


Abstract
Predicting drug-drug interactions requires an assessment of the drug concentration available to the enzyme active site, both in vivo, and within an in vitro incubation. These predictions are confounded when the inhibitor accumulates within the liver, either as a result of active transport processes or intracellular binding (including lysosomal trapping). In theory, hepatocytes should provide a more accurate estimation of inhibitory potency compared with microsomes for those compounds that undergo hepatic accumulation. However, they are not routinely used for Ki determination and there is limited comparative information available. Therefore, the aims of this study were to compare Ki values determined in rat microsomes and freshly isolated hepatocytes using six cytochrome P450 inhibitors (miconazole, fluconazole, ketoconazole, quinine, fluoxetine, and fluvoxamine) with a range of uptake properties (cell-to-medium concentration ratios 4.2-6000). Inhibition studies were performed using four probe substrates for CYP2C, CYP2D, and CYP3A enzymes (tolbutamide and phenytoin, dextromethorphan and midazolam, respectively). Comparison of unbound Ki values (range 0.05-30 μM) showed good agreement between microsomes and hepatocytes for inhibition of 18 pathways of metabolism. In addition to this, there was no relationship between the cell-to-medium concentration ratios (covering over 3 orders of magnitude) and the microsomal to hepatocyte Ki ratio of these inhibitors. These data suggest that the hepatic accumulation of these inhibitors results from intracellular binding rather than the involvement of uptake transporters and indicate that microsomes and hepatocytes appear to be equivalent for determining the inhibitory potency of the six inhibitors investigated in the present study.
Quantitative prediction of drug-drug interactions (DDIs) requires an accurate estimation of drug concentration at the enzyme active site, both in vivo within the liver and within an in vitro incubation. Since it is impossible to directly measure this liver concentration in humans, plasma concentrations have been used as an in vivo surrogate, but with varying degrees of success (Ito et al., 2002, 2004). Although the use of plasma inhibitor concentration has accurately predicted the degree of in vivo DDIs for several compounds (Brown et al., 2005, 2006; Obach et al., 2006), even the use of total plasma concentrations may underestimate hepatocellular concentrations for inhibitors that are accumulated within the liver.
Drug can be sequestered within the hepatocyte via several mechanisms; for example, lysosomal uptake, intracellular protein binding, or the involvement of hepatic uptake transporters. Lysosomes represent 1% of the hepatocyte volume; therefore, trapping is an important distribution process for weak bases, which can accumulate within this compartment due to the pH gradient between the acidic lysosome compartment (pH 5), the hepatic cytosol (pH 7.2), and the plasma (pH 7.4) (Daniel, 2003). Drug accumulation resulting from this process or from intracellular binding to cytosolic proteins will not result in a drug concentration within the hepatic cytosol that differs from the unbound concentration in the external incubation medium. These processes should be of little consequence since only the unbound intracellular drug will be available for enzyme inhibition. In contrast, however, hepatic uptake transporters located on the basolateral (sinusoidal) membrane may result in unbound cellular concentrations in excess of that in the incubation medium, which will have implications for the prediction of DDIs from in vitro data.
Hepatic microsomes are typically used for the determination of in vitro Ki or IC50 values; however, the inhibition data obtained in this in vitro system could significantly underestimate the in vivo potency if drug accumulation occurs. Therefore, one could reasonably expect inhibition data obtained in hepatocytes to be more representative of the in vivo situation, due to the presence of intracellular proteins, organelles, and an intact plasma membrane containing both hepatic uptake (basolateral) and efflux (apical) transporter proteins. Thus, a comparison of Ki data between microsomes and hepatocytes could theoretically indicate the mechanism of inhibitor accumulation. After appropriate binding corrections, more potent hepatocyte inhibition would suggest the involvement of hepatic transporters since a higher concentration of unbound drug available for enzyme inhibition can be achieved compared with a similar incubation concentration in microsomes (Ito et al., 1998a). Alternatively, similar values would indicate that hepatic accumulation results from intracellular binding or lysosomal trapping. Although hepatocytes are used for the prediction of metabolic clearance (Hallifax et al., 2005; Brown et al., 2007), published literature comparing the use of hepatocytes as an alternative system to microsomes for Ki determination remains somewhat limited (Di Marco et al., 2003; Jones et al., 2004; Engtrakul et al., 2005; Mano et al., 2007). In addition, these previous investigations are limited since they are restricted to one substrate-inhibitor pair (Di Marco et al., 2003; Jones et al., 2004; Engtrakul et al., 2005), or inhibitors, which are not expected to undergo hepatic accumulation (Mano et al., 2007).
Lipophilic bases such as fluoxetine and fluvoxamine have previously been shown to extensively partition within the liver (von Moltke et al., 1994, 1995), and the accumulation of these compounds (in addition to that of quinine) is thought to be attributable to lysosomal uptake (Hallifax and Houston, 2007). Previous investigations with ketoconazole have also demonstrated a concentrative uptake effect in isolated rat hepatocytes; however, the mechanism for this accumulation has yet to be established (Yamano et al., 1999). For these reasons, we have selected the aforementioned four inhibitors, fluoxetine, fluvoxamine, quinine, and ketoconazole, for further study, along with two additional azole antifungals, miconazole and fluconazole, to form a set of six compounds with a range of hepatic uptake and binding characteristics. The aims of the present study are to compare P450 inhibition properties in both rat microsomes and freshly isolated hepatocytes following corrections for the projected unbound drug within the incubation, to make an assessment of the inhibitor concentration within the hepatocyte, and to identify the relative contributions of intracellular binding and active transport. To this end, tolbutamide and phenytoin, dextromethorphan and midazolam, were studied as appropriate P450 probes for the rat P450s in the CYP2C, CYP2D, and CYP3A families, respectively (Soucek and Gut, 1992; Kobayashi et al., 2003; Chovan et al., 2007), with kinetic and inhibition studies performed for each compound.
Materials and Methods
Chemicals. Dextrorphan and 3-methoxymorphinan were purchased from Hoffman La Roche (Basel, Switzerland), fluoxetine and fluvoxamine were purchased from Tocris Cookson Ltd. (Bristol, UK), ketoconazole was purchased from ICN Biomedicals Inc. (Irvine, CA), and collagenase H was purchased from Roche Molecular Biochemicals (Mannheim, Germany). Fluconazole and hydroxy-tolbutamide were generous gifts from Pfizer (Sandwich, UK) and Aventis (Strasbourg, France), respectively. Midazolam, 1-hydroxymidazolam, 4-hydroxymidazolam, and diazepam were generous gifts from Roche Products Limited (Welwyn, UK). All other chemicals and reagents used were of the highest grade available and were purchased from Sigma Chemical (Poole, Dorset, UK) or BDH (Poole, Dorset, UK).
Animal Source, Housing, and Diet. Male Sprague-Dawley rats (240-260g) were obtained from the Biological Sciences Unit, Medical School, University of Manchester. They were housed in groups of two to four, in opaque boxes on a bedding of sawdust in rooms maintained at a temperature of 20 ± 3°C, with a relative humidity of 40 to 70% and a 12 h light/dark cycle. The animals were allowed free access to CRM diet and fresh drinking water.
Microsomal Studies. Rats were sacrificed by cervical dislocation and washed microsomes were prepared as described previously (Hayes et al., 1995). Microsomal protein content was determined using the method of Lowry et al., (1951). All kinetic and inhibition studies were performed in duplicate under initial rate conditions with respect to incubation time and microsomal protein concentration.
Kinetic studies. Dextromethorphan (0.05-1000 μM), midazolam (0.5-150 μM), and phenytoin (0.1-100 μM) were preincubated with microsomal protein and phosphate buffer (0.1 M, pH 7.4) for 5 min in a shaking water bath at 37°C. Reactions were initiated by the addition of an NADPH regenerating system, and were terminated by the addition of ice-cold acetonitrile (dextromethorphan), 0.1 M carbonate buffer (pH 10) (midazolam), and tert-butyl methyl ether (phenytoin). Incubation conditions for each substrate are shown in Table 1.
Incubation conditions for substrates and inhibitors in rat microsomal and hepatocyte studies
Inhibition studies. Tolbutamide, dextromethorphan, midazolam, and phenytoin were incubated at concentrations equivalent to 0.5 KM, KM, and 2 KM with a range of inhibitor concentrations (Table 1), with incubations performed as described for the microsomal kinetic studies. Tolbutamide concentrations were based upon KM data previously obtained within this laboratory (Ashforth et al., 1995), with reactions terminated by the addition of ice-cold acetonitrile.
Microsomal Binding Studies. Equilibrium dialysis was used to determine the extent of binding of fluconazole, ketoconazole, and miconazole to rat liver microsomes using methods described previously (Brown et al., 2006). The microsomal binding of quinine, fluoxetine, and fluvoxamine was determined by microfiltration as described previously (Brown et al., 2006).
Hepatocyte studies. Rats were sacrificed by cervical dislocation and hepatocytes were prepared using an adaptation of the collagenase perfusion method (Berry and Friend, 1969) as described previously (Hayes et al., 1995). Hepatocyte viability was determined using the trypan blue exclusion test, and only those hepatocyte preparations with viabilities greater than 85% were used. All kinetic and inhibition studies were performed in duplicate under initial rate conditions with respect to incubation time and hepatocyte density.
Kinetic studies. Dextromethorphan (0.005-1000 μM), midazolam (0.1-100 μM), and phenytoin (0.1-50 μM) were preincubated with Williams Medium E (pH 7.4) in 1.5-ml Eppendorf tubes for 5 min in a Thermo mixer (Eppendorf AG, Hamburg, Germany) set at 37°C and 900 rpm. Reactions were initiated by the addition of prewarmed hepatocytes (37°C) and were terminated by snap-freezing in liquid nitrogen. Incubation conditions for each substrate are shown in Table 1.
Inhibition studies. Tolbutamide, dextromethorphan, midazolam, and phenytoin were incubated with a range of inhibitor concentrations (Table 1), with incubations performed as described for the hepatocyte kinetic studies. Tolbutamide concentrations were based upon KM data previously obtained within this laboratory (Ashforth et al., 1995).
Determination of drug uptake into hepatocytes. The cellular protein, cellular volume, and adherent water layer volume were determined as described previously (Hallifax and Houston, 2006). The uptake of ketoconazole (0.005-100 μM), fluconazole (0.5-250 μM), and miconazole (0.1 and 1 μM) into rat hepatocytes was determined by centrifugation through silicone oil using methods described previously (Hallifax and Houston, 2006). The cell-to-medium concentration ratios of quinine, fluoxetine, and fluvoxamine were taken from a previous study within this laboratory (Hallifax and Houston, 2007).
Sample Hydrolysis, Extraction and Analysis. LC-MS/MS was used for the analysis of all tolbutamide and dextromethorphan microsomal and hepatocyte studies. HPLC was used for the analysis of all the remaining studies.
Microsomal sample preparation. After termination of the tolbutamide and dextromethorphan studies, microsomal samples were centrifuged at 11,600g (Eppendorf Centrifuge 5413; Eppendorf International, Hamburg, Germany) for 10 min and the supernatant was analyzed by LC-MS/MS using methods described previously (Brown et al., 2006). After the addition of internal standard, HPLC samples were extracted with ethyl acetate (midazolam, fluconazole, and ketoconazole) or TBME (phenytoin) by rotary mixing for 30 min and centrifugation at 900g (MSE, Mistral 2000; Fisher Scientific, Loughborough, UK) for 10 min. The organic layer was removed and evaporated to dryness at 50°C. Samples were reconstituted in mobile phase and analyzed by HPLC.
Hepatocyte Sample Preparation. After termination of the reaction, hepatocyte samples were thawed and incubated with β-glucuronidase with sulfatase activity (200 units/ml in sodium acetate, 60 mM, pH 4.5) for 60 min in a shaking water bath at 37°C. Tolbutamide and dextromethorphan incubations were then terminated by the addition of ice-cold acetonitrile containing internal standard. Samples were centrifuged at 11,600g (Eppendorf Centrifuge 5413, Eppendorf International) for 10 min and an aliquot of the supernatant was analyzed by LC-MS/MS (Brown et al., 2006). Midazolam, phenytoin, fluconazole, and ketoconazole samples were extracted as described for microsomes and were analyzed by HPLC.
HPLC methods. The metabolites of midazolam and phenytoin, in addition to those of fluconazole and ketoconazole, were analyzed by HPLC as follows. The HPLC system used in the analysis of midazolam consisted of a Supelco Supelcosil LC-ABZ (150 × 4.6 mm) column, and a mobile phase of 70% ammonium acetate (0.025 M) adjusted to pH 5 and 30% acetonitrile, delivered by an LDC/Milton Roy Constametric 3000 pump, at a flow rate of 1 ml/min, and at a UV wavelength of 240 nm measured by an Applied Biosystems 783 detector (Applied Biosystems, Foster City, CA). The HPLC system used in the analysis of phenytoin consisted of a Zorbax RX-C18 (250 × 4.6 mm) column, and a mobile phase of 70% water containing 0.02% (v/v) triethylamine, adjusted to pH 5.5, and 30% acetonitrile, delivered by an LDC/Milton Roy Constametric 3000 pump, at a flow rate of 1 ml/min, and at a UV wavelength of 214 nm, measured by an Applied Biosystems 783 detector. The HPLC system used in the analysis of fluconazole consisted of a Phenomenex Prodigy ODS2 (150 × 4.6 mm) column (Phenomenex, Torrance, CA), and a mobile phase of 75% water and 25% acetonitrile, delivered by an LDC/Milton Roy Constametric 3000 pump, at a flow rate of 0.8 ml/min, and at a UV wavelength of 260 nm measured by a Applied Biosystems 783 detector. The HPLC system used in the analysis of ketoconazole consisted of a Zorbax RX-C8 (250 × 4.6 mm) column, and a mobile phase of 40% sodium hydrogen orthophosphate (0.02M) containing 0.1% triethylamine, adjusted to pH 6.8 and 60% acetonitrile, delivered by a LDC/Milton Roy Constametric 3000 pump, at a flow rate of 0.8 ml/min, with fluorescence detection at excitation and emission wave-lengths of 260 and 370 nm, respectively, measured by a Jasco FP-920 Intelligent Fluorescence detector (Jasco, Tokyo, Japan). All samples were quantified using the peak height ratio with the following internal standards: diazepam, MPPH, phenacetin, and codeine for midazolam; and phenytoin, fluconazole, and ketoconazole, respectively.
Data Analysis.Kinetic data. All data were analyzed by nonlinear regression analysis using Grafit 4 (Erithacus Software Ltd., Horley, Surrey, UK) using models for Michaelis-Menten kinetics, biphasic kinetics with substrate inhibition (eq. 1), or sigmoidal kinetics. The goodness of fit criteria used to select the model with the most appropriate fit comprised visual inspection, consideration of the randomness of residuals, and the standard error of the parameters. 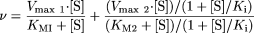
Inhibition Data. IC50 plots were used to distinguish between competitive and noncompetitive inhibition. Using nonlinear regression, a model for either competitive or noncompetitive inhibition was fitted to the data using Grafit 4 (Erithacus Software Ltd.) to determine the Ki value of each inhibitor. However, the classic inhibition models cannot be used for substrates for which metabolism is described by non-Michaelis-Menten kinetics; therefore Ki values for inhibition of midazolam and phenytoin were determined by plotting the IC50 versus the S/KM or S/S50 allowing linear regression, where the intercept is equivalent to the Ki value.
Binding data. The fraction unbound of fluconazole, ketoconazole, miconazole, quinine, fluoxetine, and fluvoxamine in rat liver microsomes was determined by the ratio of the concentration in the filtrate to the initial total concentration as described previously (Jones et al., 2004). The fraction unbound of each inhibitor was determined at different microsomal protein concentrations as described previously (Brown et al., 2006). Total microsomal Ki values were corrected for the fuinc to give the unbound Ki value.
Cellular uptake data. The concentration of drug in the cells (Ccell) was calculated using the total amount of drug in cells and the cell volume for each hepatocyte preparation as described previously (Hallifax and Houston, 2006). The hepatocyte/medium partition coefficient (Kp) was calculated based upon the ratio of the concentration of drug within the hepatocyte to the concentration of drug in the incubation medium. Based upon the total cell and incubation volumes, along with the Kp values, the unbound fraction of inhibitor within the whole incubation was calculated using eq. 2, where Vinc represents the volume of the incubation. Equation 2 was derived from mass balance considerations, assuming that the unbound concentration in the cell is equal to the unbound concentration in the incubation, and that no active transport occurs. Total hepatocyte Ki values were corrected by the fuinc (eq. 2) to give the unbound Ki value. 
Statistical analysis. Inhibition data were compared between rat liver microsomes and hepatocytes to make an assessment of the unbound inhibitor concentration available to the enzyme within the hepatocyte. A paired t test was used to assess any statistically significant differences between systems.
To expand upon the database, inhibition data for omeprazole and diazepam in rat microsomes and hepatocytes were included from a previous publication within this laboratory (Jones et al., 2004). In addition, KM values for a number of substrates in both rat microsomes and hepatocytes were also compared from data previously obtained within this laboratory (Ashforth et al., 1995; Hayes et al., 1995; Worboys et al., 1996; Carlile et al., 1998; Jones et al., 2004).
Results
Substrate Kinetic Characteristics. The metabolism of midazolam, phenytoin, and dextromethorphan was investigated in rat liver microsomes (n = 3-7) and in freshly isolated rat hepatocytes (n = 3-5). In rat liver microsomes, the formation of both 1-hydroxymidazolam and 4-hydroxymidazolam displayed sigmoidal or autoactivation kinetics, which were best described using the Hill equation. In rat hepatocytes, however, the formation of both metabolites displayed typical Michaelis-Menten kinetics, with no sigmoidal behavior in evidence. Whereas in rat liver microsomes, the formation of 4-hydroxymidazolam predominated, 1-hydroxymidazolam was the major pathway in rat hepatocytes. Kinetic parameters for both rat microsomes and hepatocytes are shown in Table 2. The metabolism of phenytoin in both in vitro systems, monitored via the formation of 5-(4-hydroxyphenyl)-5-phenylhydantoin, was best described by biphasic kinetics with a high-affinity, low-capacity site and a low-affinity, high-capacity site. Kinetic parameters for phenytoin hydroxylation in both rat microsomes and hepatocytes are shown in Table 2.
Kinetic parameters for metabolism of dextromethorphan, midazolam, phenytoin, and tolbutamide in rat liver microsomes and freshly isolated hepatocytes Data are expressed as the mean of n = 3 (microsomes) and n = 4 (hepatocytes) ± S.D.
In both rat liver microsomes and freshly isolated hepatocytes, the formation of dextrorphan from dextromethorphan displayed biphasic kinetics with a high-affinity, low-capacity site and a low-affinity, high-capacity site, with substrate inhibition also evident. As a result, the two-site Michaelis-Menten model was modified to incorporate substrate inhibition at the low-affinity site, and this model was fitted to the data. The formation of 3-methoxymorphinan in both rat microsomes and hepatocytes displayed hetero-activation kinetics, again with evidence of substrate inhibition. Kinetic parameters for both rat microsomes and hepatocytes are shown in Table 2. No models were fitted to the 3-methoxymorphinan data, and kinetic parameters were determined by visual inspection of the data. As previously reported in human liver microsomes (Brown et al., 2006), the formation of dextrorphan accounted for more than 95% of dextromethorphan metabolism, with no difference in the contribution of the 3-methoxymorphinan pathway in both microsomes and hepatocytes.
Inhibitor Uptake and Binding Characteristics. The cell-to-medium concentration ratios indicated that the majority of the inhibitors studied displayed significant accumulation into hepatocytes, with values ranging from 143 to 6000, for quinine and miconazole, respectively (Table 3). Fluconazole, however, displayed little hepatic accumulation, with a cell-to-medium concentration ratio of 4.2. The hepatic uptake of quinine, fluoxetine, and fluvoxamine has previously been shown to display concentration-dependent uptake into isolated rat hepatocytes (Hallifax and Houston, 2007), whereas the uptake of ketoconazole, fluconazole and miconazole was found to be concentration-independent over the concentration range studied. The binding of each inhibitor within the hepatocyte incubation was determined using eq. 2, and these values along with microsomal binding data are shown in Table 3.
In vitro inhibition data in rat microsomes and hepatocytes obtained for substrates and inhibitors of CYP2C9, CYP2D6, and CYP3A4
In the present study microsomal values represent mean (n = 3) ± S.D. and hepatocyte values represent mean (n = 4) ± S.D.
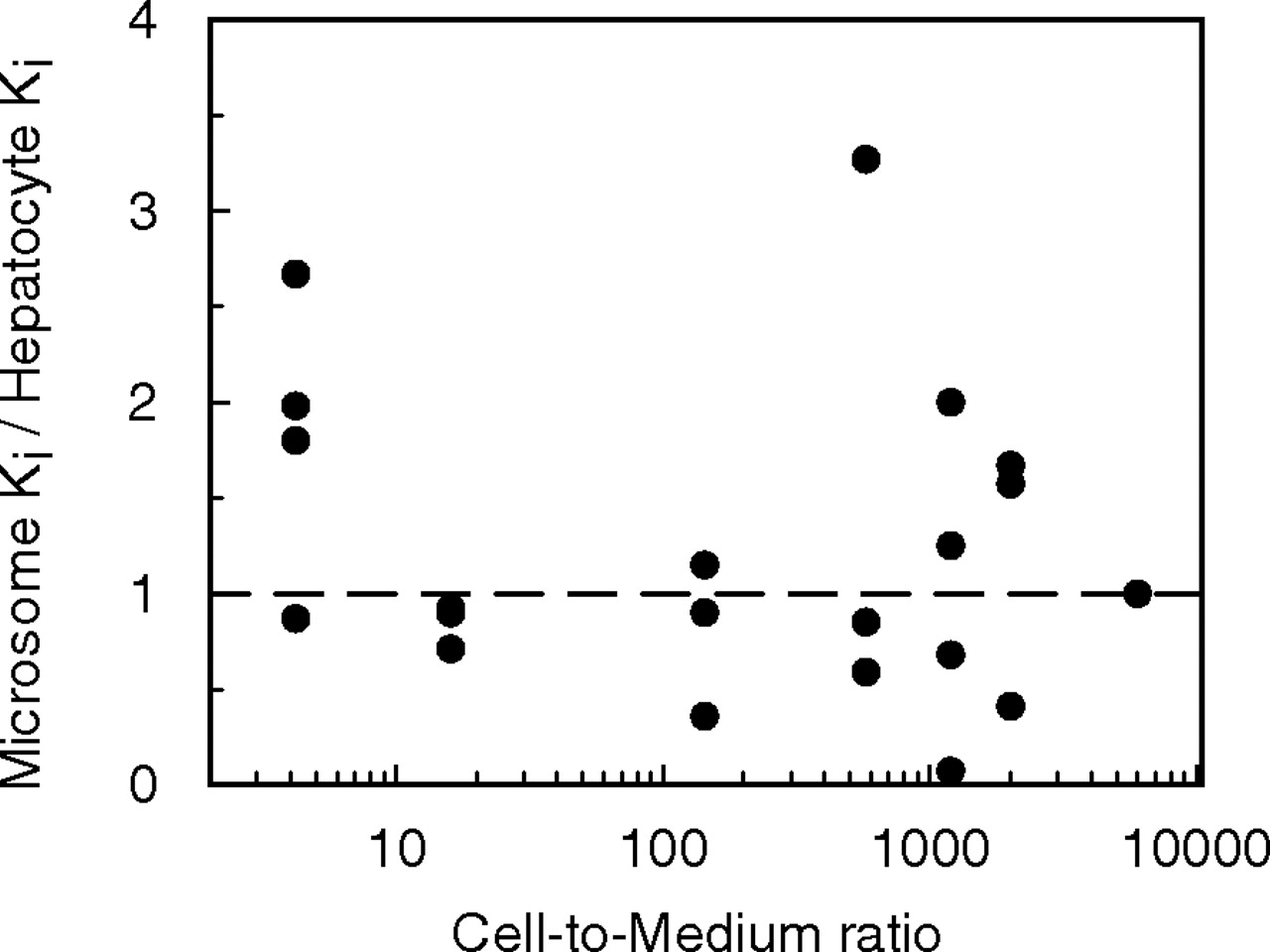
Inhibition Studies in Microsomes and Hepatocytes. The in vitro Ki values for miconazole, fluconazole, ketoconazole, quinine, fluoxetine, and fluvoxamine were determined using P450 probes tolbutamide, phenytoin, dextromethorphan, and midazolam. Table 3 shows the Ki,u value for each inhibitor, corrected for microsomal binding (fu range 0.06-1) and binding within the hepatocyte incubation (fu range 0.06-0.99). The unbound Ki values obtained for each of the seven inhibitors (including omeprazole data taken from Jones et al., 2004) in rat microsomes and hepatocytes were compared, and a good agreement between the parameters from both in vitro systems resulted for the inhibition of 21 pathways of metabolism (Fig. 1), with 16 Ki values within 2-fold in both microsomes and hepatocytes. Analysis of these data with a paired t test resulted in no statistically significant difference (p > 0.05) between microsomes and hepatocytes. However, although the data indicate a good correlation for Ki values above 0.1 μM, there were some outliers observed below this value, notably for ketoconazole and fluoxetine. Microsomal Ki values were more than 2-fold lower than the hepatocyte Ki values for inhibition of 1-hydroxymidazolam and high-affinity dextrorphan by ketoconazole and fluoxetine, respectively. However, it is evident that there is an inconsistency in these inhibition data, since lower microsomal Ki values (<2-fold hepatocyte Ki values) were not observed for the inhibition of 4-hydroxymidazolam, phenytoin, or tolbutamide by ketoconazole, or the inhibition of low-affinity dextrorphan or 3-methoxymorphinan by fluoxetine. The cell-to-medium concentration ratios of the seven inhibitors span over 3 orders of magnitude, but there appeared to be no clear relationship between hepatic accumulation and the unbound microsomal to hepatocyte Ki ratios observed for these inhibitors (Fig. 2).

Comparison of unbound Ki values between rat microsomes and hepatocytes involving inhibition data obtained with miconazole (•), fluconazole (□), ketoconazole (▵), quinine (▪), fluoxetine (▾), fluvoxamine (○), and omeprazole (▴) for 21 pathways of metabolism. The solid line represents the line of unity, whereas the dashed lines represent the 2-fold limits.
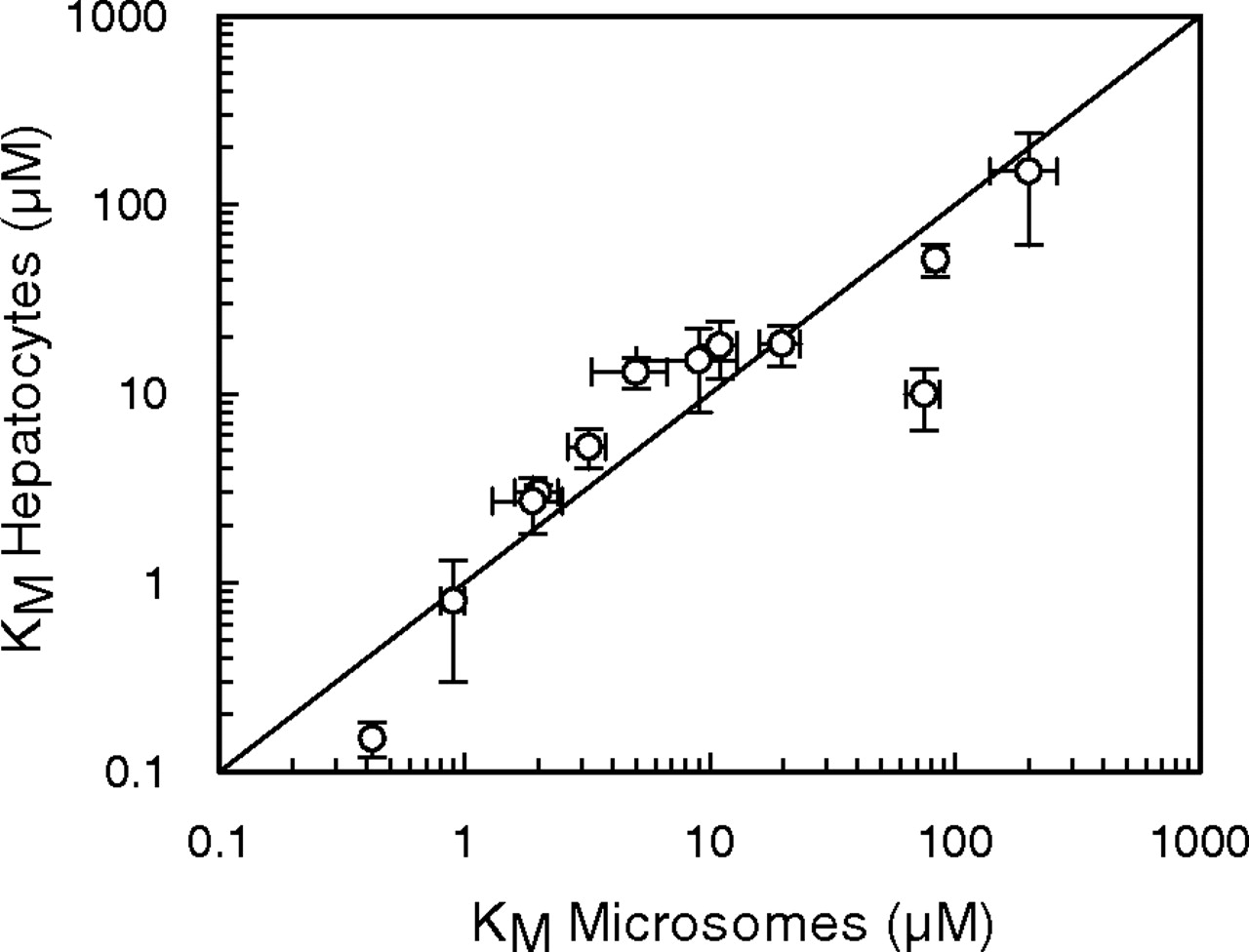
In addition, a comparison of the KM values obtained in rat microsomes and freshly isolated rat hepatocytes for tolbutamide (Ashforth et al., 1995), diazepam (Jones et al., 2004), ethoxycoumarin (Carlile et al., 1998), ondansetron (Worboys et al., 1996), and caffeine (Hayes et al., 1995), in addition to dextromethorphan, midazolam, and phenytoin, was made. It showed good agreement between systems for 14 pathways involved in the metabolism of the eight substrates (Fig. 3), with no statistically significant differences between microsomes and hepatocytes when analyzed by a paired t test.

Relationship between the cell-to-medium concentration ratio and the microsomal to hepatocyte Ki ratios for seven inhibitors and 21 pathways of metabolism.
Discussion
The use of hepatocytes is increasing, particularly for metabolic stability studies (Lau et al., 2002; Shibata et al., 2002; Soars et al., 2002), and for predictions of human metabolic clearance (Hallifax et al., 2005; Brown et al., 2007). However, there are limited data on the use of hepatocytes to determine in vitro inhibitory potency, particularly for those compounds known to accumulate within the liver (see introduction). Therefore, the aims of the present study were to compare in vitro Ki values obtained in rat microsomes and hepatocytes using four P450 probes for the CYP2C, 2D, and 3A enzymes (tolbutamide, phenytoin, dextromethorphan, and midazolam), and a range of P450 inhibitors (miconazole, ketoconazole, fluconazole, quinine, fluoxetine, and fluvoxamine), with differing hepatic uptake and binding characteristics. To enable the comparison to be valid, unbound Ki values were obtained following correction for binding and hepatic uptake of each inhibitor. The use of rat tissue, rather than human tissue, for these studies removes the issue of interdonor variability, which can confound the comparison of hepatic microsomal and hepatocellular kinetic parameters (Hallifax et al., 2005).

Comparison of KM values in rat microsomes and hepatocytes for 14 pathways involved in the metabolism of tolbutamide, phenytoin, dextromethorphan, diazepam, ethoxycoumarin, ondansetron, and caffeine. The solid line represents the line of unity.
The metabolism of midazolam in the rat is less well characterized than in humans. In rat, CYP3A1 and CYP3A2 are the major isoforms responsible for metabolizing midazolam (Chovan et al., 2007), and in contrast to humans, 4-hydroxymidazolam is the major metabolite (Ghosal et al., 1996). In the present study, the formation of both 1-hydroxy and 4-hydroxymidazolam displayed sigmoidal kinetics in rat microsomes; however, in rat hepatocytes, the formation of both metabolites was best described by Michaelis-Menten kinetics (Table 2). These microsomal data are consistent with those previously described in the literature (Ghosal et al., 1996); however, the kinetics of metabolite formation have not previously been described for midazolam in rat hepatocytes. The metabolism of phenytoin to HPPH in both rat microsomes and hepatocytes displayed biphasic kinetics, consistent with literature data (Ashforth et al., 1995).
Formation of dextrorphan from dextromethorphan in both rat microsomes and hepatocytes displayed biphasic kinetics, with substrate inhibition in evidence at higher substrate concentrations (Table 2). Although substrate inhibition has not previously been reported for dextrorphan formation in rat microsomes, it has been observed in humans (Lin et al., 2001). However, previous studies in dexamethasone-induced rat microsomes have indicated the involvement of CYP3A in addition to CYP2D in dextrorphan formation (Witherow and Houston, 1999), which is consistent with the substrate inhibition involved at the higher dextromethorphan concentrations. The biphasic microsomal data observed for dextrorphan are consistent with those described previously by Witherow and Houston (1999); however, they also reported a sigmoidal kinetic profile for dextrorphan formation in hepatocytes, as did the study by Griffin and Houston (2004), in contrast to the biphasic kinetics observed in the present study. These differences probably result from the use of both a wider substrate concentration range and an increase in analytical sensitivity and selectivity via the use of LC-MS/MS in the present study, compared with HPLC with fluorescence detection used previously. 3-Methoxymorphinan formation in both microsomes and hepatocytes was best described by sigmoidal or autoactivation kinetics, consistent with previous literature (Witherow and Houston, 1999; Griffin and Houston, 2004).
Uptake into isolated rat hepatocytes differed widely for each inhibitor, as indicated by cell-to-medium concentration ratios of 6000, 2010, 1200, 577, 143, 16, and 4.2 for miconazole, fluoxetine, ketoconazole, fluvoxamine, quinine, omeprazole, and fluconazole, respectively. Drug accumulation within hepatocytes can occur as a result of active transport processes, intracellular binding, or a combination of both. Intracellular binding to sites not involved in metabolism will reduce the inhibitor concentration within the hepatocyte; however, this should be of little consequence since the unbound concentration within the cell should be in equilibrium with the unbound concentration within the medium (Austin et al., 2005). Active transport processes, however, will raise the intracellular concentration of unbound drug in excess of that external medium. The Ki values of these seven inhibitors (following correction for binding) showed a good correlation between rat microsomes and hepatocytes (Fig. 1) over 3 orders of magnitude. This finding suggests that intracellular binding is the likely explanation for the high cell-to-medium concentration ratios observed, since the potency in both systems is comparable. In contrast however, the human hepatocyte Ki value of ibuprofen toward zidovudine (AZT) glucuronidation was significantly more potent than in human microsomes by approximately 5-fold (Engtrakul et al., 2005). Although these results would appear to indicate the involvement of hepatic transporters in the uptake of ibuprofen, previous studies have indicated only a minimal contribution of the organic anion-transporting polypeptides (OATPs) for ibuprofen (Kouzuki et al., 1999), suggesting that this discrepancy may result from an issue unrelated to transport.
Although microsomal and hepatocyte data were in good agreement for most inhibitors, significantly more potent microsomal inhibition (<2-fold hepatocyte Ki) was observed for inhibition of 1-hydroxymidazolam and high-affinity dextrorphan formation, by ketoconazole and fluoxetine, respectively. We currently have no explanation for these findings since microsomal and hepatocyte Ki values were comparable for inhibition of other metabolic pathways by the same inhibitors (Table 3); therefore, the mechanism responsible for these differences remains unclear. However, determining Ki values for pathways that display atypical kinetics can be problematic, and indeed, the 1-hydroxymidazolam and high-affinity dextrorphan pathways are sigmoidal and biphasic, respectively. Although 1-hydroxymidazolam inhibition by fluconazole along with the inhibition of dextromethorphan pathways by quinine and fluvoxamine would appear comparable in both systems, it is feasible to suggest that the resolution of multisite complexities may be more challenging for potent rather than more moderate P450 inhibitors.
The microsomal to hepatocyte Ki ratio has been proposed to be equivalent to the unbound tissue-to-plasma partition coefficient (Ito et al., 1998b); however, in the present study there was a lack of correlation between the cell-to-medium concentration ratios and the Ki ratio (Fig. 2). The cell-to-medium concentration ratio represents both the total and unbound drug within the hepatocyte; thus, the current findings would appear to support the use of the unbound, rather than the total partition coefficient.
Previous studies in this laboratory (Hallifax and Houston, 2007) have shown that the hepatic uptake of quinine, fluoxetine, and fluvoxamine is composed of a saturable and a nonsaturable component, of which the saturable component is thought to be attributed to lysosomal uptake. This finding supports the close comparability between microsomal and hepatocyte Ki values in the present study. Although there is evidence to suggest that quinine is a substrate for the rat organic cation transporter (rOCT1) expressed in the renal proximal tubule and hepatocytes (Busch et al., 1996), it is classified as a class I compound in the Biopharmaceutical Classification System (Wu and Benet, 2005); therefore, transporter effects are thought to be minimal. The hepatic uptake of ketoconazole has been previously shown to be both temperature- and concentration-dependent, with uptake inhibited by the ATP inhibitor, rotenone (Yamano et al., 1999), suggesting that a saturable carrier-mediated process may be involved in uptake. This concentration dependence is in contrast to our results and may result from the confounding issue of ketoconazole metabolism. This was avoided in the present study via the use of aminobenzotriazole, a mechanism-based inhibitor of P450.
A good correlation was also observed between KM values in rat microsomes and hepatocytes for 14 pathways involved in the metabolism of eight substrates (Fig. 3), used both in the present study and compiled from previous publications from this laboratory (Ashforth et al., 1995; Hayes et al., 1995; Worboys et al., 1996; Carlile et al., 1998; Jones et al., 2004). These data suggest that for these compounds, the extracellular drug concentration is in equilibrium with the intracellular drug concentration, since there were no observed differences in substrate-enzyme affinity between both in vitro systems. These results are in contrast to previously observed human data, in which KM values in cryopreserved human hepatocytes were found to be, on average, 2-fold lower than human microsomal KM values, for 29 pathways involved in the metabolism of 15 substrates (Brown et al., 2007), four of which were also present in the current analysis. Since there appeared to be no involvement of transporters in the elimination of these substrates, the reasons for the altered KM values remain unclear, and suggest that these observations could result from the interindividual variability between the livers selected for study, a factor which is avoided via the use of rat hepatocytes in the present study.
The present study represents the first, to our knowledge, to compare Ki values between microsomes and hepatocytes using a series of P450 inhibitors with a wide range of inhibitory potencies and accumulation properties. These data illustrate that microsomes are equivalent to hepatocytes for determining the in vitro inhibitory potency for several compounds that accumulate as a result of intracellular binding. However, for inhibitors that undergo active transport processes, more potent hepatocyte Ki values would be expected, compared with those in microsomes, since the unbound intracellular and extracellular inhibitor concentrations will not be equivalent (Hewitt et al., 2007). These studies provide a basis for further studies with inhibitors/substrates that are known substrates of hepatic uptake transporters to establish the utility of hepatocyte incubation for in vivo prediction studies.
Acknowledgments
We thank Sue Murby, Beverley Brennan, and David Hallifax for valuable assistance.
Footnotes
-
doi:10.1124/dmd.107.017095.
-
This work was partially funded by a consortium of pharmaceutical companies (Bristol-Myers Squibb, GlaxoSmithKline, F. Hoffmann La Roche, Lilly, Novartis, Pfizer, and Servier) within the Centre for Applied Pharmacokinetic Research at the University of Manchester. H.S.B was financially supported by a Bristol-Myers Squibb studentship. A.C. was financially supported by AstraZeneca BBSRC CASE studentship. Part of this study was presented at the 8th European ISSX Meeting (Dijon, France) 2003 and the 16th International Symposium on Microsomes and Drug Oxidations (Budapest, Hungary) 2006.
-
ABBREVIATIONS: DDI, drug-drug interaction; P450, cytochrome P450; LC-MS/MS, liquid chromatography-tandem mass spectrometry; AZT, 3′-azido-2′,3′-dideoxythymidine; HPPH, 5-(4-p-hydroxyphenyl)-5-phenylhydantoin; TBME, tert-butyl methyl ether; MPPH, 5-(4-methylphenyl)-5-phenylhydantoin; CRM, Chow Rat Mouse.
-
↵1 Current affiliation: Department of Metabolism, Covance Laboratories Ltd., Otley Road, Harrogate, HG3 1PY, UK.
- Received June 8, 2007.
- Accepted August 24, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}