


Abstract
Rosuvastatin is an HMG-CoA reductase inhibitor and one of the most hydrophilic among the commercially available statins. It is efficiently accumulated in the liver and excreted into the bile in an unchanged form in rats, suggesting that hepatic transporters play a major role in its clearance. Therefore, we investigated the transporters responsible for the hepatic uptake and biliary excretion of rosuvastatin. Uptake studies revealed that human organic anion transporting polypeptide (OATP) 1B1, OATP1B3, and OATP2B1 accept rosuvastatin as a substrate. Among the OATP family transporters, OATP1B1 contributes predominantly to the hepatic uptake of rosuvastatin, as estimated with the previously published relative activity factor method, and OATP1B3 is also partly involved. Significant vectorial basal-to-apical transport was observed in OATP1B1/multidrug resistance-associated protein 2 (MRP2), OATP1B1/multidrug resistance protein 1 (MDR1), and OATP1B1/breast cancer resistance protein (BCRP) double transfectants compared with that in an OATP1B1 single transfectant or in vector-transfected control cells. The ATP-dependent uptake of rosuvastatin by human BCRP-expressing membrane vesicles was significantly higher than the uptake by green fluorescent protein-expressing control vesicles, suggesting that MRP2, MDR1, and BCRP can transport rosuvastatin. Under in vivo conditions, the biliary excretion clearances based on the intrahepatic concentration of the parent rosuvastatin in Eisai hyperbilirubinemic rats and Bcrp1 knockout mice were reduced to 53% and 12% of those in the control Sprague-Dawley rats and FVB mice, respectively, indicating that rat Mrp2 and mouse Bcrp1 are both partly involved in the biliary excretion of rosuvastatin. These results suggest that multiple transporters are involved in the hepatic uptake and efflux of rosuvastatin.
Statins are potent inhibitors of HMG-CoA reductase. They reduce the plasma concentration of low-density lipoprotein cholesterol in a dose-dependent manner. Rhabdomyolysis is a rare but life-threatening adverse effect of statins, and high plasma levels of statins are a risk factor for this complication (Hamilton-Craig, 2001). Because the liver is the pharmacological target as well as the clearance organ for statins, it is essential to clarify the mechanisms underlying the hepatobiliary transport of rosuvastatin to predict both the desired pharmacological effects and the unwanted adverse effects.
Of all the commercially available statins, rosuvastatin exhibits the greatest inhibitory effect on cholesterol biosynthesis in primary cultured rat hepatocytes (McTaggart et al., 2001). Rosuvastatin is relatively hydrophilic compared with other statins (McTaggart et al., 2001) and is anionic at neutral pH. After its oral administration to rats, it is efficiently concentrated in the rat liver and collected in the bile, mostly in an unchanged form (Nezasa et al., 2002). In humans, its hepatic elimination accounts for approximately 70% of its total clearance (Martin et al., 2003). Metabolic clearance is thought to be minor (Fujino et al., 2004). It has been suggested that the major clearance mechanism of rosuvastatin is biliary excretion of the unchanged form, indicating that several transporters might be involved in that step.
Many uptake and efflux transporters of organic anions in the human liver have been characterized. Organic anion transporting polypeptide (OATP) 1B1 (Abe et al., 1999; Hsiang et al., 1999), OATP1B3 (König et al., 2000), OATP2B1 (Kullak-Ublick et al., 2001), and organic anion transporter 2 (Babu et al., 2002), on the sinusoidal membrane, are thought to be involved in the hepatic uptake of a wide variety of anions, and sodium-taurocholate cotransporting polypeptide (NTCP) is responsible for the uptake of bile acids (Trauner and Boyer, 2003). Among these, OATP1B1 and OATP1B3 are selectively expressed in the human liver and exhibit broad substrate specificities, suggesting the importance of these transporters in the hepatic uptake of many anions, including clinically important drugs.
Multidrug resistance-associated protein 2 (MRP2) (König et al., 1999), multidrug resistance 1 (MDR1) (Tanigawara, 2000), breast cancer resistance protein (BCRP) (Staud and Pavek, 2005), and the bile salt export pump (Trauner and Boyer, 2003) are expressed on the canalicular membrane. MRP2 is thought to be responsible for the biliary excretion of various organic anions, including glutathione and glucuronide conjugates. Recently, other ATP-binding cassette transporters have also been shown to accept some organic anions (Hirano et al., 2005), so it is possible that multiple efflux transporters are cooperatively involved in their biliary excretion.
In general, several drug transporters expressed on the same membrane act to transport substrates in the same direction, with overlapping substrate specificities. Thus, membrane penetration of substrates is often achieved by multiple transporters. We consider it necessary therefore to determine the contribution of each transporter to the hepatic transport of drugs because such information enables us to predict changes in hepatic intrinsic clearance when the functions of transporters are altered by genetic polymorphisms, disease, or drug-drug interactions.
Hirano et al. (2004) have already established methodologies for estimating the contribution of transporters to the hepatic uptake of compounds using transporter-specific substrates and Western blot analysis. Using these approaches, we have clarified the differences in the relative contributions of OATP1B1 and OATP1B3 to the hepatic uptake of some drugs (Maeda and Sugiyama, 2007). We have established a set of double transfectants expressing uptake (OATP1B1) and efflux transporters (MDR1, MRP2, or BCRP) to evaluate the efflux transporters in humans (Sasaki et al., 2002; Matsushima et al., 2005). Using these cell lines, we have shown that MDR1 and BCRP as well as MRP2 can transport several organic anions. These cell lines are useful for the easy identification of uptake and efflux transporters and for comparing the relative transport clearance mediated by each transporter. It has recently been reported that OATP1B1, OATP1B3, and OATP2B1 all transport rosuvastatin (Ho et al., 2006), but their relative contributions have not been clarified yet.
In this study, we focused on OATP1B1, OATP1B3, and OATP2B1 as transporters responsible for the hepatic uptake of rosuvastatin and MRP2, MDR1, and BCRP as transporters responsible for its biliary excretion. We estimated the relative contribution of these transporters to the hepatic uptake and biliary excretion of rosuvastatin using several approaches established in our laboratory.
Materials and Methods
Animals. Male Sprague-Dawley (SD) rats (9 weeks old) and Eisai hyperbilirubinemic rats (EHBRs) (9 weeks old) were purchased from Japan SLC, Inc. (Shizuoka, Japan). Male wild-type FVB mice and Bcrp1 knockout mice (13–15 weeks old) were also used in this study (Jonker et al., 2002). All animals were maintained under standard conditions with a reverse dark-light cycle and were treated humanely. Food and water were available ad libitum. The studies reported in this article were performed in accordance with the guidelines provided by the Institutional Animal Care Committee, Graduate School of Pharmaceutical Sciences, The University of Tokyo.
Materials. Rosuvastatin, calcium bis-[(+)-(3R,5S,6E)-7-[4-(p-fluorophenyl)-6-isopropyl-2-(N-methylmethanesulfonamido)-5-pyrimidinyl]-3,5-dihydroxy-6-hepteonate], and [3H]rosuvastatin (79 Ci/mmol) were kindly donated by AstraZeneca (Macclesfield, UK). [3H]Estrone-3-sulfate (E1S) (45 Ci/mmol) and [3H]17β-estradiol-17β-d-glucuronide (E217βG) (46 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). [3H]Cholecystokinin octapeptide (CCK-8) was purchased from GE Healthcare (Piscataway, NJ). Unlabeled E1S, CCK-8, and E217βG were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were of analytical grade and commercially available.
Establishment of Cell Lines and Culture Conditions. Construction of OATP1B1 (OATP1B1*1a)-, OATP1B3-, and OATP2B1-expressing HEK293 cells and single- and double-transfected MDCKII cells expressing human OATP1B1, MDR1, MRP2, or BCRP was as described previously (Hirano et al., 2004; Matsushima et al., 2005). The transporter-expressing or vector-transfected HEK293 cells and MDCKII cells were grown in low-glucose Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C under 5% CO2 and 95% humidity.
Uptake Study. The uptake study was performed as described previously (Hirano et al., 2004). Briefly, transporter-expressing or vector-transfected HEK293 cells were seeded onto 12-well plates at a density of 1.2 × 105 cells/well and grown for 2 days. The culture medium was then replaced with medium supplemented with 5 mM sodium butyrate for 24 h before the transport study to increase the expression of transporters. The cells were washed twice and preincubated with Krebs-Henseleit buffer (118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2 adjusted to pH 7.4) at 37°C for 15 min. Uptake was initiated by the addition of Krebs-Henseleit buffer containing radiolabeled and unlabeled rosuvastatin and was terminated at the designated time by the addition of ice-cold Krebs-Henseleit buffer after the removal of the incubation buffer. The cells were then washed three times with 1 ml of ice-cold Krebs-Henseleit buffer, solubilized in 500 μl of 0.2 N NaOH, and incubated overnight at 4°C. Aliquots (500 μl) were transferred to scintillation vials after the addition of 250 μl of 0.4 N HCl. The radioactivity associated with the cells and with the incubation buffer was measured in a liquid scintillation counter (LS6000SE; Beckman Coulter, Fullerton, CA) after the addition of 2 ml of scintillation cocktail (Clear-sol I; Nacalai Tesque, Kyoto, Japan) to the scintillation vials. The remaining 50 μl of cell lysate was used to determine the protein concentration by the method of Lowry et al. (1951), with bovine serum albumin as the standard. Ligand uptake was normalized to the amount of cellular protein and expressed as the uptake volume (microliters per milligram of protein), calculated as the amount of radioactivity associated with the cells (disintegrations per minute per milligram of protein) divided by its concentration in the incubation medium (disintegrations per minute per microliter). Transporter-mediated uptake was calculated by subtracting the uptake of control cells from that of transporter-expressing cells.
Estimation of the Contribution of OATP1B1 and OATP1B3 to the Uptake of Compounds in Human Hepatocytes. The detailed methodology has been described previously (Hirano et al., 2004). In brief, the relative activity factor (R value) for OATP1B1 or OATP1B3 was calculated as the ratio of the uptake clearance of E1S or CCK-8, respectively, in human hepatocytes (CLhepatocyte) to that in transporter-expressing cells (CLtransporter). Then the contribution of OATP1B1 or OATP1B3 to the hepatic uptake of the test compound in human hepatocytes was calculated using the following equations: 
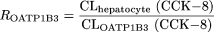
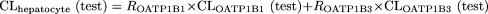
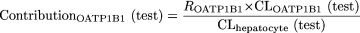

Transcellular Transport Study. The transcellular transport study was carried out as reported previously (Sasaki et al., 2002; Matsushima et al., 2005). In brief, transfected MDCKII cells were seeded onto Transwell membrane inserts (6.5 mm diameter, 0.4 μm pore size; Corning Costar, Bodenheim, Germany) at a density of 1.4 × 105 cells per well and grown to confluence for 3 days. The culture medium was replaced with medium supplemented with 5 mM sodium butyrate 24 h before the transport study to induce the expression of exogenous transporters. The cells were washed twice and preincubated with Krebs-Henseleit buffer at 37°C for 20 min. Transcellular transport was initiated by the addition of Krebs-Henseleit buffer containing radiolabeled and unlabeled rosuvastatin either to the apical compartment (250 μl) or to the basal compartment (1000 μl). At the designated times, 100 μl of buffer in the opposite compartment was transferred to a scintillation vial. After the addition of 3 ml of scintillation fluid (Clear-sol I), the radioactivity was measured with a liquid scintillation counter (LS6000SE). At the end of the experiments, the cells were washed three times with 1.5 ml of ice-cold Krebs-Henseleit buffer after removal of the incubation buffer and were solubilized in 500 μl of 0.2 N NaOH. After the addition of 100 μl of 1 N HCl, an aliquot (450 μl) was transferred to a liquid scintillation vial. The radioactivity was measured in a liquid scintillation counter after the addition of 2 ml of scintillation cocktail (Clear-sol I) to the scintillation vial. An aliquot (50 μl) of cell lysate was used to determine the protein concentration, as described above. The transcellular transport clearance of rosuvastatin (microliters per milligram of protein) from the basal to the apical side (PSnet, b to a) and from the apical to the basal side (PSnet, a to b) was calculated by dividing the basal-to-apical or apical-to-basal transcellular transport velocity by the initial concentration in the basal or the apical compartment, respectively. The efflux clearance across the apical membrane (PSapical) (microliters per milligram of protein) was calculated by dividing the transcellular transport velocity from the basal to the apical side by the steady-state intracellular concentration of rosuvastatin, assuming that the cell volume was 4 μl/mg of protein (Sasaki et al., 2002).
Transport Study with Human BCRP-Expressing Membrane Vesicles. The construction and infection of recombinant adenovirus carrying human BCRP/ABCG2 and GFP and the preparation of membrane vesicles have been described previously (Kondo et al., 2004). The transport studies were performed using a rapid filtration technique (Niinuma et al., 1999). Briefly, 15 μl of transport buffer (10 mM Tris-HCl, 250 mM sucrose, and 10 mM MgCl2, pH 7.4) containing radiolabeled and unlabeled rosuvastatin was preincubated at 37°C for 3 min and then rapidly mixed with 5 μl of membrane vesicle suspension (2 μg of protein). The reaction mixture contained 5 mM ATP or AMP, together with an ATP-regenerating system (10 mM creatine phosphate and 100 μg/ml creatine phosphokinase). The transport reaction was terminated by the addition of 1 ml of ice-cold stop buffer (10 mM Tris-HCl, 250 mM sucrose, and 0.1 M NaCl, pH 7.4). The stopped reaction mixture was filtered through a 0.45-μm JH filter (Millipore Corporation, Bedford, MA) and then washed twice with 5 ml of stop buffer. The radioactivity retained on the filter was determined in a liquid scintillation counter (LS6000SE) after the addition of 2 ml of scintillation cocktail (Clear-sol I) with shaking overnight. The ATP-dependent uptake of rosuvastatin mediated by BCRP was estimated by subtracting the ATP-dependent uptake into the control vesicles from that into the BCRP-expressing vesicles, and ATP-dependent uptake was calculated by subtracting the uptake into the vesicles in the presence of AMP from that in the presence of ATP.
Kinetic Analysis. The kinetic parameters were calculated using the following equations: 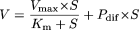
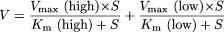 where V is the uptake velocity of the substrate (picomoles per minute per milligram of protein), S is the substrate concentration in the medium (micromolar), Km is the Michaelis constant (micromolar concentration), Vmax is the maximum uptake velocity (picomoles per minute per milligram of protein), and Pdif is the nonsaturable uptake clearance (microliters per minute per milligram of protein). “High” and “low” in eq. 7 indicate parameters of the high- and low-affinity components, respectively. Fitting was performed with the nonlinear least-squares method (the Damping Gauss-Newton method algorithm) using the MULTI program (Yamaoka et al., 1981).
where V is the uptake velocity of the substrate (picomoles per minute per milligram of protein), S is the substrate concentration in the medium (micromolar), Km is the Michaelis constant (micromolar concentration), Vmax is the maximum uptake velocity (picomoles per minute per milligram of protein), and Pdif is the nonsaturable uptake clearance (microliters per minute per milligram of protein). “High” and “low” in eq. 7 indicate parameters of the high- and low-affinity components, respectively. Fitting was performed with the nonlinear least-squares method (the Damping Gauss-Newton method algorithm) using the MULTI program (Yamaoka et al., 1981).
In Vivo Infusion Study in Rats. Male SD rats and EHBRs (Mrp2-deficient rats) weighing approximately 300 to 350 g were used for these experiments. Under pentobarbital anesthesia (40 mg/kg), the femoral vein was cannulated with a polyethylene catheter (PE-50) for the injection of rosuvastatin. The bile duct was cannulated with a polyethylene catheter (PE-10) for bile collection. The rats received a constant infusion of rosuvastatin at a dose of 667 pmol/min/kg. Blood samples were collected from the femoral artery cannulated with a polyethylene catheter (PE-50), and the plasma was prepared by centrifugation of the blood samples (10,000g; Microfuge, Beckman Coulter). The bile was collected in preweighed test tubes at 60-min intervals throughout the experiments. The radioactivity in 100 μl of plasma and in 20 μl of bile was determined in a liquid scintillation counter after the addition of 5 ml of scintillation cocktail (Hionic-Fluor; PerkinElmer Life and Analytical Sciences). The rats were killed after 300 min, and the entire liver was excised immediately. The liver was weighed and minced, and 200 μl of hydrogen peroxide and 400 μl of isopropanol were added to approximately 200 mg of minced liver. This sample was incubated at 55°C for 3 h after the addition of 2 ml of Soluene 350 (PerkinElmer Life and Analytical Sciences) to dissolve the tissues. The radioactivity was determined in a liquid scintillation counter after the addition of 10 ml of scintillation cocktail (Hionic-Fluor).
In Vivo Infusion Study in Mice. Male FVB and Bcrp1 knockout mice weighing approximately 28 to 33 g were used throughout these experiments. Under pentobarbital anesthesia (40 mg/kg), the jugular vein was cannulated with a polyethylene catheter (PE-10) connected to a 30-gauge needle for the injection of rosuvastatin. The bile duct was cannulated with a polyethylene catheter (UT-03) for bile collection. The mice received a constant infusion of rosuvastatin at a dose of 100 pmol/min/kg after a bolus i.v. administration of rosuvastatin (3 nmol/kg). Blood samples were collected from the contralateral jugular vein, and the plasma was prepared by centrifugation of the blood samples (10,000g). The bile was collected in preweighed test tubes at 30-min intervals throughout the experiments. The radioactivity in 20 μl of plasma and in 10 μl of bile was determined in a liquid scintillation counter after the addition of 5 ml of scintillation cocktail (Hionic-Fluor). The mice were killed after 120 min, and the entire liver was excised immediately. The liver was weighed and minced, and 100 μl of hydrogen peroxide and 200 μl of isopropanol were added to approximately 100 mg of minced liver. The sample was incubated at 55°C for 3 h after the addition of 2 ml of Soluene 350 to dissolve the tissues. The radioactivity was then determined in a liquid scintillation counter after the addition of 10 ml of scintillation cocktail (Hionic-Fluor).
Quantification of the Percentage of Parent Rosuvastatin. Nonmetabolized rosuvastatin was separated, and the percentage of parent rosuvastatin relative to the total radioactivity was quantified with a thin-layer chromatography (TLC) method (Nezasa et al., 2002). Plasma and liver samples were obtained after 300 min from rats or after 120 min from mice. The samples were mixed with 2 and 3 volumes of methanol, respectively, and were centrifuged for 3 min to separate the supernatants. These methanol extracts were evaporated to dryness under reduced pressure in a rotary evaporator. The residues were reconstituted in a small volume of methanol and subjected to TLC. Portions of the bile samples, collected at 240 to 300 min from rats and 90 to 120 min from mice in each infusion study, were applied directly to a TLC plate (LK6DF; Whatman, Brentford, UK). Unlabeled rosuvastatin (50 nmol) was also applied to the plate to overlap each sample. The TLC plates were developed with chloroform-methanol-acetic acid (20:10:1, by volume) for 60 min (Nezasa et al., 2002). After development, the silica gel on the plates was scraped off on the basis of the locations of the standard bands. The radioactivity was measured after the addition of 10 ml of scintillation cocktail (Clear-sol I) with shaking overnight, to determine the relative amounts (percentages) of unchanged rosuvastatin to the total radioactivity in each sample.
Pharmacokinetic Analysis. Total plasma clearance relative to plasma concentration (CLtot, p) and biliary excretion clearance relative to plasma concentration (CLbile, p) and relative to hepatic concentration (CLbile, H) were calculated with the following equations: 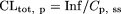

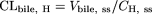 where Inf represents the infusion rate (picomoles per minute per kilogram), Cp, ss is the plasma concentration at steady state (nanomolar), Vbile, ss is the biliary excretion rate at steady state (picomoles per minute per kilogram), and CH, ss is the hepatic concentration at steady state (nanomolar). In rats, Cp, ss was calculated as the plasma concentration of the parent rosuvastatin at 300 min and Vbile, ss as the biliary excretion rate of the parent rosuvastatin from 240 to 300 min. In mice, Cp, ss was calculated as the plasma concentration of the parent rosuvastatin at 120 min and Vbile, ss as the biliary excretion rate of the parent rosuvastatin from 90 to 120 min. In both animals, CH, ss was calculated as the hepatic rosuvastatin concentration at the end of the in vivo experiment. To calculate CH, ss, the specific gravity of the liver was assumed to be 1. Thus, the amount in the liver (picomoles per minute per gram of liver) can be regarded as the hepatic concentration (picomoles per milliliter = nanomolar concentration).
where Inf represents the infusion rate (picomoles per minute per kilogram), Cp, ss is the plasma concentration at steady state (nanomolar), Vbile, ss is the biliary excretion rate at steady state (picomoles per minute per kilogram), and CH, ss is the hepatic concentration at steady state (nanomolar). In rats, Cp, ss was calculated as the plasma concentration of the parent rosuvastatin at 300 min and Vbile, ss as the biliary excretion rate of the parent rosuvastatin from 240 to 300 min. In mice, Cp, ss was calculated as the plasma concentration of the parent rosuvastatin at 120 min and Vbile, ss as the biliary excretion rate of the parent rosuvastatin from 90 to 120 min. In both animals, CH, ss was calculated as the hepatic rosuvastatin concentration at the end of the in vivo experiment. To calculate CH, ss, the specific gravity of the liver was assumed to be 1. Thus, the amount in the liver (picomoles per minute per gram of liver) can be regarded as the hepatic concentration (picomoles per milliliter = nanomolar concentration).

Time profiles of rosuvastatin uptake in human OATP1B1-, OATP1B3-, and OATP2B1-expressing HEK293 cells. •, ▪, ▴, and ○ represent the uptake of rosuvastatin (0.1 μM) by human OATP1B1-, OATP1B3-, OATP2B1- and vector-transfected HEK293 cells, respectively. Each point represents the mean ± S.E. (n = 3). Where vertical bars are not shown, the S.E. values are within the limits of the symbols.
Statistical Analysis. Statistically significant differences were determined using Student's t test. Differences were considered to be significant at P < 0.05.
Results
Uptake of Rosuvastatin and Reference Compounds (E1S andCCK-8) into OATP1B1-, OATP1B3-, and OATP2B1-Expressing HEK293 Cells. The time profiles for the uptake of rosuvastatin are shown in Fig. 1. The uptake of rosuvastatin (0.1 μM) into OATP1B1-, OATP1B3-, and OATP2B1-expressing HEK293 cells was significantly higher than that into vector-transfected control cells. In parallel, we measured the uptake of transporter-selective reference compounds (E1S for OATP1B1 and CCK-8 for OATP1B3) mediated by OATP1B1 and OATP1B3. The uptake clearance of the reference compounds and rosuvastatin in the OATP1B1- and OATP1B3-expressing cells is summarized in Table 1.
Uptake clearance of reference compounds (E1S and CCK-8) and rosuvastatin in OATP1B1- and OATP1B3-expressing HEK293 cells (our results) and human hepatocytes (reported values), and the relative contribution of OATP1B1 and OATP1B3 to the overall hepatic uptake of rosuvastatin in human hepatocytes
In the experiments using transporter expression systems, the uptake of E1S (reference compound for OATP1B1) for 0.5 min, CCK-8 (reference compound for OATP1B3) for 5 min, and rosuvastatin for 1 min were simultaneously determined, respectively. OATP1B1- and OATP1B3-mediated transport was calculated by subtracting the uptake in vector-transfected control cells from that in OATP1B1- and OATP1B3-expressing cells. The method is described in detail under Materials and Methods.
Concentration-Dependent Uptake of Rosuvastatin Mediated by OATP1B1, OATP1B3, and OATP2B1. The transporter-mediated uptake of rosuvastatin for 1 min (0.1–100 μM) was estimated, and Eadie-Hofstee plots for the uptake of rosuvastatin mediated by OATP1B1, OATP1B3, and OATP2B1 are shown in Fig. 2, A, B, and C, respectively. The concentration-dependent uptake of rosuvastatin mediated by each transporter can be explained by one saturable and one nonsaturable component. Their kinetic parameters were calculated using eq. 6 and are summarized in Table 2.
Kinetic parameters of the uptake of rosuvastatin in OATP1B1-, OATP1B3-, and OATP2B1-expressing HEK293 cells
Data shown in Fig. 2 were used to determine the Km, Vmax, and Pdif values. These parameters were determined by fitting the data using the nonlinear least-squares method. Each point represents the mean ± computer-calculated S.D. (n = 6).
Transcellular Transport of Rosuvastatin across Double Transfectants. Time profiles for the transcellular transport of rosuvastatin (0.1 μM) across double-transfected MDCKII cells expressing an uptake transporter (OATP1B1) and an efflux transporter (MDR1, MRP2, or BCRP) were compared with those across single-transfected cells or vector-transfected control cells. As shown in Fig. 3, a symmetrical flux of rosuvastatin was observed across the control, OATP1B1-, MRP2-, MDR1-, and BCRP-expressing MDCKII cells (Fig. 3, A–E). In contrast, the basal-to-apical fluxes of rosuvastatin were approximately 8, 25, and 1.7 times higher than those in the opposite direction in OATP1B1/MRP2, OATP1B1/MDR1, and OATP1B1/BCRP double-transfected cells, respectively (Fig. 3, F–H). To confirm whether rosuvastatin is a substrate of MRP2, MDR1, or BCRP, the efflux clearance of rosuvastatin (0.1 μM) across the apical membranes of MDCKII cells (PSapical) was also calculated at steady state by dividing the basal-to-apical transport rate by the intracellular concentration of rosuvastatin measured at 120 min. The PSapical values for rosuvastatin in OATP1B1/MRP2, OATP1B1/MDR1, and OATP1B1/BCRP double-transfected cells were approximately 10, 5.4, and 2.7 times higher, respectively, than that in OATP1B1 single-transfected cells (data not shown). We also checked the transcellular transport and PSapical value for E217βG in parallel as a positive control and obtained results similar to those measured previously (Matsushima et al., 2005) (data not shown).
Concentration-Dependent Transcellular Transport of Rosuvastatin in OATP1B1/MRP2 and OATP1B1/MDR1 Double Transfectants. PSnet, b to a and PSapical of rosuvastatin showed concentration dependence in both OATP1B1/MRP2- and OATP1B1/MDR1-expressing cell lines (Fig. 4). Kinetic analyses revealed that the saturation is best described by assuming the presence of one saturable and one nonsaturable component. The analysis produced the Km, Vmax, and Pdif values shown in Table 3.
Kinetic parameters PSnet, b to a and PSapical of rosuvastatin in OATP1B1/MRP2- and OATP1B1/MDR1 double-transfected MDCKII cells
Data shown in Fig. 4 were used to determine the Km, Vmax, and Pdif values. These parameters were determined by fitting the data using the nonlinear least-squares method. Each point represents the mean ± computer-calculated S.D. (n = 3).
ATP-Dependent Uptake of Rosuvastatin into BCRP-Expressing Membrane Vesicles. The time profiles and Eadie-Hofstee plots for the uptake of rosuvastatin by BCRP-expressing membrane vesicles are shown in Fig. 5. The uptake of rosuvastatin (0.1 μM) into membrane vesicles prepared from human BCRP-expressing HEK293 cells was markedly stimulated in the presence of ATP but not in vesicles prepared from GFP-expressing control cells (Fig. 5A). The concentration dependence (0.1–100 μM) of the ATP-dependent uptake of rosuvastatin mediated by BCRP (1 min) was also examined. The saturable uptake can be explained by two saturable components (Fig. 5B), and by fitting these data to eq. 7, the Km and Vmax values were calculated to be 2.02 ± 1.12 μM and 304 ± 195 pmol/min/mg of protein, respectively, for the high-affinity component and 60.9 ± 40.1 μM and 3320 ± 937 pmol/min/mg of protein, respectively, for the low-affinity component (mean ± computer-calculated S.D.).

Eadie-Hofstee plots of the uptake of rosuvastatin in human OATP1B1-, OATP1B3-, and OATP2B1-expressing HEK293 cells. The concentration dependence of the (A) OATP1B1-, (B) OATP1B3-, and (C) OATP2B1-mediated uptake of rosuvastatin is shown in the Eadie-Hofstee plots. The uptake clearance in 1 min was determined at various concentrations (0.1–100 μM). The OATP1B1-, OATP1B3-, and OATP2B1-mediated transport was calculated by subtracting the uptake in vector-transfected control cells from that in transporter-expressing cells. The data were fitted to eq. 6 (described under Materials and Methods) by the nonlinear least-squares method, and each solid line represents the fitted curve. Each point represents the mean ± S.E. (n = 6). Where vertical and/or horizontal bars are not shown, the S.E. values are within the limits of the symbols.
Kinetic Parameters of Unchanged Rosuvastatin in SD Rats and EHBRs. The plasma concentration and biliary excretion rate, estimated from the total tritium radioactivity derived from the administered rosuvastatin, reached a plateau at 240 min during constant infusion (Fig. 6). The percentage of parent rosuvastatin in plasma, bile, and liver at 300 min was estimated by TLC, as described under Materials and Methods. The percentages of unchanged rosuvastatin relative to the total radioactivity in the plasma, bile, and liver were 34, 85, and 50%, respectively, for SD rats, and 48, 84, and 51%, respectively, for EHBRs. The clearance of parent rosuvastatin in SD rats and EHBRs was calculated using eqs. 8 to 10, and these parameters are summarized in Table 4. The CLtot, p, CLbile, p, and CLbile, H values for parent rosuvastatin in SD rats were significantly lower than those for the control SD rats. The Kp, H value for parent rosuvastatin in EHBRs was also significantly lower than that in SD rats.
Pharmacokinetic parameters of rosuvastatin during constant i.v. infusion into SD rats and EHBRs
Each point represents the mean ± S.E. (n = 3).
Kinetic Parameters of Parent Rosuvastatin in FVB Mice andBcrp1Knockout Mice. The plasma concentration and biliary excretion rate, estimated using the total tritium radioactivity derived from the administered rosuvastatin, almost reached a plateau at 90 min during constant infusion after i.v. bolus administration (Fig. 7). The percentages of parent rosuvastatin in the plasma, bile, and liver at 120 min were estimated by TLC, as described under Materials and Methods. The percentages of unchanged rosuvastatin relative to the total radioactivity in the plasma, bile, and liver were 42, 69, and 25%, respectively, for the FVB mice and 42, 11, and 9%, respectively, for the Bcrp1 knockout mice. In plasma, the percentage of parent rosuvastatin in the Bcrp1 knockout mice was approximately the same as that in FVB mice, whereas in bile and liver that in the Bcrp1 knockout mice was significantly lower than that in the FVB mice. The pharmacokinetic parameters for the parent rosuvastatin in FVB mice and Bcrp1 knockout mice were calculated using the equations described under Materials and Methods, and these parameters are summarized in Table 5. The Vbile, ss, CLbile, p, and CLbile, H values for the parent rosuvastatin in the Bcrp1 knockout mice were much lower than those in the control mice. The CLtot, p and Kp, H values for parent rosuvastatin in the Bcrp1 knockout mice seemed to be lower than those in the FVB mice, although the difference was not statistically significant.
Pharmacokinetic parameters of rosuvastatin during constant intravenous infusion into FVB mice and Bcrp1 knockout mice
Each point represents the mean ± S.E. (n = 3).

Time profiles for the transcellular transport of rosuvastatin across MDCKII monolayers expressing human transporters. Transcellular transport of rosuvastatin (0.1 μM) across MDCKII monolayers expressing OATP1B1 (B), MRP2 (C), MDR1 (D), BCRP (E), both OATP1B1 and MRP2 (F), both OATP1B1 and MDR1 (G), or both OATP1B1 and BCRP (H) was compared with that across a vector-transfected control MDCKII monolayer (A). • and ○ represent the transcellular transport (PSnet) in the basal-to-apical and apical-to-basal directions, respectively. Each point represents the mean ± S.E. (n = 3). Where vertical bars are not shown, the S.E. values are within the limits of the symbols. **, P < 0.01; ***, P < 0.001.
Discussion
Rosuvastatin is marginally metabolized in the human liver and is thought to be excreted into the bile mainly in an intact form. In this study, we focused on the involvement of the OATP family transporters in the sinusoidal uptake and MRP2, MDR1, and BCRP in the biliary excretion of rosuvastatin.
Significant uptake of rosuvastatin in OATP1B1-, OATP1B3-, and OATP2B1-expressing cells was observed (Fig. 1). The uptake clearances for the reference compounds measured simultaneously were similar to the reported values (Hirano et al., 2004) (Table 1). Concentration-dependent rosuvastatin uptake via OATP1B1, OATP1B3, and OATP2B1 was described by assuming one saturable and one nonsaturable component (Fig. 2; Table 2). This kind of saturation pattern was not due to the cytotoxicity induced by high concentrations of rosuvastatin, because the protein amount was not decreased after exposure to 100 μM rosuvastatin (data not shown). Similar saturation patterns have also been observed in the uptake of other compounds (CCK-8 and valsartan) via the OATP family transporters (Hirano et al., 2004; Yamashiro et al., 2006). We think this phenomenon does not have any direct clinical impact on the transport properties of rosuvastatin because its plasma concentration in the clinical situation is much lower than the Km value for each transporter.
Although one compound can be recognized by several transporters, we have reported that the major transporter responsible for hepatic uptake varies among substrates (Maeda and Sugiyama, 2007). We estimated the relative contributions of the OATP family transporters to the hepatic uptake of rosuvastatin using a previously reported methodology (Hirano et al., 2004). The contribution of OATP2B1 was deemed to be negligible because the ratio of the protein expression in human hepatocytes to that in our OATP2B1-expressing cells was 6 to 15 times lower than that of cells expressing OATP1B1 or OATP1B3 (Hirano et al., 2006). We measured the uptake clearance of transporter-selective reference compounds (E1S and CCK-8) and of rosuvastatin mediated by OATP1B1 and OATP1B3. Using these values and the reported uptake clearances for E1S and CCK-8 in three different batches of human hepatocytes (Hirano et al., 2004), we estimated the relative contributions of OATP1B1 and OATP1B3 to the hepatic uptake of rosuvastatin in human hepatocytes (Table 1). This result suggests that OATP1B1 is mainly involved in the uptake of rosuvastatin, with 16 to 34% attributable to OATP1B3.
By measuring the Na+-dependent uptake of NTCP, a recent study estimated that its contribution to the uptake of rosuvastatin in human hepatocytes is approximately 35% (Ho et al., 2006). Although we did not consider the contribution of NTCP, assuming that Na+-independent uptake is dominated by OATP family transporters, the relative contribution of OATP1B1 is deemed to be 43 to 55% of its total uptake. It can be said that OATP1B1, as well as NTCP, is important in the uptake of rosuvastatin.
Among other statins, OATP1B1 is the predominant transporter in the hepatic uptake of pravastatin and pitavastatin (Nakai et al., 2001; Hirano et al., 2004). The recent clinical studies demonstrated that a single nucleotide polymorphism (SNP) (T521C) in OATP1B1 caused a significant increase in the area under the plasma concentration-time curves of pravastatin and pitavastatin (Nishizato et al., 2003; Chung et al., 2005) but a relatively small increase in that of rosuvastatin (Lee et al., 2005). This difference might be partly explained by the large contribution of other transporters, such as NTCP and OATP1B3, to its hepatic uptake compared with other statins.

Concentration dependence of the transcellular transport clearance (PSnet) (A and B) and apical transmembrane clearance (PSapical) (C and D) of rosuvastatin across MDCKII monolayers expressing OATP1B1/MRP2 (A and C) or OATP1B1/MDR1 (B and D). The concentration dependence of PSnet from the basal to the apical side (A and B) and of PSapical (C and D) across MDCKII monolayers expressing both OATP1B1 and MRP2 (▪) or both OATP1B1 and MDR1 (▴) were compared with those across an OATP1B1-expressing monolayer (□) or a vector-transfected control MDCKII monolayer (○). PSnet from the basal to the apical side and PSapical for 120 min were determined at various concentrations of rosuvastatin (0.1–300 μM). The data were fitted to eq. 6 (described under Materials and Methods) by the nonlinear least-squares method, and each solid line represents the fitted curve. Each point represents the mean ± S.E. (n = 3). Where vertical and/or horizontal bars are not shown, the S.E. values are within the limits of the symbols.
Next, we examined the candidate transporters responsible for the biliary excretion of rosuvastatin using double transfectants. A significant basal-to-apical flux was observed in all the double transfectants (Fig. 3), suggesting that MRP2, MDR1, and BCRP recognize rosuvastatin as a substrate. Huang et al. (2006) reported previously that no vectorial transcellular transport of rosuvastatin was observed in MDR1-expressing MDCK cells. This may be explained by the inefficient penetration of rosuvastatin across the basal membrane in single transfectants, whereas OATP1B1 accelerates its accumulation into double transfectants. Huang et al. (2006) also observed no significant transport in MRP2-expressing membrane vesicles. We have no good explanation for the different outcomes in these different experimental systems with the different exposure of rosuvastatin to the transporter. However, from our experiments, MRP2 could also be involved in the biliary excretion of rosuvastatin in humans.
In both OATP1B1/MRP2 and OATP1B1/MDR1 double transfectants, the PSnet and PSapical were observed (Fig. 4; Table 3). The Km values for rosuvastatin uptake for OATP1B1 (0.80 μM) and OATP1B3 (14.2 μM) were different from those of PSnet, suggesting that the transcellular transport clearance of rosuvastatin might not be determined solely by the uptake clearance.
Although the transcellular transport in the OATP1B1/BCRP double transfectant was too small to perform kinetic analyses, its transport was clearly observed in BCRP-expressing membrane vesicles, which is consistent with a previous report (Huang et al., 2006). The low transcellular transport and efflux clearance of rosuvastatin across the apical membrane in OATP1B1/BCRP double transfectants might partly result from the difference in the protein expression levels of BCRP in the BCRP-expressing vesicles and the double transfectant. The BCRP-mediated transport of rosuvastatin was well explained by two saturable components. This may be due to the existence of two binding sites in BCRP, at least for the recognition of rosuvastatin, although the exact reason is still unknown.
To investigate further whether an efflux transporter is involved in the biliary excretion of rosuvastatin in vivo, we observed the biliary excretion of rosuvastatin in Mrp2-deficient EHBRs and Bcrp1 knockout mice. CLbile, H decreased to 53% in EHBRs compared with that of the control SD rats (Table 4) and to 12% in Bcrp1 knockout mice compared with that of the control FVB mice (Table 5). These results suggest that Mrp2 and Bcrp1 play important roles in the biliary excretion of rosuvastatin, with relative contributions of 50 and 90% in rats and mice, respectively. This result implies a species difference in the relative contributions of MRP2 and BCRP to the biliary excretion of rosuvastatin. Similarly, the biliary excretions of 2-amino-1-methyl-6-phenylimidazo[4,5-β]pyridine decreased to 23 and 17% of the control value in TR– rats (Mrp2-deficient) and Bcrp1 knockout mice, respectively (Dietrich et al., 2001; van Herwaarden et al., 2003). Previous reports have suggested that Mrp2 expression is relatively high in rats compared with that in other animals (Ishizuka et al., 1999). This species difference in the contribution of efflux transporters to biliary excretion must be clarified to interpret the results of animal experiments appropriately.

Time profiles and Eadie-Hofstee plots of the transport of rosuvastatin by human BCRP- or GFP-expressing membrane vesicles. A, time profile of the uptake of rosuvastatin (0.1 μM) was examined at 37°C in medium containing 5 mM ATP (• and ▴) or AMP (○ and ▵). Circles and triangles represent the uptake in BCRP- and GFP-expressing membrane vesicles, respectively. Each point represents the mean ± S.E. (n = 3). B, the concentration dependence of the ATP-dependent transport of rosuvastatin mediated by BCRP is shown in the Eadie-Hofstee plots. The transport clearance for 1 min was determined at various concentrations (0.1–100 μM). The ATP-dependent transport mediated by BCRP was calculated by subtracting the ATP-dependent transport in GFP-expressing control vesicles from that in BCRP-expressing vesicles. The data were fitted to eq. 7 (described under Materials and Methods) by the nonlinear least-squares method, and each solid line represents the fitted curve. Each point represents the mean ± S.E. (n = 3). Where vertical and/or horizontal bars are not shown, the S.E. values are within the limits of the symbols.

Time profiles for plasma concentrations and biliary excretion rates for rosuvastatin in control SD rats and Mrp2-deficient EHBRs. The plasma concentrations (A) and biliary excretion rates (B) of the total radioactivity during the constant intravenous infusion of rosuvastatin into SD rats (•) and EHBRs (○) were determined. Each point represents the mean ± S.E. (n = 3). Where vertical bars are not shown, the S.E. values are within the limits of the symbols.
If the biliary excretion of rosuvastatin is decreased by a functional deficiency in efflux transporters, the Kp,H value should increase, which is inconsistent with our results (Tables 4 and 5). The decrease in the Kp,H value in transporter-deficient animals suggests that the hepatic uptake of rosuvastatin might have decreased and/or the sinusoidal efflux increased. Indeed, it has been reported that the up-regulation of Mrp3 and the down-regulation of Oatp1a1 and Oatp1a4 expression were observed in EHBRs (Akita et al., 2001; Kuroda et al., 2004). This kind of compensation might change the hepatic clearance of rosuvastatin and its distribution.
Currently, we have no methodology for predicting the quantitative contributions of efflux transporters to the biliary excretion of compounds in human hepatocytes. However, in comparing the PSapical value for rosuvastatin for each transporter with the reported values for other compounds, we can discuss the differences in the relative importance of three efflux transporters. The PSapical of pravastatin in the OATP1B1/MRP2 double transfectant was much larger than that in the OATP1B1/MDR1 or OATP1B1/BCRP transfectants (Matsushima et al., 2005), which is consistent with a previous result showing that biliary excretion was dramatically reduced in EHBRs (Yamazaki et al., 1996). Conversely, the relative contribution of MDR1 and BCRP to the biliary excretion of rosuvastatin in humans should be larger than that of pravastatin. Recent reports have suggested that a SNP in BCRP (C421A) affects the pharmacokinetics of rosuvastatin (Zhang et al., 2006). Future studies are required to predict the quantitative contribution of efflux transporters to the overall biliary excretion of compounds in humans.
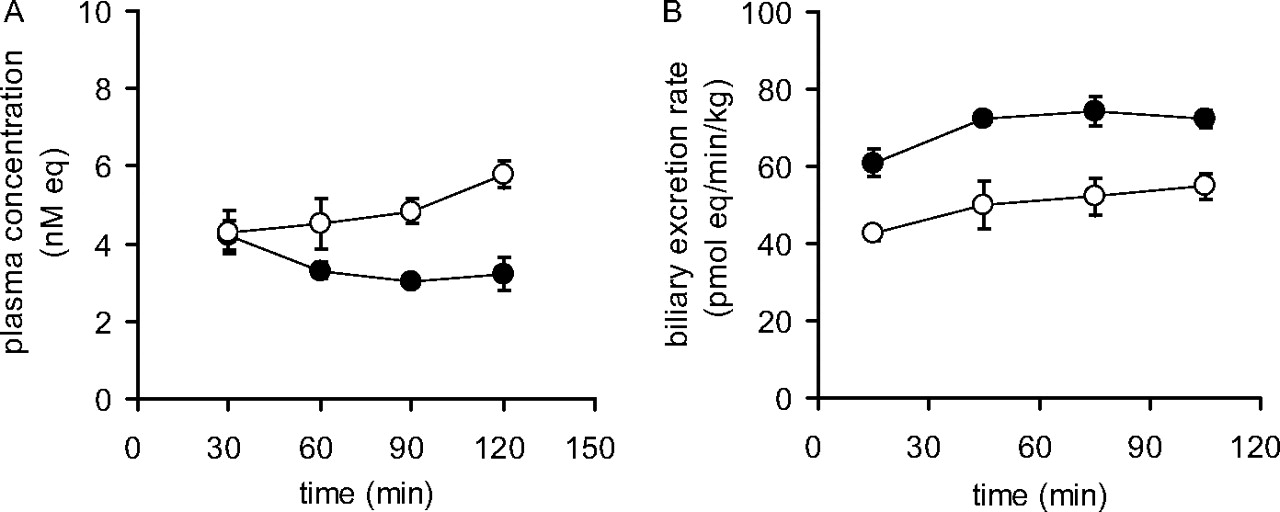
Time profiles for plasma concentrations and biliary excretion rates of rosuvastatin in control FVB mice and Bcrp1 knockout mice. The plasma concentrations (A) and biliary excretion rates (B) of the total radioactivity during constant i.v. infusion of rosuvastatin into FVB mice (•) or Bcrp1 knockout mice (○) were determined. Each point represents the mean ± S.E. (n = 3). Where vertical bars are not shown, the S.E. values are within the limits of the symbols.
In this study, we have demonstrated that rosuvastatin is a substrate of OATP1B1, OATP1B3, and OATP2B1 in sinusoidal uptake and of MRP2, MDR1, and BCRP in biliary excretion. We suggest that OATP1B1 as well as NTCP plays an important role in rosuvastatin uptake into human hepatocytes and that OATP1B3 is also partly involved in its uptake. This kind of information will make it possible to predict changes in the pharmacokinetics of rosuvastatin under special circumstances, such as SNPs, disease, and/or drug-drug interactions.
Acknowledgments
We thank Dr. Alfred H. Schinkel (The Netherlands Cancer Institute, Amsterdam, The Netherlands) for supplying the Bcrp1 knockout mice and Dr. Piet Borst (The Netherlands Cancer Institute) for providing the MDCKII cells expressing MRP2 and MDR1.
Footnotes
-
This study was supported by Labor Sciences Research Grants from the Ministry of Health, Labor, and Welfare for research on toxicogenomics and a Grant-in-Aid for Young Scientists (B) (19790119) from the Ministry of Education, Culture, Sports, Science and Technology.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.021410.
-
ABBREVIATIONS: OATP, organic anion transporting polypeptide; NTCP, sodium-taurocholate cotransporting polypeptide; MRP, multidrug resistance-associated protein; MDR, multidrug resistance protein; BCRP/Bcrp, breast cancer resistance protein; SD, Sprague-Dawley; EHBR, Eisai hyperbilirubinemic rat; E1S, estrone-3-sulfate; E217βG, 17β-estradiol-17β-d-glucuronide; CCK-8, cholecystokinin octapeptide; HEK, human embryonic kidney; MDCK, Madin-Darby canine kidney; GFP, green fluorescent protein; TLC, thin-layer chromatography; SNP, single nucleotide polymorphism.
- Received March 11, 2008.
- Accepted July 7, 2008.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}