


Abstract
Hepatocytes and liver microsomes are considered to be useful for investigating drug metabolism catalyzed mainly via glucuronidation. However, there have been few reports comparing the glucuronidation inhibition potentials of drug in hepatocytes to those in liver microsomes. 3′-Azido-3′-deoxythymidine (AZT, zidovudine) glucuronidation (AZTG) is the major metabolic pathway for AZT. In this study, the inhibition potentials of drugs against UDP-glucuronosyltransferase (UGT)-catalyzed AZTG in the hepatocytes and liver microsomes of rats are compared. The AZTG inhibition potentials of diclofenac, diflunisal, fluconazole, indomethacin, ketoprofen, mefenamic acid, naproxen, niflumic acid, and valproic acid in liver microsomes and hepatocytes were investigated using liquid chromatography with tandem mass spectrometry. Diflunisal (inhibition type: noncompetitive) inhibited AZTG most potently in rat liver microsomes (RLMs) with an IC50 value of 34 μM. The IC50 values of diclofenac, fluconazole, indomethacin, ketoprofen, mefenamic acid, naproxen, niflumic acid, and valproic acid against AZTG in RLMs ranged from 34 to 1791 μM. Diclofenac, diflunisal, indomethacin, ketoprofen, naproxen, and valproic acid inhibited AZTG in hepatocytes with IC50 values of 58, 37, 88, 361, 486, and 281 μM, respectively. These values were similar to those obtained in RLMs. In conclusion, the AZT glucuronidation inhibition potentials of drugs in the hepatocytes and liver microsomes of rats were found to be similar, and liver microsomes can be useful for evaluating UGT isozyme inhibition potentials.
Glucuronidation catalyzed by UDP-glucuronosyltransferase (UGT) is one of the major steps in the metabolism of endogenous substances and xenobiotics. It is generally recognized that UGTs are important isozymes in the glucuronidation of nonsteroidal anti-inflammatory drugs. If UGTs are the primary isozymes involved in the clearance of drugs, their inhibition may cause enhanced drug exposure, perhaps even reaching toxic levels. It has been shown that 3′-azido-3′-deoxythymidine (AZT) glucuronidation (AZTG) is catalyzed primarily by UGT2B7 (Court et al., 2003; Mano et al., 2007), and that concomitant dosages of fluconazole and valproic acid increase plasma AZT levels (Lertora et al., 1994; Sahai et al., 1994). This increase is attributed to the inhibition of UGT2B7 by these drugs. To predict in vivo drug-drug interactions, in vitro inhibition studies using matrix like liver microsomes and hepatocytes are very valuable.
The majority of inhibition studies on metabolic enzymes such as cytochrome P450 (P450) and UGT have been performed using liver microsomes. Although the present popular opinion may be that hepatocytes yield the most reliable values for estimating the metabolizing enzyme inhibition potential for drugs, there has been no evidence presented to demonstrate that hepatocytes are the best predictors of inhibition potential (Jones et al., 2004). A recent report by Engtrakul et al. (2005) showed that the IC50 value of ibuprofen against AZT glucuronidation in human hepatocytes (170 μM) was significantly lower than that in human liver microsomes (HLMs) (975 μM). In addition, the authors caution against relying on inhibition potentials extrapolated from data obtained using liver microsomes. However, their study was performed with only one substrate-inhibitor pair, which indicates that studies with more pairs are necessary to determine whether hepatocytes generally yield more potent inhibition values than do liver microsomes. Although few studies have been reported in terms of comparing inhibitory potential of drug against glucuronidation, a number of reports comparing P450 isozyme-mediated metabolism inhibition in liver microsomes and hepatocytes are available. Di Marco et al. (2003) reported that pyrilamine, propafenone, verapamil, ketoconazole, and terfenadine inhibited dextromethorphan demethylation in rat liver microsomes and rat hepatocytes with similar potency. Jones et al. (2004) demonstrated that omeprazole inhibited diazepam metabolism to 4′-hydroxydiazepam, 3-hydroxydiazepam, and nordiazepam with similar IC50 values in rat liver microsomes (RLMs) and hepatocytes.
In this study, the AZTG inhibition potencies of nine drugs, diclofenac, diflunisal, fluconazole, indomethacin, ketoprofen, mefenamic acid, naproxen, niflumic acid, and valproic acid (Fig. 1), in RLMs and cryopreserved rat hepatocytes were investigated. The glucuronidation inhibition potential in both liver matrices was then compared. This study provides data on the issue of whether in vitro testing of inhibitory potentials of drugs against drug metabolism in liver microsomes is representative of hepatic metabolism exemplified by the use of hepatocytes.

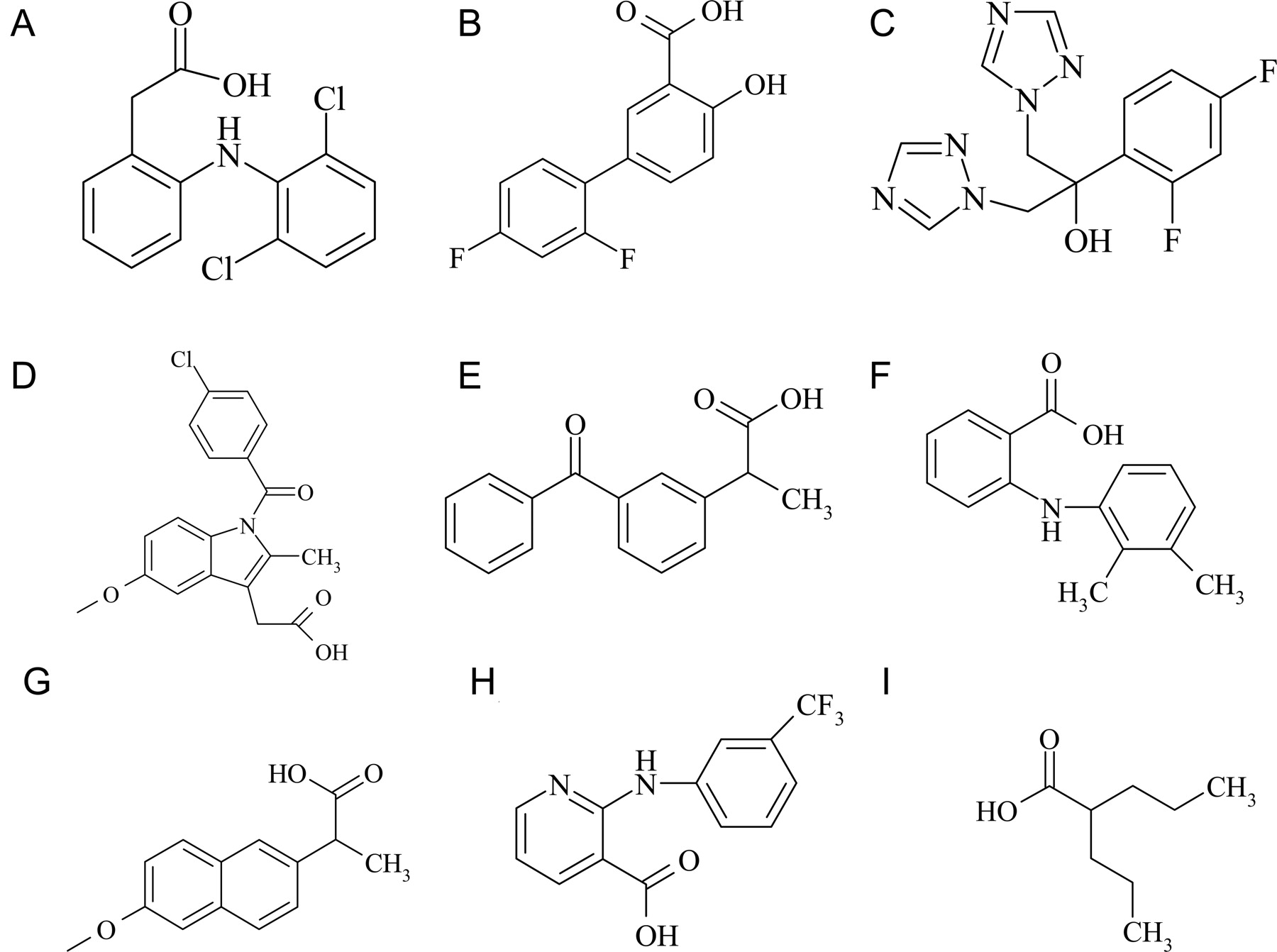
Chemical structures of drugs investigated. A, diclofenac; B, diflunisal; C, fluconazole; D, indomethacin; E, ketoprofen; F, mefenamic acid; G, naproxen; H, niflumic acid; and I, valproic acid.
Materials and Methods
Chemicals and Reagents. AZT, diclofenac, fluconazole, indomethacin, ketoprofen, mefenamic acid, naproxen, and niflumic acid were purchased from Sigma (St. Louis, MO). Diflunisal and valproic acid were purchased from ICN Biomedicals (Aurora, OH) and Wako Pure Chemicals (Osaka, Japan), respectively. AZT glucuronide was obtained from Toronto Research Chemicals Inc. (Toronto, ON, Canada). RLMs and cryopreserved rat hepatocytes were purchased from Xenotech (Kansas City, KS). All other chemicals were of analytical grade.
Glucuronidation in RLMs. AZT (2 mM) was incubated with RLMs (0.1 mg protein/ml) for 20 min in a final volume of 0.25 ml of Tris-HCl buffer (50 mM, pH 7.5) containing 8 mM MgCl2, 25 μg/ml alamethicin, and 5 mM UDPGA in both the presence and absence of drugs (diclofenac, diflunisal, fluconazole, indomethacin, ketoprofen, mefenamic acid, naproxen, niflumic acid, and valproic acid). The AZT concentration (2 mM) was below its Km value (9.1 mM; Cretton et al., 1990). In a separate study, AZT (0.2–10 mM) was incubated with RLMs under the same conditions described above in the presence and absence of diflunisal (40 μM) to evaluate the type of inhibition diflunisal exerted against AZTG. Kinetic studies done in RLMs using different buffers were performed as stated above. The buffers tested were 100 mM sodium carbonate and Williams' Medium E (containing 2 mM l-glutamine) (WME), both of which apparently altered AZTG in HLMs, compared with the effects of Tris-HCl (Engtrakul et al., 2005). All buffers were prepared at pH 7.5. After the reaction mixture had been preincubated at 37°C for 5 min, the reaction was started by adding UDPGA at a final concentration of 5 mM. After incubating for 20 min in RLMs, the reaction was terminated by adding 0.05 ml of acetonitrile to precipitate the protein. Formic acid (10%, 0.01 ml) and the internal standard [0.02 ml of the deuterium isotope of AZT glucuronide (5 μg/ml)] were then added. Next, the samples were centrifuged at 1870g for 10 min to obtain the supernatant, aliquots (20 μl) of which were injected into a liquid chromatography with tandem mass spectrometry (LC-MS/MS) system. The substrate and inhibitors were dissolved in dimethyl sulfoxide to make an incubation mixture with a final dimethyl sulfoxide concentration of 1% (v/v).
Glucuronidation in Hepatocytes. Cryopreserved rat hepatocytes were stored in liquid nitrogen until use. Thawing was done according to the instructions supplied by Xenotech, after which the hepatocytes were suspended in enough WME to make a concentration of 2 × 106 cells/ml. The viability, determined using the trypan blue exclusion method, was found to be >68.5%. The time profile for AZTG was assessed first. The hepatocyte suspension (2.5 × 105 cells in 125 μl) was preincubated at 37°C for 3 min, after which the reactions were initiated by adding 125 μl of WME containing AZT (2 mM). After incubation at 37°C for the designated time, the reaction was terminated by adding acetonitrile (50 μl), followed by 10% formic acid (10 μl) and the internal standard (100 ng). The reaction mixtures were centrifuged at 1870g for 10 min, and aliquots of the supernatant (20 μl) were injected into the LC-MS/MS apparatus to determine the levels of AZT glucuronide. The kinetic study for AZTG and the inhibition effects of drugs on AZTG in rat hepatocytes were then addressed. AZT (0.2–10 mM) was incubated with hepatocytes (2.5 × 105 cells) for 30 min using the same procedures described above. The inhibitory potentials of drugs (diclofenac, diflunisal, indomethacin, ketoprofen, naproxen, and valproic acid) were then evaluated. AZT (1 mM) was incubated for 30 min with hepatocytes (2.5 × 105 cells) spiked with varying concentrations of these drugs. As a separate study, the effect of UDPGA levels on AZTG in hepatocytes was investigated using externally added UDPGA with a final concentration of 5 mM. The kinetic study for AZTG was then performed as described above.
Assay. The peak areas of AZT glucuronide and its internal standard were analyzed using LC-MS/MS (Mano et al., 2007). A TSQ7000 triple quadrupole mass spectrometer with an atmospheric pressure ionization source (Thermo Electron, Waltham, MA) was used. The atmospheric pressure ionization source was fitted with an electrospray ionization inlet for ionizing the analytes. Nitrogen was used as both the sheath and auxiliary gas with pressures of 80 psi and 40 units, respectively. Electron spray voltage was set at 4.5 kV and the heated capillary temperature was maintained at 350°C. AZT glucuronide and its internal standard were monitored in the negative ion mode using selected reaction monitoring by transmitting the molecular ions at m/z 442 and 445, respectively. These ions were subjected to collision-activated dissociation using argon (2.0 mtorr) at 20 eV, and then, the product ions were monitored at m/z 442 and 445 for AZT glucuronide and the internal standard, respectively. Chromatographic separation was achieved using a Capcellpak UG80 column (4.6 mm × 150 mm, 5 μm; Shiseido, Tokyo, Japan) with a flow rate of 0.5 ml/min. The mobile phase was composed of 0.1% formic acid/acetonitrile (7:3, v/v). The standard curve for the AZT glucuronide was linear from 0.1 to 10 μM, and the correlation coefficient was >0.99. The relative errors of the back-calculated values at each concentration were less than 15%.
Data Analysis. The IC50 value of each inhibitor was estimated from the inhibition against AZTG in the presence of inhibitors with varying concentrations by fitting the data to eq. 1, where I is the inhibitor concentration. 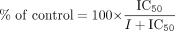 The Ki value of diflunisal was determined using eq. 2.
The Ki value of diflunisal was determined using eq. 2. 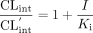 where CLint′ and CLint represent the intrinsic clearance of AZTG in RLMs in the presence and absence of diflunisal, respectively. Prism Ver. 3.02 software (GraphPad Software, San Diego, CA) was used for the calculations.
where CLint′ and CLint represent the intrinsic clearance of AZTG in RLMs in the presence and absence of diflunisal, respectively. Prism Ver. 3.02 software (GraphPad Software, San Diego, CA) was used for the calculations.
Results
Glucuronidation in RLMs. The effect on AZTG inhibition caused by diclofenac, diflunisal, fluconazole, indomethacin, ketoprofen, mefenamic acid, naproxen, niflumic acid, and valproic acid in RLMs was investigated. Diflunisal had the most potent inhibitory effect with an IC50 value of 34 ± 7.1 μM (mean ± computer-calculated S.E.). The IC50 values of diclofenac, fluconazole, indomethacin, ketoprofen, naproxen, mefenamic acid, niflumic acid, and valproic acid were 40 ± 1.8, 1791 ± 226, 178 ± 37, 103 ± 3.9, 276 ± 7.7, 332 ± 9.5, 37 ± 0.9, 102 ± 5.8, and 538 ± 35 μM (Table 1), respectively.
IC50 values for drugs inhibiting AZT glucuronidation in the liver microsomes and hepatocytes of rats
The data represent the mean ± computer-calculated S.E.
The type of inhibition diflunisal exerts against AZTG was investigated in RLMs. In the absence of diflunisal, AZTG exhibited Michaelis-Menten kinetics with Km and Vmax values of 10 ± 1.8 mM and 1345 ± 152 pmol/min/mg protein, respectively (Fig. 2). In the presence of 40 μM diflunisal, the Vmax value was reduced to 666 ± 75 pmol/min/mg protein, whereas the Km value (8.2 ± 1.6 mM) was similar to that without diflunisal. The inset in Fig. 2, an Eadie-Hofstee plot, suggests that one or more isozymes with similar Km values are involved in AZTG, and that diflunisal inhibits AZTG in a noncompetitive manner with a Ki value of 61 μM.
The effects of different buffer conditions on AZTG with RLMs were determined. The kinetics for AZTG in both carbonate buffer and WME obeyed Michaelis-Menten kinetics, and kinetic analysis revealed the Km values to be 5.6 ± 0.5 and 6.6 ± 0.4 mM, respectively. The respective Vmax values were 483 ± 20 and 528 ± 18 pmol/min/mg protein.
Glucuronidation in Hepatocytes. The time profile for the formation of AZT glucuronide was investigated in cryopreserved rat hepatocytes. The data showed that the glucuronidation velocity was linear for at least 60 min during incubation (Fig. 3A). The kinetics of AZTG was next evaluated using varying concentrations of AZT as a substrate for glucuronidation. The AZT concentration-velocity curve adheres to typical Michaelis-Menten kinetics, and the Eadie-Hofstee plot implies that more than one UGT isozyme is involved in AZTG. The Km value for the high-affinity isozyme was estimated to be 1.0 ± 0.4 mM, and the Vmax and CLns (clearance for low-affinity isozymes) values were 64 ± 15 pmol/min/106 cells and 4.4 ± 1.3 nl/min/106 cells, respectively (Fig. 3B). The impact of UDPGA levels on AZTG was also assessed in cryopreserved hepatocytes. When hepatocytes were incubated with externally added UDPGA (5 mM), the resulting Eadie-Hofstee plot suggested that one or more isozymes with similar Km values were involved in AZTG as they were in RLMs, with Km and Vmax values of 3.2 ± 0.4 mM and 275 ± 12 pmol/min/106 cells, respectively.
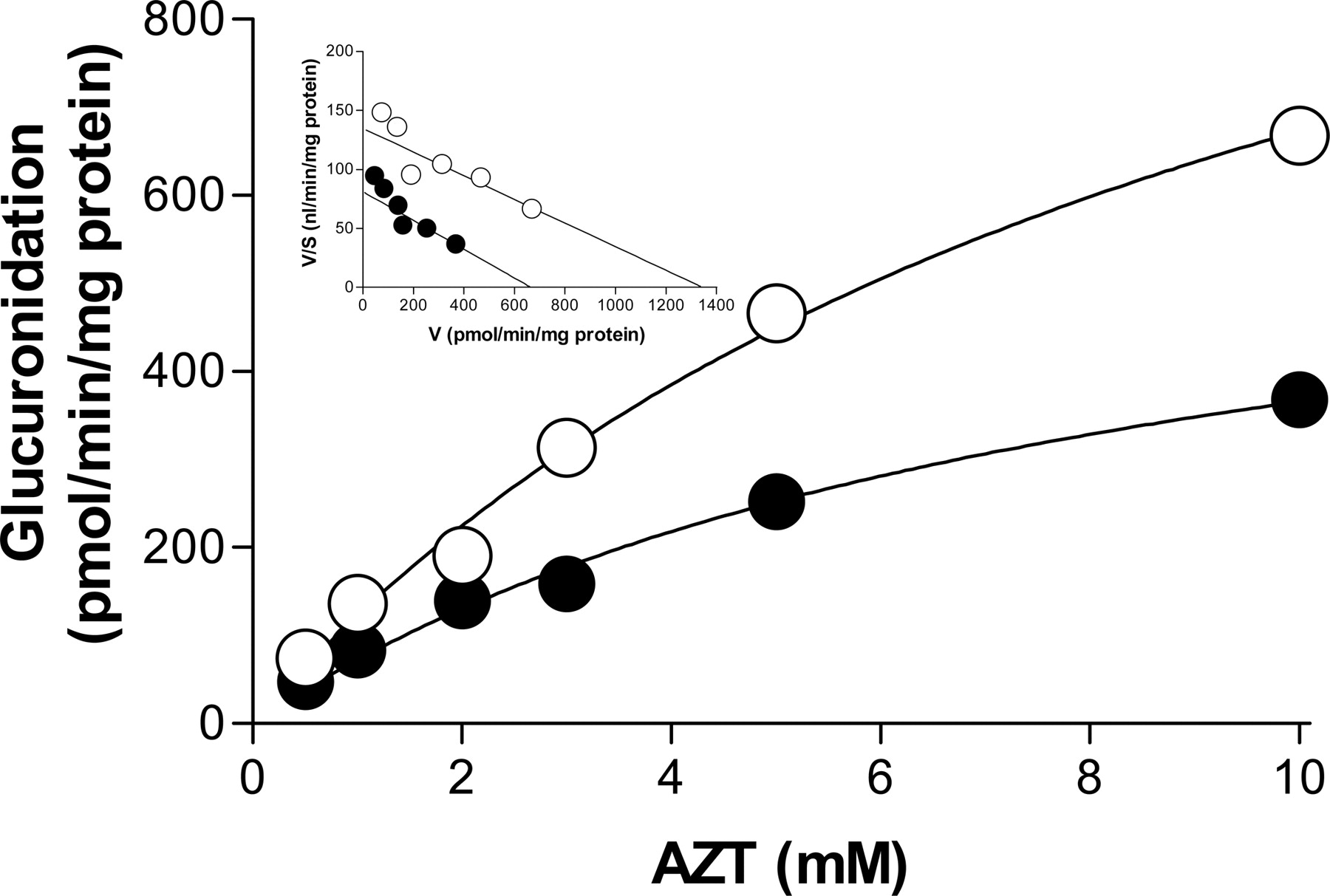
Inhibitory effects of diflunisal on AZT glucuronidation in RLMs. AZT (0.2–10 mM) was incubated in the presence (•) and absence (○) of diflunisal (40 μM) in RLMs (0.1 mg protein/ml) for 20 min. The x and y axes represent the substrate concentration and AZT glucuronidation velocity, respectively. The Eadie-Hofstee plot is presented as an inset. Each incubation was performed in duplicate, and the data represent the mean.
The inhibitory effects of diclofenac, diflunisal, indomethacin, ketoprofen, naproxen, and valproic acid on AZTG were then assessed in rat hepatocytes. Diclofenac, diflunisal, indomethacin, ketoprofen, naproxen, and valproic acid inhibited AZTG with IC50 values of 58 ± 2.2, 37 ± 2.2, 88 ± 16, 361 ± 34, 486 ± 52, and 281 ± 52 μM, respectively (Table 1). The IC50 values for these drugs against AZTG in hepatocytes were compared with those in RLMs. Representative AZTG inhibition profiles (indomethacin) in RLMs and hepatocytes are presented in Fig. 4. The AZTG inhibition potentials of the drugs investigated were found to be similar in both types of matrices (Fig. 5).
Discussion
This article describes the AZTG inhibition potentials of drugs in RLMs and cryopreserved hepatocytes. AZTG is mainly catalyzed by UGT2B7 in humans, and clinical drug interactions via UGT2B7 have been reported when AZT is concomitantly administered with fluconazole and valproic acid (Lertora et al., 1994; Sahai et al., 1994). When trying to predict in vivo drug interaction via glucuronidation, animal models (e.g., rats) may be valuable tools for bridging in vitro and in vivo data. A recent article by Engtrakul et al. (2005) showed that the Km of AZTG and the IC50 value of ibuprofen against AZTG in human hepatocytes are lower than those in HLMs and suggests that liver microsomes appear to underestimate inhibitory potentials of drugs (Engtrakul et al., 2005). Thus, in this study, the AZTG inhibition potentials of drugs in rats in both RLMs and hepatocytes were investigated, and the results were compared to determine whether the IC50 values in the hepatocytes are lower than those in liver microsomes.
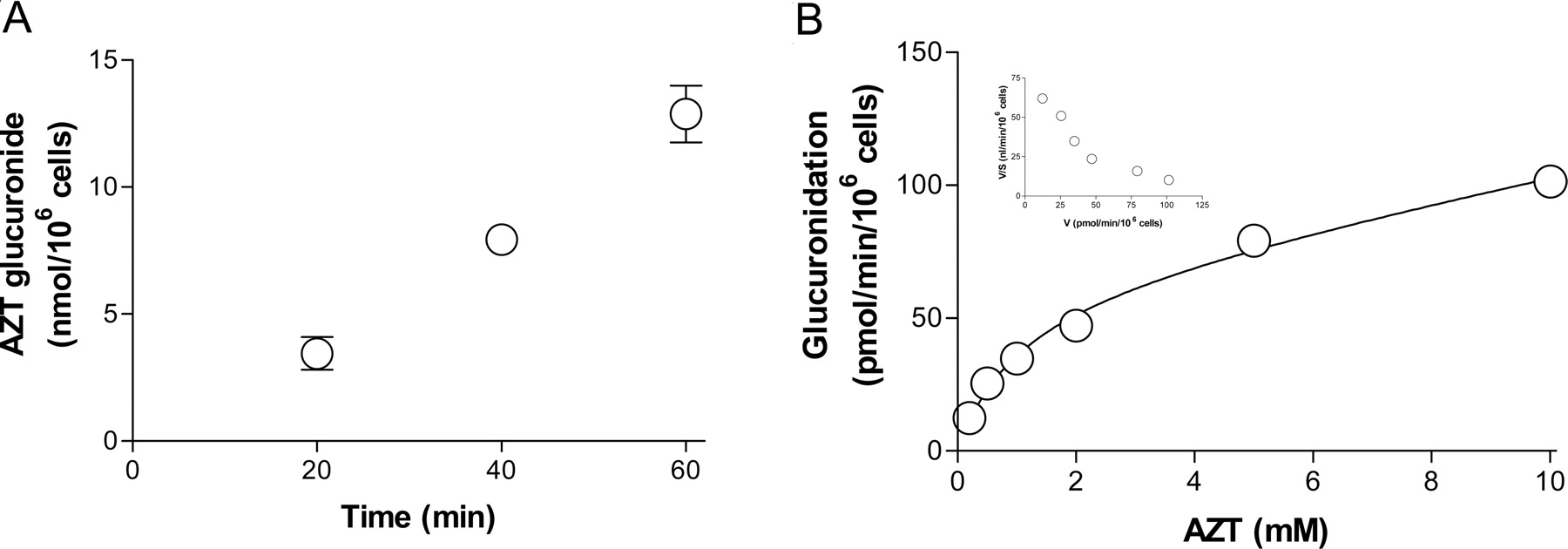
AZT glucuronidation in cryopreserved rat hepatocytes. Time profiles for the formation of AZT glucuronide (A) and kinetics for AZT glucuronidation (B) are presented. The Eadie-Hofstee plot is presented as an inset (B). AZT (2 mM) was incubated with rat hepatocytes (2.5 × 105 cells) for the times designated (A), and AZT (0.2–10 mM) was incubated with rat hepatocytes (2.5 × 105 cells) for 30 min (B). Each incubation was performed in triplicate, and the data represent the mean ± S.D.

Inhibition of AZT glucuronidation by indomethacin in the liver microsomes (A) and cryopreserved hepatocytes (B) of rats. AZT (2 mM) was incubated with liver microsomes (0.1 mg protein/ml) for 20 min (A), and AZT (1 mM) was incubated with hepatocytes (2.5 × 105 cells) for 30 min (B), with varying concentrations of indomethacin. Each incubation was performed in duplicate, and data represent the mean.

Comparison of the AZT glucuronidation inhibition potentials of drugs in RLMs and cryopreserved rat hepatocytes. AZT (2 or 1 mM) was incubated separately with RLMs (0.1 mg protein/ml) and hepatocytes (2.5 × 105 cells) for 20 and 30 min, respectively, in the presence or absence of inhibitors. The x and y axes represent the IC50 values in RLMs and hepatocytes, respectively, and the solid and dashed lines represent the line of unity and 2-fold differences, respectively. Dic, diclofenac; Dif, diflunisal; In, indomethacin; K, ketoprofen; Na, naproxen; V, valproic acid.
Although AZTG is catalyzed mainly by UGT2B7 in humans, the UGT isozyme responsible for catalysis in rats remains to be investigated. Despite this deficit, the Eadie-Hofstee plot in Fig. 2 suggests that one or more isozymes with similar Km values are involved in AZTG. The Km value for AZT in RLMs (10 mM) is consistent with that in the previous report (9.1 mM; Cretton et al., 1990). It should be noted that the solubility of AZT is less than that necessary to obtain accurate kinetic parameters in RLMs; however, the Km value obtained is similar to the one reported. The Km value for AZTG in rat hepatocytes (1.0 mM; Fig. 3B) was lower than that in RLMs (10 mM). This is a phenomenon similar to one that occurred in the previous study; the Km value for AZTG in human hepatocytes was much lower than that in HLMs (87 versus 760 μM; Engtrakul et al., 2005). The reason for this discrepancy remains to be determined, although the active transport systems involved in AZT uptake in the hepatocytes might be responsible. It has also been reported that AZT is a substrate for rat organic anion transporter 2, which is abundantly expressed in the liver (Morita et al., 2001). Buffer systems in HLMs (Engtrakul et al., 2005) and differences in the environment surrounding the UGT isozymes might contribute to the differences in the Km values. The levels of UDPGA, a glucuronidation cofactor, might also be behind the differences between the two matrices. In RLMs, enough UDPGA was added to the reaction mixtures to make a concentration of 5 mM, but no cofactor was added to the rat hepatocytes. Therefore, the UDPGA levels in the hepatocytes might have been insufficient, given the high concentration of AZT. To find potential factors that may contribute, at least partially, to the differences in the kinetic parameters of AZTG between hepatocytes and liver microsomes, the impact of UDPGA levels and buffer incubation conditions on AZTG in hepatocytes and RLMs was determined. Externally added 5 mM UDPGA significantly increased the Km value to 3.2 mM, which is closer to that in RLMs, compared with that in hepatocytes that did not have UDPGA added (Km 1.0 mM). In addition, the Eadie-Hofstee plot suggests that one or more isozymes with similar Km values are involved in AZTG in hepatocytes, as was observed in RLMs (data not shown). Next, the effects of incubation buffers on AZTG were assessed in RLMs. Incubation in sodium carbonate buffer and WME resulted in a Km value that decreased to 5.6 and 6.6 mM in RLMs, which is closer to that in hepatocytes incubated with 5 mM UDPGA (Km 3.2 mM). A decrease in the Km value for AZTG in sodium carbonate buffer was also observed in HLMs (Engtrakul et al., 2005). These findings suggest that buffer systems and UDPGA levels are at least partially involved in the differences in the Km values for AZTG between RLMs and hepatocytes.
To compare the AZTG inhibition potentials of drugs in liver microsomes with those in hepatocytes, the IC50 values were also determined in rat hepatocytes. Representative AZTG inhibition profiles (indomethacin) in RLMs and hepatocytes are presented in Fig. 4. The IC50 values of diclofenac, diflunisal, indomethacin, ketoprofen, naproxen, and valproic acid in hepatocytes were similar to those in RLMs (Fig. 5). This finding differs from the previous report in that the IC50 value of ibuprofen against AZTG in human hepatocytes was significantly lower than that in HLMs (170 versus 975 μM; Engtrakul et al., 2005). However, the study in human hepatocytes was carried out using only one substrate-inhibitor pair; thus, studies with more pairs are necessary to determine whether hepatocytes generally yield more potent inhibition values than liver microsomes. In addition, the inhibition potentials of drugs against various UGT isozymes should also be investigated using probe substrates for each isozyme. This could yield insight into whether care should be taken when liver microsomes are used to assess metabolizing enzyme inhibition potentials for drugs.
When the inhibitory potentials of the drugs are estimated, the nonspecific binding of the inhibitors to the incubation matrix should be taken into account. McGinnity et al. (2004) reported that the unbound fraction is a major contributing factor in the differences in apparent IC50 values for recombinant CYP2C9 and hepatocytes in the incubation matrix. However, acidic drugs such as nonsteroidal anti-inflammatory drugs tend to exhibit low nonspecific binding to the matrix (Austin et al., 2002; McGinnity et al., 2004). Thus, the nonspecific binding of inhibitors is considered to have only a slight effect on the apparent IC50 value.
The previous report that similar IC50 values of drugs are obtained between liver microsomes and hepatocytes against P450 isozyme-mediated metabolism (Di Marco et al., 2003; Jones et al., 2004) and the current study findings suggest that liver microsomes could be used as an in vitro system for assessing the metabolizing enzyme inhibition potentials of drugs.
In summary, the AZTG inhibition potentials of several drugs were investigated in RLMs and cryopreserved rat hepatocytes. The IC50 values in liver microsomes and hepatocytes were found to be similar. These findings show that liver microsomes can be used to quantitatively determine the glucuronidation inhibition potentials of drugs.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.014225.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; AZT, 3′-azido-3′-deoxythymidine; AZTG, AZT glucuronidation; P450, cytochrome P450; HLM, human liver microsome; LC-MS/MS, liquid chromatography with tandem mass spectrometry; RLM, rat liver microsome; UDPGA, UDP-glucuronic acid; WME, Williams' Medium E.
- Received December 6, 2006.
- Accepted January 25, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}