissimilar compounds. In humans, the CYP3A enzymes are collectively responsible for the oxidation of greater than 60% of all clinically prescribed drugs, as well as many steroids and bile acids (Guzelian, 1988; Maurel, 1996).
Early research on PCN revealed its ability to induce hepatomegaly in rodents (Garg et al., 1970). Subsequent reports indicated that this marked increase in liver size was due to increases in both cell number (hyperplasia) and cell size (hypertrophy) (Japundzic et al., 1974). This research raises questions about the role of PXR in mediating these two processes.
The Oatp2 gene-product, in part, mediates the hepatic uptake of cardiac glycosides, such as ouabain and digoxin in rodents (Abe et al., 1998;Reichel et al., 1999). Insights regarding the involvement of drug transporter systems in the regulation of drug disposition came when it was noted that newborn mammals exhibit a marked sensitivity to ouabain, a nonmetabolized cardiac glycoside, when compared with adults (Klaassen, 1972, 1973). This increased susceptibility of newborn rats to ouabain appeared to be due to the inability of the liver of newborn animals to concentrate ouabain. Additionally, pretreatment of both newborn and adult rodents with PCN markedly increases hepatic uptake and biliary excretion of ouabain (Klaassen, 1974a,b). PCN treatment also markedly enhanced the uptake of [3H]digoxin, an Oatp2 substrate, in isolated hepatocytes (Eaton and Klaassen, 1979). This research strongly implies the existence of a xenobiotic-inducible drug transporter system separate from the xenobiotic-inducible drug-metabolizing systems and suggests that these two systems could in principle be coregulated.
PCN treatment has been shown to enhance cholesterol turnover (Honohan and Parkinson, 1975), decrease cholesterol hydroxylase activity (Mason and Boyd, 1978), and decrease biliary bile acid excretion (Turley and Dietschy, 1984). More recent evidence reveals that bothCyp7a1 transcription and cholesterol 7α-hydroxylase activities are dramatically reduced following administration of PCN in rodents (Li et al., 1990; Stahlberg, 1995). These data suggest a role for the PXR in the regulation of Cyp7a1 transcription, although it is not currently clear whether this is a direct or an indirect effect.
In this study, wild-type mice and mice lacking functional PXR (PXR-KO) were used to examine specific functional endpoints following PCN treatment. As an indication of cell proliferation, the presence of proliferating cell nuclear antigen (PCNA) immunoreactivity was measured. The number of cells per high power visual field (light microscopy, 400× magnification) was determined as a reflection of cell size. As a measure of CYP3A activity, both testosterone 6β-hydroxylation and zoxazolamine-induced paralysis time were measured. Hepatic uptake of [3H]digoxin and bile acid excretion into bile were measured as markers for Oatp2 and cholesterol 7α-hydroxylase activities, respectively. Taken together, these experiments demonstrate that, in addition to regulating the transcription of CYP3A, Oatp2, andCyp7a1, PXR activation by PCN regulates the activity of their gene-products in vivo. Thus, PXR is involved in regulating both xenobiotic homeostasis and bile acid homeostasis in vivo, and these two processes appear to be coupled, at least in part, by PXR activation. Furthermore, these experiments demonstrate a pivotal role for PXR in mediating PCN-induced hepatomegaly through induction of cellular hypertrophy and hyperplasia.
Experimental Procedures
Maintenance and Treatment of PXR-KO and Wild-Type Mouse Populations.
Generation of the PXR-KO mice was previously described (Staudinger et al., 2001). Adult male wild-type mice and PXR-KO mice were maintained on standard laboratory chow and were allowed food and water ad libitum. All mice were treated once a day i.p. with either vehicle (corn oil) or PCN at the indicated doses for 4 days.
Hepatocyte Proliferation and Cell Size Following PCN Treatment.
Four mice were randomly allocated per treatment group. Livers from mice pretreated with corn oil or PCN were removed and weighed on the morning of day 5 following 4 days of treatment. Portions of the liver were fixed and immunostained for the presence of PCNA, as described previously (Hood et al., 1999). The cell size was determined by counting the total number of nuclei in a given field at 400× magnification following hematoxylin and eosin staining. The slides were blinded and randomized, and the nuclei were counted in 15 to 20 fields/treatment group. The data are expressed as cells per high power field.
RNA Isolation and Northern Blot Analysis.
Total RNA was isolated from liver using a commercially available reagent (Trizol; Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Total RNA (10 μg) was resolved on a 1% agarose/2.2 M formaldehyde-denaturing gel and transferred to a nylon membrane (Hybond N+; Amersham Pharmacia Biotech, Inc., Piscataway, NJ). Blots were hybridized with32P-labeled cDNAs corresponding to the rodent cDNA sequences for Cyp3a11 (bases 69–1609; GenBank no. NM 007818), Oatp2 (bases 1–240; GenBank no. AB031814), Cyp7a1 (bases 235–460; GenBank no. J05460), and β-actin (CLONTECH, Palo Alto, CA). Northern blots were scanned, and band intensities were quantified using ImagQuant software (Molecular Dynamics, Sunnyvale, CA).
Preparation of Microsomes.
Liver microsomes were prepared as previously described (Pearce et al., 1996). Briefly, 3 ml of homogenization buffer (50 mM Tris-HCl, pH 7.4, 150 mM KCl, and 2 mM EDTA)/gram of liver on ice was used to homogenize samples using a motor driven liver homogenizer and a Teflon pestle. The liver homogenates were centrifuged at 12,000g for 20 min at 4°C. The resulting fraction was centrifuged at 104,000gfor 60 min at 4°C. The pellets were fully resuspended in ice-cold wash buffer (150 mM KCl and 10 mM EDTA, pH 7.4) and centrifuged again at 104,000g for 60 min at 4°C. The pellets were resuspended in 0.4 ml of 0.25 M sucrose/gram of starting liver weight and frozen at −80°C.
Protein Concentration and High-Pressure Liquid Chromatography Testosterone 6β-Hydroxylation Assay.
The protein concentration of isolated microsomal preparations was determined with the bicinchoninic acid protein assay reagent kit (Pierce, Rockford, IL), as described by the manufacturer. Microsomal testosterone 6β-hydroxylase activities were determined as described previously (Pearce et al., 1996).
Zoxazolamine Paralysis.
All mice were injected i.p. with 112 mg/kg zoxazolamine in 2% Tween 80 in 0.9% saline solution. Paralysis time was measured as the time between loss and return of the righting reflex (Selye, 1971).
[3H]Digoxin Uptake.
Hepatic uptake of [3H]digoxin was assayed as previously described (Klaassen, 1972). Briefly, each experimental group was administered a digoxin solution at a dose of 1 mg/kg i.v. dissolved in 10% ethanol and 5% glucose with 10 μCi/kg [3H]digoxin. Livers were removed after 5 min, and a portion of the tissue was digested in 10% (w/v) tissue and gel solubilizer (Packard Instruments, Meriden, CT) at 60°C until dissolved. A 200-μl aliquot was removed and placed in a scintillation vial with 15 ml of scintillation fluid (Research Product International Corp., Mount Prospect, IL) and quantified in a scintillation counter.
Biliary Bile Acid Excretion.
Mice were anesthetized using pentobarbital (60 mg/kg). The bile duct was exposed using a midline incision and cannulated with polyethylene tubing (PE-90). Bile samples were collected for 30 to 60 min. The animal's body temperature was maintained at 37°C throughout the procedure. Bile acid concentrations were determined using the bile acids kit (Sigma Diagnostics, St. Louis, MO). Briefly, bile was diluted in fetal bovine serum, and biliary bile acid concentration was determined using a standard curve generated using a bile acid calibrator set (Sigma Diagnostics; cat no. 450-11).
Statistical Analysis.
Differences between liver mass, messenger RNA levels, the numbers of PCNA positive nuclei, cells per field, and enzymatic activities in vehicle and PCN-treated animals were determined using a one-way analysis of variance followed by the Duncan's multiple range posthoc test.
Results
PXR Is Required for PCN-Induced Hepatomegaly.
Acute treatment of rodents with PCN causes an increase in liver mass and is thought to be due to increases in cellular hypertrophy and hyperplasia (Japundzic et al., 1974). To determine the role of PXR in this process, liver and body weight were quantified in the wild-type and PXR-KO mice following treatment with PCN. As expected, the wild-type mice exhibited approximately a 40% increase in the relative liver weight (grams of liver per 100 g of total body weight) from 4.5 ± 0.1 to 6.3 ± 0.4, whereas the PXR-KO mice did not exhibit any increase following PCN treatment (Fig.1A). The wild-type mice exhibited about a 10-fold increase in the percentage of total PCNA immunopositive nuclei following PCN treatment from 0.7 ± 0.2 to 11.4 ± 1.0, whereas the PXR-KO mice did not (Fig. 1B). When cell size was determined using the number of cells per high power field, a decrease of about 23% from 69.7 ± 1.2 to 53.6 ± 0.6 in the number of cells per field was observed in PCN-treated wild-type mice, indicating an increase in cell size; however, this effect was totally absent in the PXR-KO mice (Fig. 1C). Photomicrographs of representative liver sections stained with anti-PCNA antibodies and hematoxylin reveal both an increase in cell division and cell size in the PCN-treated wild-type mice but not in the PXR-KO mice (Fig.2). These results are similar to those published by Xie et al. (2001a), except hepatomegaly was observed in the PXR-null mice expressing the constitutively active human SXR/PXR transgene under the control of the albumin promoter.
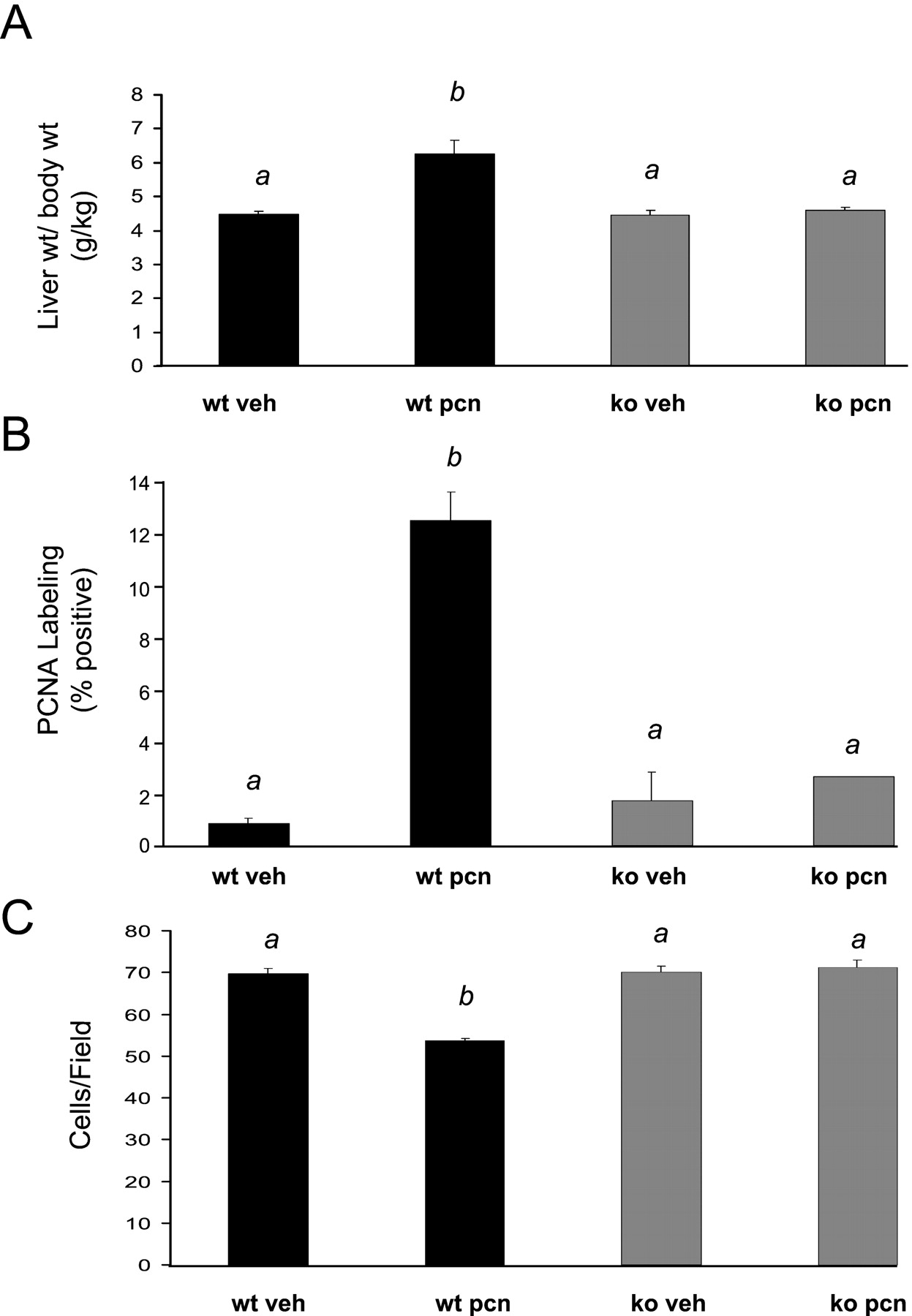
Liver enlargement and hepatocyte proliferation caused by PCN.
A, mice were treated with vehicle (corn oil) or PCN (400 mg/kg) for 4 days. On the morning of the day 5, the body weight and the liver weight were measured. The percentage of liver weight relative to total body weight is represented as mean ± S.E.M. and was determined in at least five animals. B; thin sections of liver from vehicle or PCN-treated mice were stained with an anti-PCNA antibody. All hepatocytes and PCNA-positive hepatocytes were counted in five randomly selected high-power fields per section. The labeling index was determined as the percentage of counted hepatocytes showing positive staining for PCNA. C, all hepatocytes in a given field were counted. Five high-power (400×) fields per liver section were counted. Minimums of three liver sections per treatment group were counted. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). wt, wild-type; veh, vehicle.

Immunohistochemistry of PCNA positive nuclei.
Wild-type and PXR-KO mice were treated for 4 days i.p. with either vehicle (corn oil) or PCN (400 mg/kg). On the morning of the day 5, livers were fixed and sectioned and stained for the presence of proliferating cell nuclear antigen and then counterstained with hematoxylin. Photomicrographs containing arrows pointing to the immunopositive nuclei of representative sections are shown from a vehicle treated wild-type mouse (A), a wild-type mouse treated with PCN (B), a vehicle treated PXR-knockout mouse (C), and a PXR-knockout mouse treated with PCN (D).
PXR Regulates the Expression and Activity of Cyp3a11.
Because PXR has been shown to mediate PCN-inducible transcription of rodent CYP3A family members (Bertilsson et al., 1998;Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998;Schuetz et al., 1998; Goodwin et al., 1999), the expression ofCyp3a11 in wild-type mice and PXR-KO mice was examined. Wild-type mice exhibited a 6-fold increase in the hepatic expression ofCyp3a11 following PCN treatment (Fig.3A). Surprisingly, the vehicle treated PXR-KO mice exhibited a 3-fold increase in the basal expression level of Cyp3a11 when compared with their wild-type vehicle treated littermates (Fig. 3B). Induction of Cyp3a11expression following PCN treatment was totally absent in the PXR-KO mice. These results are consistent with those previously published (Staudinger et al., 2001).

Induction of Cyp3a11expression.
A, wild-type and PXR-KO mice were treated for 4 days i.p. with either vehicle (corn oil) or PCN (400 mg/kg). On the morning of the day 5, total liver RNA was isolated and run on a 1% agarose gel. After transferring to a nylon membrane, the blot was probed with a32P-labeled cDNA probe for mouse Cyp3a11. B, the level ofCyp3a11 induction was quantified and graphed. The data are expressed as mean ± S.E.M. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). wt, wild-type; veh, vehicle.
Because PCN treatment is known to induce both the expression (Gonzalez et al., 1985) and activity (Eberhart et al., 1992) of hepaticCYP3A family members, it was of interest to measure CYP3A activity as reflected by testosterone 6β-hydroxylation. Wild-type mice treated with PCN exhibited a 5- to 7-fold increase in liver microsomal testosterone 6β-hydroxylation, whereas the PXR-KO mice exhibited no such increase in testosterone 6β-hydroxylation following PCN treatment (Fig. 4A). Notably, the basal level of testosterone 6β-hydroxylation was increased approximately 2-fold in the vehicle treated PXR-KO liver microsomes when compared with testosterone 6β-hydroxylation levels in the vehicle treated wild-type liver microsomes.
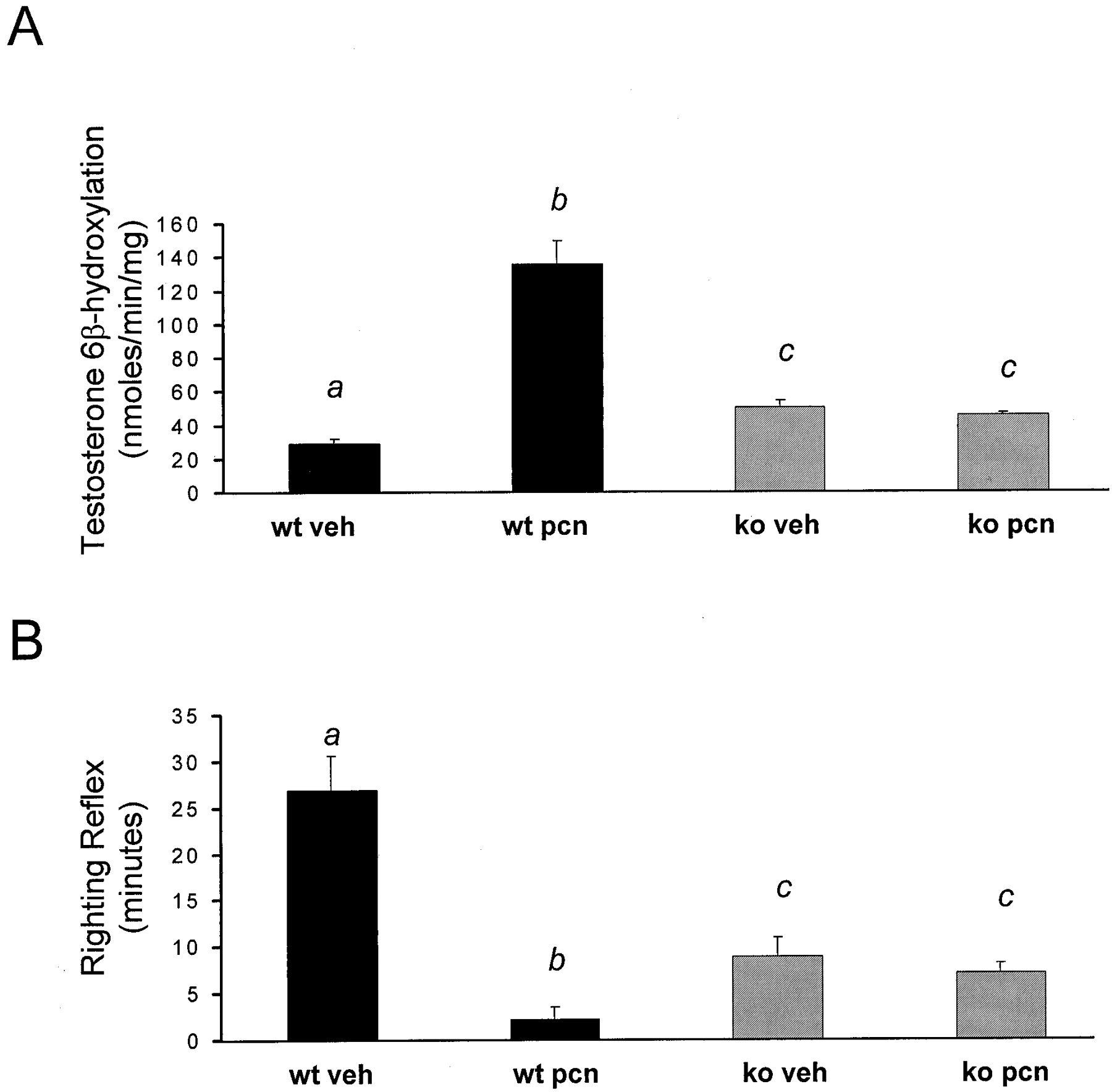
Induction of microsomal testosterone 6β-hydroxylation and zoxazolamine resistance.
A, wild-type mice and PXR-KO mice were treated i.p. with either vehicle (corn oil) or PCN (400 mg/kg) for 4 days. Livers were collected on the morning of the day 5, and microsomal preparations were isolated. The data are expressed as mean ± S.E.M. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). B, wild-type mice and PXR-KO mice were treated i.p. with either vehicle (corn oil) or PCN (400 mg/kg) for 4 days. On the morning of the day 5, all of the mice were injected i.p. with 112 mg/kg zoxazolamine, and the duration of the loss of the righting reflex was measured. The data are expressed as mean ± S.E.M. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). wt, wild-type; veh, vehicle.
Reduction in zoxazolamine-induced paralysis time following treatment is one of the classical parameters used to identify “catatoxic” steroids (Selye, 1971). Therefore, it was of interest to measure zoxazolamine-induced paralysis time following PCN administration in wild-type and PXR-KO mice in an effort to dissect the possible role of PXR in mediating PCN-inducible resistance to the classic cytochrome P450 substrate and muscle relaxant zoxazolamine. The vehicle treated wild-type mice were paralyzed for approximately 26 min following administration of zoxazolamine, whereas the PCN-treated wild-type mice were paralyzed for approximately 2 min following zoxazolamine administration (Fig. 4B). The PXR-KO mice were paralyzed for 6 to 8 min in both vehicle and PCN-treated experimental groups following zoxazolamine administration. The vehicle treated PXR-KO mice exhibited a significant decrease in zoxazolamine-induced paralysis time when compared with their vehicle treated wild-type littermates. These results are similar to those previously published by Xie et al. (2000a), although the line of PXR-null mice that exhibited a decrease in zoxazolamine-induced paralysis time expressed the constitutively active SXR/PXR transgene under the control of the albumin promoter.
PXR Regulates the Expression and Activity of Oatp2.
Because PCN treatment is known to increase hepatic uptake of cardiac glycosides in rodents and hepatocytes (Klaassen, 1972, 1973; Eaton and Klaassen, 1979), Oatp2 expression was measured following PCN administration in the wild-type mice and PXR-KO mice. Oatp2expression was induced in a dose-dependent manner following PCN administration in the wild-type mice but was not induced in the PXR-KO mice (Fig. 5A), as was previously demonstrated (Staudinger et al., 2001).

Dose-dependent induction of hepatic Oatp2 expression and [3H]digoxin uptake.
A, wild-type mice and PXR-KO mice were treated i.p. with increasing dosages of PCN dissolved in corn oil [vehicle (V), 25, 50, 100, and 400 mg/kg)] for 4 days. Total liver RNA was isolated on the day 5, pooled, and run on a 1% agarose gel. After transferring to a nylon membrane, the blot was probed with a 32P-labeled cDNA probe for mouse Oatp2. B, wild-type mice and PXR-KO mice were treated i.p. with either vehicle (corn oil) or PCN (400 mg/kg) for 4 days. On the morning of the day 5, the animals were injected i.v. with [3H]digoxin. Livers were collected 5 min after the injection, and hepatic uptake was calculated as a percentage of the total dose. The data are expressed as mean ± S.E.M. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). wt, wild-type; veh, vehicle.
Because Oatp2 mediates the hepatic uptake of digoxin and ouabain (Abe et al., 1998; Reichel et al., 1999), hepatic uptake of [3H]digoxin was measured in the wild-type mice and PXR-KO mice following PCN treatment. The wild-type mice treated with PCN exhibited a 2-fold increase in hepatic [3H]digoxin content following PCN treatment (Fig. 5B). Although the basal level of [3H]digoxin content was higher in the PXR-KO mice when compared with the wild-type mice, PCN treatment did not increase the hepatic [3H]digoxin in the PXR-KO mice.
PXR Regulates the Expression of Cyp7a1 and the Activity of Cholesterol 7α-Hydroxylase.
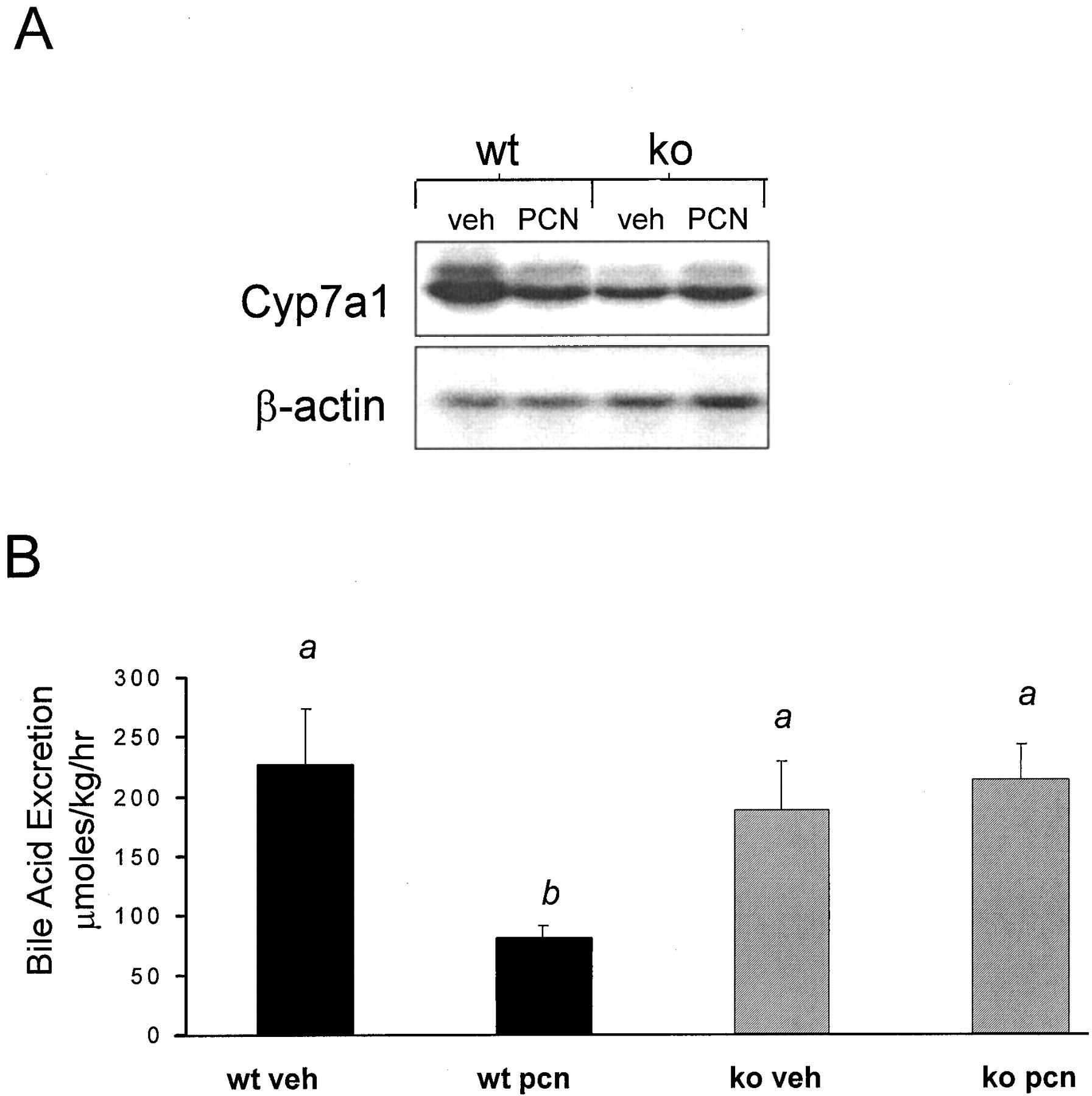
The rodent Cyp7a1 gene encodes cholesterol 7α-hydroxylase, the first and rate-limiting enzyme in the biochemical pathway that converts cholesterol into bile acids. Because PCN treatment is known to down-regulate Cyp7a1 expression in rodents (Li et al., 1990), it was of interest to determine whether PCN treatment also led to down-regulation of Cyp7a1 expression in the PXR-KO mice. The wild-type mice exhibited a reduction in hepatic Cyp7a1expression following PCN treatment, whereas the PXR-KO mice were refractory to Cyp7a1 down-regulation by PCN treatment (Fig.6A), as was previously shown (Staudinger et al., 2001).

Repression of Cyp7a1 expression and bile acid excretion.
A, wild-type mice and PXR-KO mice were treated for 4 days i.p. with either vehicle (corn oil) or PCN (400 mg/kg). RNA was isolated on the day 5 and pooled. Following transfer, the blot was probed with a32P-labeled cDNA probe for Cyp7a1. B, 24 h after 4 days of i.p. vehicle (corn oil) and PCN (400 mg/kg) treatment, bile was collected, and bile acid excretion was measured. Letters different from each other indicate a statistical difference between treatment groups (p < 0.05). wt, wild-type; veh, vehicle.
Because PCN and bile acid treatment has been shown to decrease the activity of cholesterol 7α-hydroxylase and excretion of bile acids into bile (Mason and Boyd, 1978; Turley and Dietschy, 1984; Li et al., 1990), it was of interest to determine whether PCN treatment had any effect upon bile acid excretion into bile in the PXR-KO mice. Treatment with PCN led to a significant decrease in bile acid excretion in wild-type mice but had no effect in PXR-KO mice (Fig. 6B).
Discussion
The response elements that mediate CYP3A induction in different species are known to bind and activate transcription through PXR (Jones et al., 2000). Species-specific CYP3A induction can be accounted for by PXR evolution because the replacement of mouse PXR with human PXR yields a “humanized” mouse that responds to human PXR activators by up-regulating mouse Cyp3a11transcription (Xie et al., 2000a). This is significant because transcriptional regulation of CYP3A family members in liver is at the center of many clinically important drug interactions (Guzelian, 1988; Maurel, 1996). PXR is a master regulator of hepaticCYP3A transcription that is activated by a number of structurally unrelated lipophilic compounds. Although much is known about the role of PXR in regulating induction of CYP3Atranscription in transfected cell lines, little is known about the role of PXR in regulating drug-induced liver cell hyperplasia and hypertrophy in vivo. In addition, little data exists regarding the physiological consequence of PXR activation in regulating CYP3A, Oatp2, and cholesterol 7α-hydroxylase activities in vivo. Thus, the work presented here seeks to elucidate the role of PXR in regulating PCN-induced hepatomegaly and the activities of CYP3A, Oatp2, and cholesterol 7α-hydroxylase in vivo in mice.
In this study, it was demonstrated that PXR-KO mice do not exhibit liver cell hypertrophy or hyperplasia following PCN treatment. These data indicate that PXR activation is required for PCN-induced hepatomegaly and suggest an important role for PXR in regulating liver cell hypertrophy and proliferation following xenobiotic and drug treatment. Activation of PXR by PCN leads to induction of the rodent hepatic Cyp3a11 gene in a PXR-dependent manner. PXR activation by PCN also increased testosterone 6β-hydroxylation and decreased the duration of zoxazolamine-induced paralysis time in a PXR-dependent manner, indicating a central role for PXR in regulating hepatic CYP3A activity in vivo. These results are similar to those reported by Xie et al. (2000a) in the PXR-null mice expressing the constitutively active human SXR/PXR transgene under the control of the albumin promoter. Ablation of PXR function in vivo surprisingly increased basal Cyp3a11 transcription and CYP3A activity, indicating a previously unrecognized role for PXR in repression and activation of CYP3A gene expression. Additionally, PCN treatment resulted in increased expression of Oatp2 and increased hepatic uptake of the Oatp2 substrate [3H]digoxin, suggesting that Oatp2 probably represents a direct PXR-target gene. By contrast, the PCN-dependent induction of CYP3A and Oatp2 expression and activity was not observed in the PXR-KO mice, further supporting an important role for PXR in the regulation of uptake and metabolism of xenobiotics in vivo. PXR activation by PCN reduced Cyp7a1 transcription and decreased bile acid excretion in a PXR-dependent manner, indicating a previously unrecognized role for PXR in cholesterol and bile acid homeostasis in vivo. The molecular mechanisms by which PXR suppressesCyp7a1 expression are yet to be elucidated. Notably, expression of the small heterodimeric partner gene, an orphan nuclear receptor implicated in the farnesoid X receptor-mediated suppression ofCyp7a1 (Goodwin et al., 2000; Lu et al., 2000), was not regulated in a PXR-dependent manner (data not shown). Thus, multiple mechanisms exist to suppress Cyp7a1 expression and thereby prevent accumulation of potentially hepatotoxic bile acids. Taken together, these data conclusively demonstrate a central role for PXR in regulating PCN-induced hepatomegaly, Cyp3a11,Oatp2, and Cyp7a1 gene expression levels and their respective activities in vivo. These studies also suggest an important role for PXR in the modulation of hepatic uptake, metabolism, and biosynthesis of xenobiotics, steroids, and bile acids.
In the present study, ablation of PXR function in vivo led to a slight but statistically significant increase in the levels ofCyp3a11 gene expression, testosterone 6β-hydroxylase activity, and zoxazolamine resistance. This contradicts earlier reports by Xie et al. (2000a,b) in which they suggested that ablation of PXR function has no effect on the basal level of Cyp3a11 gene transcription and CYP3A activity. Because PXR is thought to interact with a corepressor, the silencing mediator of repression and thyroid receptors protein, it is possible that the significant increase inCyp3a11 transcription and activity in the PXR-KO mice is due to the removal of corepressor activity. Alternatively, the increased level of Cyp3a11 expression and CYP3A activity could be due to the action of the constitutive androstane receptor (NR1I3) in the absence of functional PXR because the constitutive androstane receptor also appears to regulate CYP3A gene expression by competing for ligands (Moore et al., 2000) and response elements (Xie et al., 2000c). Another explanation for this disparity could include strain or allelic differences between the two lines of PXR-null mice.
In summary, these studies indicate a pivotal role for PXR activation in PCN-induced hepatomegaly. These studies also further our understanding regarding the regulation of biochemically linked genes involved in xenobiotic, steroid, and bile acid homeostasis in vivo. PXR activation in vivo regulates key steps involved in the hepatic uptake, metabolism, and biosynthesis of steroids and bile acids in a coordinate manner. In conclusion, the PXR-KO animals used in this study will be a valuable tool for the discovery of additional PXR target genes and ligands, as well as in elucidating mechanisms of drug interactions and cell proliferation following xenobiotic treatment.
Footnotes
-
Research supported by National Institutes of Health Grant NIEHS ES-09649. J. S. supported by National Institutes of Health Training Grant NIEHS ES-07079.
- Abbreviations used are::
- PXR
- pregnane X receptor
- Oatp2
- organic anion transporting polypeptide
- PCN
- pregnenolone-16α-carbonitrile
- PXR-KO
- PXR-knockout mouse
- PCNA
- proliferating cell nuclear antigen
- SXR
- steroid and xenobiotic receptor
- Received June 28, 2001.
- Accepted August 3, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}