


Abstract
It has already been demonstrated that pitavastatin, a novel potent HMG-coenzyme A reductase inhibitor, is taken up into human hepatocytes mainly by organic anion transporting polypeptide (OATP) 1B1. Because OATP2B1 is also localized in the basolateral membrane of human liver, we took two approaches to further confirm the minor contribution of OATP2B1 to the hepatic uptake of pitavastatin. Western blot analysis revealed that the ratio of the band density of OATP2B1 in human hepatocytes to that in our expression system is at least 6-fold lower compared with OATP1B1 and OATP1B3. The uptake of pitavastatin in human hepatocytes could be inhibited by both estrone-3-sulfate (OATP1B1/OATP2B1 inhibitor) and estradiol-17β-d-glucuronide (OATP1B1/OATP1B3 inhibitor). These results further supported the idea that OATP1B1 is a predominant transporter for the hepatic uptake of pitavastatin. Then, to explore the possibility of OATP1B1-mediated drug-drug interaction, we checked the inhibitory effects of various drugs on the pitavastatin uptake in OATP1B1-expressing cells and evaluated whether the in vitro inhibition was clinically significant or not. As we previously reported, we used the methodology for estimating the maximum unbound concentration of inhibitors at the inlet to the liver (Iu,in,max). Judging from Iu,in,max and inhibition constant (Ki) for OATP1B1, several drugs (especially cyclosporin A, rifampicin, rifamycin SV, clarithromycin, and indinavir) have potentials for interacting with OATP1B1-mediated uptake of pitavastatin. The in vitro experiments could support the clinically observed drug-drug interaction between pitavastatin and cyclosporin A. These results suggest that we should pay attention to the concomitant use of some drugs with pitavastatin.
The liver is one of the organs responsible for the elimination of xenobiotics including many kinds of drugs. In some cases, although the compounds were not supposed to easily penetrate the plasma membrane from the viewpoint of their physicochemical properties, they were efficiently taken up into liver and excreted into bile. Recently, it has been found that several kinds of transporters greatly help the efficient membrane transport of several compounds. It has been characterized that hepatic uptake of some of the compounds is mediated by organic anion-transporting polypeptide (OATP) family transporters, organic anion transporter 2, Na+-taurocholate cotransporting polypeptide, and organic cation transporter 1 (Mizuno et al., 2003). Among these transporters, especially OATP1B1 and OATP1B3 are specifically expressed in liver and show broad substrate specificities (Hagenbuch and Meier, 2003). In contrast, OATP2B1 is also expressed in the basolateral membrane of human liver (Tamai et al., 2001). Previous reports indicated that the low pH facilitates the OATP2B1-mediated uptake of several organic anions, implying its involvement in the intestinal absorption of anions (Kobayashi et al., 2003). Although the uptake clearance at pH 7.4 was lower than that at pH 5.0, OATP2B1 could transport some organic anions such as estrone-3-sulfate, fexofenadine, benzylpenicillin, and dehydroepiandros-terone sulfate, even at pH 7.4 (Kobayashi et al., 2003; Nozawa et al., 2004). Therefore, it is possible that OATP2B1 is also involved in the hepatic uptake of anionic drugs.
Pitavastatin is a highly potent inhibitor of HMG-coenzyme A reductase, the rate-limiting enzyme in cholesterol biosynthesis (Aoki et al., 1997; Kajinami et al., 2003). Previously, Kimata et al. (1998) have revealed that [14C]pitavastatin is selectively distributed to the liver in rats with the liver-to-plasma concentration ratio of more than 50, suggesting that active transport systems can be involved in the uptake of pitavastatin. We have already demonstrated that pitavastatin is taken up into human hepatocytes predominantly by OATP1B1, although it was a substrate of both OATP1B1 and OATP1B3 (Hirano et al., 2004). We also showed that the contribution of other transporters such as OATP2B1 to the pitavastatin uptake was theoretically small, but we have not experimentally shown the minor importance of OATP2B1. Therefore, we tried to confirm that OATP1B1 is a responsible transporter for the pitavastatin uptake by two kinds of approaches: the comparison of the expression level of each transporter in human hepatocytes and expression systems by Western blot analysis, and the inhibitory effects of transporter-selective inhibitors on the uptake of pitavastatin in human hepatocytes.
The combination therapy of statins and various compounds is widely used in clinical practice. Coadministration of various drugs sometimes causes an increase in the plasma concentration of statins (Williams and Feely, 2002), which may occasionally lead to severe side effects such as myopathy and rhabdomyolysis (Evans and Rees, 2002). In the case of simvastatin, which is relatively lipophilic and metabolized by CYP3A4, itraconazole, cyclosporin A, and erythromycin were reported to increase plasma area under the plasma concentration-time curve of simvastatin by inhibition of CYP3A4-mediated metabolism (Kantola et al., 1998; Neuvonen et al., 1998; Ichimaru et al., 2001). In contrast, cyclosporin A also interacted with the nonmetabolized type of statins such as pravastatin, pitavastatin, and rosuvastatin in the clinical situation (Olbricht et al., 1997; Hasunuma et al., 2003; Simonson et al., 2004). Shitara et al. (2003) clarified that drug-drug interaction (DDI) between cyclosporin A and cerivastatin is caused by the inhibition of OATP1B1-mediated cerivastatin uptake by cyclosporin A. Because pitavastatin was reported to be taken up into hepatocytes mainly by OATP1B1 (Hirano et al., 2004), we should pay attention to the OATP1B1-mediated DDI of pitavastatin in coadministration with other drugs that can inhibit the function of OATP1B1. However, the inhibitors of OATP1B1 identified by in vitro analyses do not always cause DDI in the clinical situation when the clinical protein unbound concentration in plasma is much lower than the in vitro inhibition constant (Ki) for OATP1B1. Ito et al. (1998) proposed the calculation method for estimating the maximum unbound concentration of inhibitors at the inlet to the liver to avoid the false-negative prediction of clinical DDI.
In the present study, we confirmed the minor contribution of OATP2B1 to the hepatic uptake of pitavastatin by two approaches. Moreover, we tried to predict the possible DDI mediated by OATP1B1 between pitavastatin and various drugs by judging from the clinical maximum unbound concentration of each inhibitor in human plasma and the Ki value for OATP1B1 obtained from the in vitro study.
Materials and Methods
Materials. Pitavastatin, monocalcium bis[(3R,5S,6E)-7-[2-cyclopropyl-4-(4-fluorophenyl)-3-quinolyl]3,5-dihydroxy-6-hepteonate], was synthesized by Nissan Chemical Industries (Chiba, Japan). [3H]Pitavastatin (16.0 Ci/mmol) was synthesized by GE Healthcare (Little Chalfont, Buckinghamshire, UK). [3H]Estradiol 17β-d-glucuronide (E217βG) and [3H]estrone-3-sulfate (E1S) (45 Ci/mmol and 46 Ci/mmol, respectively) were purchased from New England Nuclear (Boston, MA). Unlabeled E217βG, E1S, and gemfibrozil were purchased from Sigma-Aldrich (St. Louis, MO). A metabolite of gemfibrozil, M3 (purity 99.6%), was chemically synthesized at KNC Laboratories, Co. Ltd. (Kobe, Japan) as shown in detail previously (Shitara et al., 2004). All other chemicals were of analytical grade and commercially available.
Uptake Study Using Transporter Expression Systems. OATP1B1-, OATP1B3-, and OATP2B1-expressing HEK293 cells and vector-transfected control cells used in this study were constructed previously (Hirano et al., 2004; Shimizu et al., 2005). Transporter-expressing or vector-transfected HEK293 cells were grown in Dulbecco's modified Eagle's medium low glucose (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% CO2 and 95% humidity. Cells were then seeded in 12-well plates coated with poly-l-lysine/poly-l-ornithine at a density of 1.5 × 105 cells per well. After 2 days, the cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before transport assay to induce the expression of exogenous transporters. The transport study was carried out as described previously (Sugiyama et al., 2001). Uptake was initiated by adding Krebs-Henseleit buffer containing radiolabeled and unlabeled substrates after cells had been washed twice and preincubated with Krebs-Henseleit buffer at 37°C for 15 min. The Krebs-Henseleit buffer consisted of 118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2 adjusted to pH 7.4. The uptake was terminated at a designated time by adding ice-cold Krebs-Henseleit buffer after removal of the incubation buffer. Then, cells were washed twice with 1 ml of ice-cold Krebs-Henseleit buffer, solubilized in 500 μl of 0.2 N NaOH, and kept overnight at 4°C. Aliquots (500 μl) were transferred to scintillation vials after adding 250 μl of 0.4 N HCl. The radioactivity associated with the cells and incubation buffer was measured in a liquid scintillation counter (LS6000SE; Beckman Coulter, Inc., Fullerton, CA) after adding 2 ml of scintillation fluid (Clear-sol I; Nacalai Tesque, Kyoto, Japan) to the scintillation vials. The remaining 50 μl of cell lysate was used to determine the protein concentration by the method of Lowry et al. (1951) with bovine serum albumin as a standard.
Uptake Study Using Human Cryopreserved Hepatocytes. This experiment was performed as described previously (Shitara et al., 2003). Cryopreserved human hepatocytes were purchased from In Vitro Technologies (Baltimore, MD). In this experiment, we selected three batches of human hepatocytes (lots OCF, 094, and ETR), which ranked in the top three of the uptake amount of E217βG, and E1S among eight independent batches of hepatocytes. Immediately before the study, the hepatocytes (1-ml suspension) were thawed at 37°C, quickly suspended in 10 ml of ice-cold Krebs-Henseleit buffer, and centrifuged (50g) for 2 min at 4°C, followed by removal of the supernatant. This procedure was repeated once more to remove cryopreservation buffer, and then the cells were resuspended in the same buffer to give a cell density of 1.0 × 106 viable cells/ml for the uptake study. The number of viable cells was determined by trypan blue staining. Before the uptake studies, the cell suspensions were prewarmed in an incubator at 37°C for 3 min. The uptake studies were initiated by adding an equal volume of buffer containing radiolabeled and unlabeled pitavastatin to the cell suspension. After incubation at 37°C for 0.5 and 2 min, the reaction was terminated by separating the cells from the buffer. For this purpose, an aliquot of 80 μl of incubation mixture was collected and placed in a centrifuge tube (450 μl) containing 50 μl of 2 N NaOH under a layer of 100 μl of oil (density, 1.015; a mixture of silicone oil and mineral oil; Sigma-Aldrich), and subsequently the sample tube was centrifuged for 10 s using a tabletop centrifuge (10,000g; Beckman Microfuge E; Beckman Coulter, Inc.). During this process, hepatocytes passed through the oil layer into the alkaline solution. After an overnight incubation in alkali to dissolve the hepatocytes, the centrifuge tube was cut and each compartment was transferred to a scintillation vial. The compartment containing the dissolved cells was neutralized with 50 μl of 2 N HCl, mixed with scintillation cocktail, and the radioactivity was measured in a liquid scintillation counter.
Antiserum and Western Blot Analysis. As shown in previous reports, anti-OATP2B1 sera were raised in rabbits against a synthetic peptide consisting of the 15 carboxyl-terminal amino acids of OATP2B1 (LLVSGPGKKPEDSRV) coupled to keyhole limpet hemocyanine at its N-terminal via an additional cysteine (Kullak-Ublick et al., 2001). Crude membrane fractions were prepared from human hepatocytes and transporter-expressing HEK293 cells as described previously (Sasaki et al., 2002). The human liver block (lot 020188) was obtained from Human and Animal Bridging Research Organization (Chiba, Japan), and crude membrane fractions were prepared as described previously (Hirano et al., 2004). The samples were diluted with 3× Red loading buffer (BioLabs, Hertfordshire, UK) and loaded onto a 7% SDSpolyacrylamide gel with a 4.4% stacking gel. Proteins were electroblotted onto a polyvinylidene difluoride membrane (Pall, East Hills, NY) using a blotter (Trans-blot; Bio-Rad, Richmond, CA) at 15 V for 1 h. The membrane was blocked with Tris-buffered saline containing 0.05% Tween 20 (TBS-T) and 5% skimmed milk for 1 h at room temperature. After washing with TBS-T, the membrane was incubated with anti-OATP2B1 serum (dilution 1:1000). The membrane was incubated with a horseradish peroxidase-labeled anti-rabbit IgG antibody (GE Healthcare) diluted 1:5000 in TBS-T for 1 h at room temperature, followed by washing with TBS-T. The band was detected and its intensity was quantified using an image analyzer (LAS-1000 plus; Fuji Film, Tokyo, Japan).
Transcellular Transport Study. OATP1B1-, OATP1B1/BCRP-, OATP1B1/MDR1-, and OATP1B1/MRP2-expressing MDCKII cells and vector-transfected control cells used in this study were constructed previously (Matsushima et al., 2005). Transporter-expressing or vector-transfected MDCKII cells were grown in Dulbecco's modified Eagle's medium low glucose supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B at 37°C with 5% CO2 and 95% humidity. MDCKII cells were seeded on Transwell membrane inserts (6.5-mm diameter, 0.4-μm pore size; Corning Costar, Bodenheim, Germany) at a density of 1.4 × 105 cells per well. After 3 days, medium was replaced with 5 mM sodium butyrate for 24 h before the transport study (Sasaki et al., 2002). The experiments were initiated by replacing the medium on the basal side of the cell layer with Krebs-Henseleiti buffer containing radiolabeled and unlabeled pitavastatin (0.3 μM). The cells were incubated at 37°C, and aliquots of medium were taken from each compartment at designated time points. Radioactivity in 100 μl of medium was measured in a liquid scintillation counter after the addition of scintillation cocktail. At the end of the experiments, the cells were washed three times with 1.5 ml of ice-cold Krebs-Henseleit buffer and solubilized in 500 μl of 0.2 N NaOH. After addition of 100 μl of 1 N HCl, 500-μl aliquots were transferred to scintillation vials. Aliquots (50-μl) of cell lysate were used to determine protein concentrations as described above. To evaluate the efflux transport clearance via recombinant BCRP, MDR1, and MRP2 in the double transfectants, the apparent efflux clearance across the apical membrane (PSapical) was calculated by dividing the steady-state velocity for the transcellular transport (Vtranscellular) of pitavastatin, determined over 3 h, by the intracellular concentration (Ccell) of pitavastatin, determined at the end of the experiments (3 h) in the absence or presence of the inhibitors. 
Kinetic Analyses. Ligand uptake in transporter cDNA-transfected cells was expressed as the uptake volume (μl/mg protein), given as the amount of radioactivity associated with the cells (dpm/mg protein) divided by its concentration in the incubation medium (dpm/μl). Transporter-specific uptake was obtained by subtracting the uptake into vector-transfected cells from the uptake into cDNA-transfected cells. Kinetic parameters were obtained using the following equation:  where v is the uptake velocity of the substrate (pmol/min/mg protein), S is the substrate concentration in the medium (μM), Km is the Michaelis constant (μM), Vmax is the maximum uptake rate (pmol/min/mg protein), and Pdif is the nonsaturable uptake clearance (μl/min/mg protein). The Damping Gauss-Newton Method algorithm was used with a MULTI program (Yamaoka et al., 1981) to perform nonlinear least-squares data fitting. The input data were weighted as the reciprocal of the observed values. Inhibition constants (Ki) of a series of compounds could be calculated by the following equation, if the substrate concentration was low enough compared with its Km value.
where v is the uptake velocity of the substrate (pmol/min/mg protein), S is the substrate concentration in the medium (μM), Km is the Michaelis constant (μM), Vmax is the maximum uptake rate (pmol/min/mg protein), and Pdif is the nonsaturable uptake clearance (μl/min/mg protein). The Damping Gauss-Newton Method algorithm was used with a MULTI program (Yamaoka et al., 1981) to perform nonlinear least-squares data fitting. The input data were weighted as the reciprocal of the observed values. Inhibition constants (Ki) of a series of compounds could be calculated by the following equation, if the substrate concentration was low enough compared with its Km value. 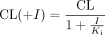 where CL represents the uptake clearance in the absence of inhibitor, CL(+I) represents the uptake clearance in the presence of inhibitor, and I represents the inhibitor concentration. When fitting the data to determine the Ki value, the input data were weighed as the reciprocal of the observed values.
where CL represents the uptake clearance in the absence of inhibitor, CL(+I) represents the uptake clearance in the presence of inhibitor, and I represents the inhibitor concentration. When fitting the data to determine the Ki value, the input data were weighed as the reciprocal of the observed values.
To determine saturable hepatic uptake clearance in human hepatocytes, we first determined the hepatic uptake clearance (CL(2 min-0.5 min)) (μl/min/106 cells) by calculating the slope of the uptake volume (Vd)(μl/106 cells) between 0.5 and 2 min as shown previously (Hirano et al., 2004) (eq. 4). The saturable component of the hepatic uptake clearance (CLhep) was determined by subtracting CL(2 min-0.5 min) in the presence of 100 μM substrate (excess) from that in the presence of 0.1 μM substrate (tracer) (eq. 5). 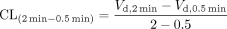
 where CL(2 min-0.5 min),tracer and CL(2 min-0.5 min),excess represent CL(2 min-0.5 min) estimated in the presence of 0.1 and 100 μM substrate, respectively.
where CL(2 min-0.5 min),tracer and CL(2 min-0.5 min),excess represent CL(2 min-0.5 min) estimated in the presence of 0.1 and 100 μM substrate, respectively.
Estimation of the Contribution of Transporters to the Hepatic Uptake in Human Hepatocytes by Western Blot Analysis. The ratio of the expression level of each transporter in human hepatocytes (per 106 cells) to that in the expression system (per mg protein) was calculated by the intensity of specific bands in Western blot analysis and defined as Rexp as shown previously (Hirano et al., 2004). The uptake clearance by each transporter in human hepatocytes was separately calculated by multiplying the uptake clearance of the pitavastatin in transporter-expressing cells (CLtest) by Rexp as described in the following equation:  The relative contribution (percentage) of each transporter to the uptake in human hepatocytes was defined by the ratio of CLhep,test for target transporter to that of the sum of CLhep,test for OATP1B1, OATP1B3, and OATP2B1.
The relative contribution (percentage) of each transporter to the uptake in human hepatocytes was defined by the ratio of CLhep,test for target transporter to that of the sum of CLhep,test for OATP1B1, OATP1B3, and OATP2B1.
Prediction of Clinical DDI between Pitavastatin and Various Drugs via OATP1B1. The degree of inhibition of OATP1B1 in humans was estimated by calculating the following R values, which represent the ratio of the uptake clearance in the absence of inhibitor to that in its presence: 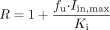 where fu represents the blood unbound fraction of the inhibitor, Iin,max represents the estimated maximum inhibitor concentration at the inlet to the liver, and Ki was obtained in the present in vitro study using OATP1B1-expressing HEK293 cells. For the estimation of R value, Iin,max was calculated by the method of Ito et al. (1998):
where fu represents the blood unbound fraction of the inhibitor, Iin,max represents the estimated maximum inhibitor concentration at the inlet to the liver, and Ki was obtained in the present in vitro study using OATP1B1-expressing HEK293 cells. For the estimation of R value, Iin,max was calculated by the method of Ito et al. (1998):  where Imax represents the reported value for the maximum plasma concentration in the systemic circulation in the clinical situation, Fa represents the absorbed fraction of inhibitor, ka is the absorption rate constant in the intestine, and Qh represents the hepatic blood flow rate in humans (1610 ml/min). To estimate the maximum Iin,max value, Fa was set at 1, ka was set at 0.1 min–1 [minimum gastric emptying time (10 min)], and the blood-to-plasma concentration ratio was assumed to be 1, if the information from the literature was not available.
where Imax represents the reported value for the maximum plasma concentration in the systemic circulation in the clinical situation, Fa represents the absorbed fraction of inhibitor, ka is the absorption rate constant in the intestine, and Qh represents the hepatic blood flow rate in humans (1610 ml/min). To estimate the maximum Iin,max value, Fa was set at 1, ka was set at 0.1 min–1 [minimum gastric emptying time (10 min)], and the blood-to-plasma concentration ratio was assumed to be 1, if the information from the literature was not available.
Results
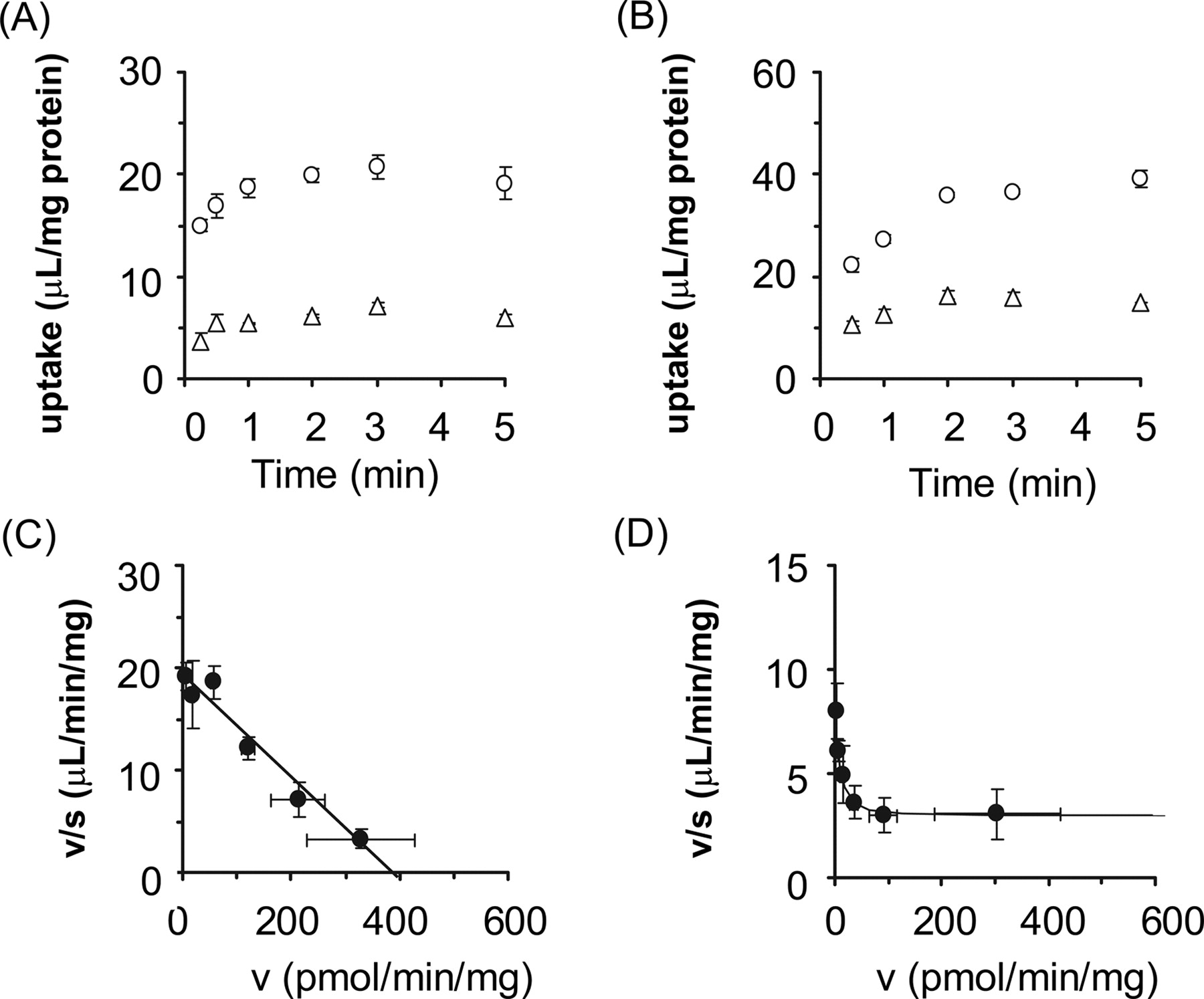
Uptake of E1S and Pitavastatin by OATP2B1-Expressing Cells. The time profiles and Eadie-Hofstee plots of the uptake of E1S and pitavastatin by OATP2B1-expressing and vector-transfected HEK293 cells are shown in Fig. 1. Pitavastatin as well as E1S was significantly taken up into OATP2B1-expressing HEK293 cells compared with vector-transfected cells (Fig. 1, A and B). The saturation kinetics of their uptake is shown in Fig. 1, C and D. The concentration dependence of the uptake of E1S could be explained by a one-saturable component (Fig. 1C). The Km and Vmax values for the OATP2B1-mediated uptake of E1S were 20.9 ± 2.0 μM and 1196 ± 40 pmol/min/mg protein, respectively. The nonsaturable component was observed in the Eadie-Hofstee plot even for the specific uptake of pitavastatin by OATP2B1 (Fig. 1D). The Km and Vmax values of pitavastatin for the saturable component and uptake clearance for the nonsaturable component were 1.17 ± 0.28 μM, 7.36 ± 1.43 pmol/min/mg protein, and 2.93 ± 0.16 μl/min/mg protein, respectively. No significant uptake of E217βG by OATP2B1 could be observed (7.51 ± 0.49 and 9.72 ± 1.29 μl/mg protein by vector-transfected and OATP2B1-expressing cells for 5 min, respectively; n = 3).
Western Blot Analysis of OATP2B1. The relative expression level of OATP2B1 in crude membrane from transfectants and human hepatocytes was estimated by Western blot analyses. An antiserum against OATP2B1 recognized approximately 85-kDa proteins in the crude membrane fractions prepared from human hepatocytes and OATP2B1-expressing cells (Fig. 2A). The molecular weight of OATP2B1 in human hepatocytes was almost the same as that prepared from human liver block, but was slightly lower than that in OATP2B1-expressing HEK293 cells. No specific band of OATP2B1 was detected in vector-transfected cells. Figure 2B showed the linear relationship between the applied protein amount of crude membrane obtained from OATP2B1-expressing cells and human hepatocytes and the intensity of the specific band measured by digital densitometer. The slope of the regression line in Fig. 2B reflected the relative expression level of OATP2B1 in transfectants and hepatocytes.

Time profiles and Eadie-Hofstee plots of the uptake of [3H]E1S and [3H]pitavastatin by OATP2B1-expressing HEK293 cells. The uptake of 0.1 μM [3H]E1S (A) and 0.1 μM [3H]pitavastatin (B) by cDNA-transfected cells was examined at 37°C. Open circles and triangles represent the uptake in OATP2B1-expressing HEK293 cells and vector-transfected control cells, respectively. The concentration dependence of OATP2B1-mediated uptake of [3H]E1S (C) and [3H]pitavastatin (D) is shown as Eadie-Hofstee plots. Closed circles represent the OATP2B1-mediated specific uptake rate, which was obtained by subtracting the initial uptake rate in vector-transfected cells from that in OATP2B1-expressing cells. The initial uptake rate calculated from the uptake of [3H]E1S and [3H]pitavastatin for 1 and 2 min, respectively, was determined at various concentrations (0.3–100 μM). Solid lines represent the fitted curves obtained by nonlinear regression analysis. Each point represents the mean ± S.E. (n = 3). Where error bars are not shown, the S.E. values are within the limits of the symbol.
Estimation of Contribution of OATP1B1, OATP1B3, and OATP2B1 in Human Hepatocytes by Western Blot Analysis. We calculated the estimated uptake clearance of pitavastatin by OATP1B1, OATP1B3, and OATP2B1 in human hepatocytes by the relative expression level of each transporter (Table 1). We obtained 62.1 μg of protein in crude membrane from 1 mg of whole cell protein in OATP2B1-expressing HEK293 cells, whereas 178, 89, and 82 μg of protein were obtained in crude membrane from 106 human hepatocytes of lot OCF, 094, and ETR, respectively. When the band density per unit protein amount in crude membrane of OATP2B1-expressing HEK293 cells is defined as 1, the relative expression levels of OATP2B1 per unit protein amount in crude membrane of hepatocytes of lots OCF, 094, and ETR are 0.200, 0.152, and 0.112 (per microgram), respectively. Using these Rexp values and our previous results (Hirano et al., 2004; shown in Table 1), we estimated the relative contribution of OATP1B1, OATP1B3, and OATP2B1 to the hepatic uptake of pitavastatin in human hepatocytes.
Contribution of OATP1B1, OATP1B3, and OATP2B1 to the hepatic uptake of pitavastatin determined by the relative expression level
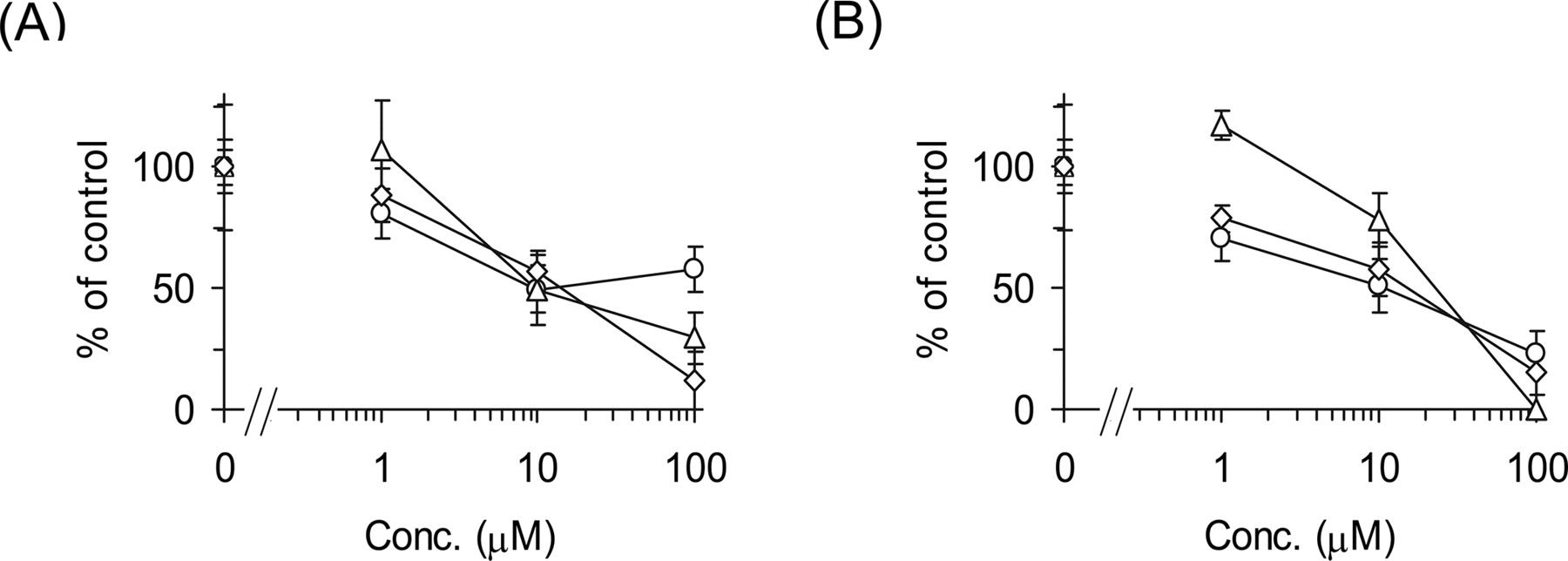
Inhibitory Effects of E217βG and E1S on the Uptake of Pitavastatin by Transporter-Expression System and Human Hepatocytes. Inhibitory effects of E217βG and E1S on the uptake of pitavastatin were examined by human cryopreserved hepatocytes (Fig. 3). E217βG (100 μM) inhibited OATP1B1- and OATP1B3-mediated transport of pitavastatin to 10.0 ± 3.2 and 21.7 ± 8.7% of control, respectively (n = 3), whereas OATP2B1-mediated transport was not affected by 100 μM E217βG (91.8 ± 16.6%). In contrast, 100 μM E1S inhibited OATP1B1- and OATP2B1-mediated transport of pitavastatin to 7.19 ± 2.94 and 56.5 ± 3.2% of control, respectively (n = 3), whereas OATP1B3-mediated transport was not affected by 100 μM E1S (102 ± 7%). In three batches of human hepatocytes, pitavastatin uptake was almost inhibited by 100 μM E217βG (Fig. 3A) and E1S (Fig. 3B).

Western blot analysis of OATP2B1. A, crude membrane fractions (2.5–40 μg) prepared from OATP2B1-expressed HEK293 cells and human hepatocytes (lot 094) were loaded and separated by SDS-polyacrylamide gel electrophoresis (7% separating gel). The sample designated as “Human liver” indicates that the crude membrane vesicles were prepared from a human frozen liver block (lot 020188) as a positive control. OATP2B1 was detected by preimmune antisera raised against the carboxyl terminus of human OATP2B1. B, comparison of the relative expression levels of OATP2B1 between transfectants and hepatocytes is shown. The x and y axes represent the amount of crude membrane obtained from transfectants and human hepatocytes and the intensity of the specific band in Western blot analysis, respectively. Closed circles and open circles indicate the band density of human hepatocytes (lot 094) and OATP2B1-expressing HEK293 cells, respectively. The solid lines represent the fitted lines obtained by linear regression analysis.
Prediction of DDI between Pitavastatin and Various Compounds by OATP1B1-Expressing HEK293 Cells. To identify clinically relevant inhibitors for OATP1B1-mediated pitavastatin uptake, inhibitory effects of several compounds on the uptake of pitavastatin were determined by OATP1B1-expressing cells. These compounds include therapeutic agents that were reported to cause DDI with statins (Williams and Feely, 2002). Cyclosporin A, fenofibrate, gemfibrozil, and gemfibrozil metabolites (gemfibrozil-M3 and gemfibrozil-1-O-glucuronide) were also investigated because drug interaction studies with pitavastatin have been previously reported (Hasunuma et al., 2003; Mathew et al., 2004). Most of the compounds we tested could inhibit OATP1B1-mediated pitavastatin uptake (Table 2). We also obtained the blood unbound fraction (fu) and calculated the estimated maximum concentration at the inlet to the liver (Iin,max) of the inhibitors from the literature information (Clark et al., 1992; Hardman et al., 2001; package insert of each drug). Inhibition constants (Ki) of various compounds for OATP1B1 obtained in the present study and the ratio of the uptake clearance in the absence of inhibitor to that in its presence (R value) are summarized in Table 2. R values of cyclosporin A, rifampicin, rifamycin SV, clarithromycin, and indinavir were higher than 2.5, suggesting that these drugs can interact with pitavastatin in a clinical situation.
The Ki values for OATP1B1-mediated pitavastatin uptake and the prediction of the possibility of DDI by considering the maximum plasma unbound concentration at the inlet to the liver
The Ki values are expressed as mean ± computer-calculated S.D. R value = 1 + fu· Iin,max/Ki.

Inhibitory effects of E217βG and E1S on the uptake of [3H]pitavastatin by human hepatocytes. The transport of [3H]pitavastatin (0.1 μM) into human hepatocytes was determined in the presence or absence of E217βG (A) and E1S (B) at the designated concentrations. Open circles, triangles, and squares represent the uptake in human hepatocytes of lots OCF, 094, and ETR, respectively. The detailed method for calculation of the uptake clearance in hepatocytes (CLhep) is described under Materials and Methods. The values are expressed as a percentage of the uptake of [3H]pitavastatin in the absence of inhibitors. Each point represents the mean ± S.E. (n = 3).
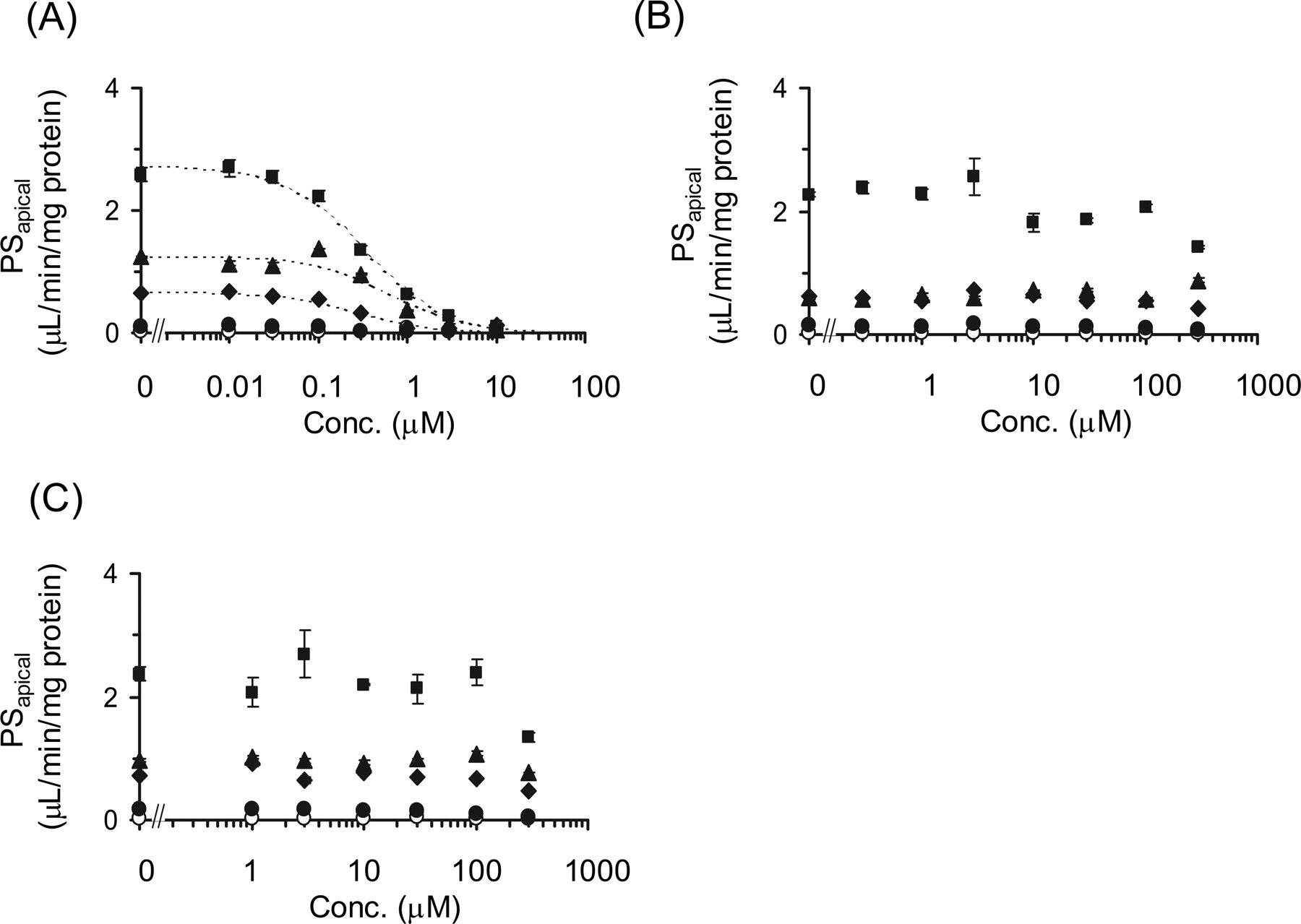
Inhibitory Effects of Cyclosporin A, Gemfibrozil, and Its Metabolites on the Transcellular Transport of Pitavastatin in OATP1B1/MRP2, OATP1B1/MDR1, and OATP1B1/BCRP Double Transfectants. The inhibitory effects of cyclosporin A, gemfibrozil, gemfibrozil-1-O-glucuronide, and gemfibrozil-M3 on the transcellular transport of pitavastatin were investigated in double-transfected cells. The transcellular transport clearance (PSnet) of pitavastatin was drastically decreased by cyclosporin A in all kinds of double transfectants (Fig. 4A). The efflux clearance from cells to the apical compartment (PSapical) was also potently reduced by cyclosporin A (Fig. 5A). In contrast, gemfibrozil and gemfibrozil-1-O-glucuronide did not change either PSnet or PSapical up to 100 μM (Figs. 4 and 5). Gemfibrozil-M3 (300 μM) could not inhibit the PSapical of pitavastatin in OATP1B1/BCRP-, OATP1B1/MDR1-, and OATP1B1/MRP2-expressing MDCKII cells (data not shown). The Ki values of these inhibitors on the PSnet and PSapical of pitavastatin are summarized in Table 3.
The Ki values of cyclosporin A, gemfibrozil, and gemfibrozil-1-O-glucuronide for the PSnet and PSapical of pitavastatin in double-transfected cells
The values are expressed as mean ± computer-calculated S.D.
Discussion
In the present study, we have excluded the possibility of a major contribution of OATP2B1 to the hepatic uptake of pitavastatin and confirmed that OATP1B1 is the most important transporter for its uptake. Next, the inhibitory effects of pitavastatin uptake by several drugs in OATP1B1-expressing cells were also investigated, and we discussed the possibility of DDI in clinical stage by considering the inhibition constant (Ki) obtained from in vitro analysis and the estimated maximum unbound concentration of each inhibitor at the inlet to the liver.
We observed the significant saturable uptake of pitavastatin in OATP2B1-expressing cells compared with control cells at pH 7.4 with a Km value of 1.17 μM (Fig. 1). It has been shown that specific uptake of pravastatin by OATP2B1 was not significantly observed at pH 7.4, whereas it can be transported at pH 5.0 (Nozawa et al., 2004), indicating that pitavastatin is preferentially recognized by OATP2B1 compared with pravastatin. The Km value of E1S for OATP2B1 from our analyses was 20.9 μM (Fig. 1), which was almost comparable to the reported values (Kullak-Ublick et al., 2001; Nozawa et al., 2004), whereas the significant uptake of E217βG at pH 7.4 was not detected as described previously (Kullak-Ublick et al., 2001; Nozawa et al., 2004).
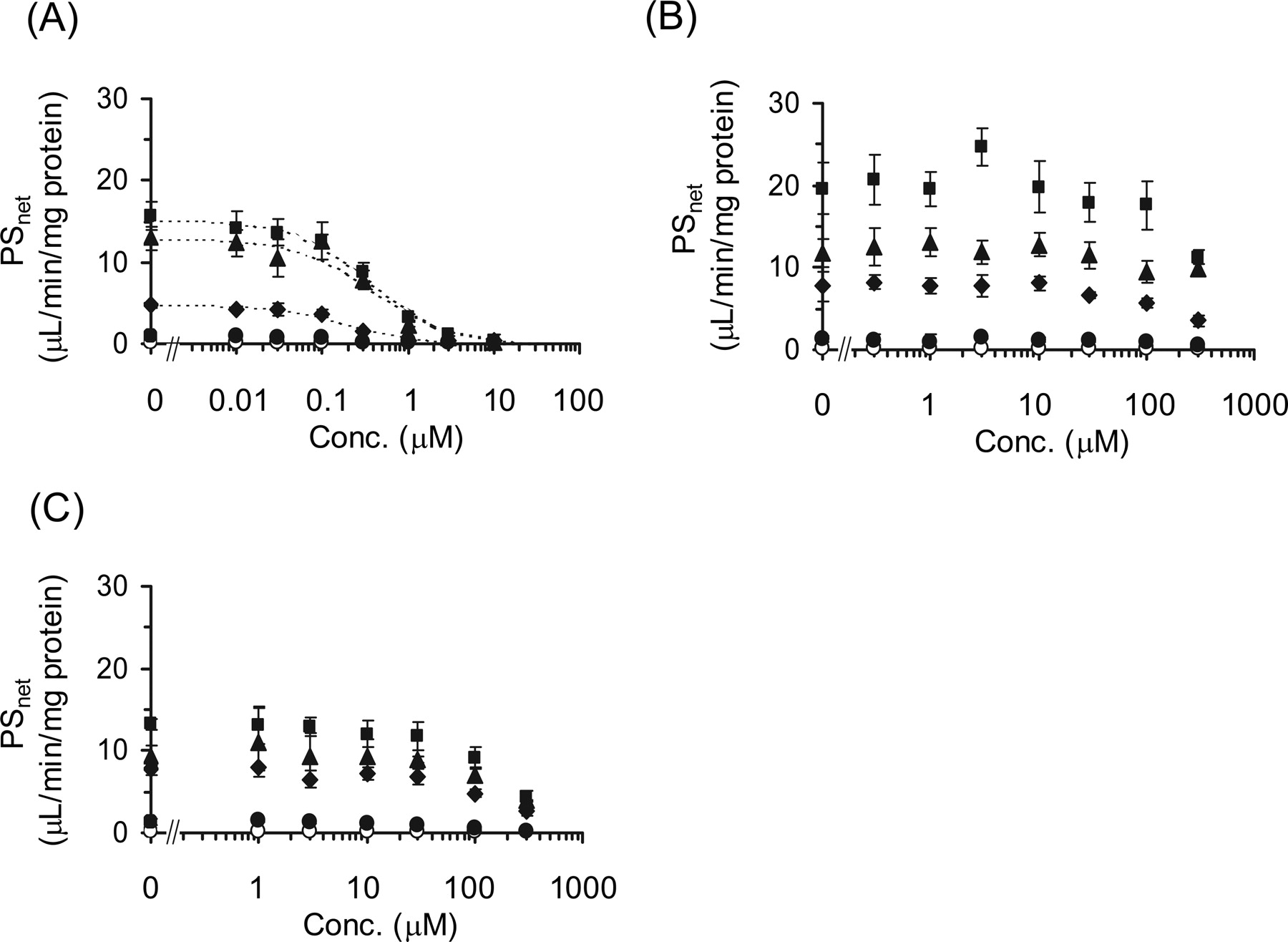
Inhibitory effects of cyclosporin A, gemfibrozil, and gemfibrozil-1-O-glucuronide on the transcellular transport of [3H]pitavastatin. The basal-to-apical flux of [3H]pitavastatin (0.3 μM) across MDCKII monolayer expressing OATP1B1 (closed circles), OATP1B1/BCRP (closed diamonds), OATP1B1/MDR1 (closed squares), and OATP1B1/MRP2 (closed triangles) was determined compared with vector-transfected control cells (open circles) in the absence and presence of cyclosporin A (A), gemfibrozil (B), or gemfibrozil-1-O-glucuronide (C). The x and y axes represent the concentration of each inhibitor in the medium at the basal compartment and the transport clearance for the transcellular transport (PS) of[3net H]pitavastatin (μl/min/mg protein). Each point and vertical bar represent the mean ± S.E. of three determinations. Where vertical bars are not shown, the S.E. values are within the limits of the symbol. Dotted lines represent the fitted curves obtained by nonlinear regression analysis.

Inhibitory effects of cyclosporin A, gemfibrozil, and gemfibrozil-1-O-glucuronide on the efflux transport of [3H]pitavastatin across the apical membrane of MDCKII cells. The efflux transport clearance of [3H]pitavastatin (0.3 μM) across the apical membrane (PSapical) of MDCKII monolayer expressing OATP1B1 (closed circles), OATP1B1/BCRP (closed diamonds), OATP1B1/MDR1 (closed squares), and OATP1B1/MRP2 (closed triangles) was determined compared with vector-transfected control cells (open circles) in the absence and presence of cyclosporin A (A), gemfibrozil (B), or gemfibrozil-1-O-glucuronide (C). The x and y axes represent the concentration of each inhibitor in the medium at the basal compartment and the transport clearance for the efflux transport across the apical membrane (PSapical) of [3H]pitavastatin (μl/min/mg protein). Each point and vertical bar represents the mean ± S.E. of three determinations. Where vertical bars are not shown, the S.E. values are within the limits of the symbol. Dotted lines represent the fitted curves obtained by nonlinear regression analysis.
Then, to refute the possibility that OATP2B1 plays an important role in the pitavastatin uptake in human hepatocytes and confirm the major contribution of OATP1B1, we took two strategies: 1) to check the inhibitable portion of pitavastatin uptake by two inhibitors in human hepatocytes, and 2) to compare the relative expression level of OATP2B1 in human hepatocytes and OATP2B1-expressing cells by Western blot analysis. As a result of the first approach, pitavastatin uptake in human hepatocytes was almost completely suppressed by both 100 μM E217βG (inhibitor of OATP1B1/OATP1B3) and E1S (inhibitor of OATP1B1/OATP2B1) (Fig. 3), suggesting that OATP1B1 mainly contributes to the hepatic uptake of pitavastatin. In contrast, the uptake of telmisartan, which can be accepted by OATP1B3, but not OATP1B1, into human hepatocytes could not be inhibited by 100 μM E1S (Ishiguro et al., 2006), supporting the validity of our approach. Moreover, from the comparison of the expression level of OATP2B1 between transfectants and hepatocytes by Western blot analysis (Fig. 2), we could calculate the OATP1B1-, OATP1B3-, and OATP2B1-mediated uptake into hepatocytes by multiplying the uptake clearance of pitavastatin in the expression system by the ratio of the expression level in these cells (Rexp). The results indicated that the contribution of OATP2B1 to the hepatic uptake of pitavastatin was less than 1%, although it is a substrate of OATP2B1, and that OATP1B1 is the most important in the hepatic uptake of pitavastatin, which is consistent with the previous results calculated from other approaches (Hirano et al., 2004).
Pitavastatin is mainly eliminated from liver in an unchanged form (Kojima et al., 2001). From the pharmacokinetic point of view, the change in the hepatic uptake clearance always directly affects the overall hepatic clearance for this type of drug (Shitara et al., 2005). Since we clarified that pitavastatin is taken up into the hepatocytes mainly by OATP1B1 in the present study, we focused on the inhibitory effects of various drugs on the OATP1B1-mediated uptake of pitavastatin. The combination therapy of statins and various drugs, such as fibrates, immune suppressants, antidiabetic drugs, antihypertensive drugs, and antibiotics, is widely used in the clinical situation (Williams and Feely, 2002). It has been reported that the plasma area under the plasma concentration-time curve of several statins was increased by coadministration of cyclosporin A and gemfibrozil (Shitara et al., 2005). Recently, Shitara et al. (2003) have demonstrated that inhibition of OATP1B1 is a major mechanism of DDI between cerivastatin and cyclosporin A. Campbell et al. (2004) also suggested that unconjugated hyperbilirubinemia induced by indinavir, rifamycin SV, and cyclosporin A is partly caused by the inhibition of OATP1B1-mediated uptake. Although pitavastatin uptake could be inhibited by several drugs in in vitro experiments, from the results of our calculation (Ito et al., 1998), the R values of most of the drugs we tested are almost equal to 1 (Table 2). To avoid the false-negative prediction of DDI, we estimated the inhibitory effects of the maximum plasma unbound concentration of inhibitors at the inlet to the liver (Iin,max). Therefore, it is unlikely that the OATP1B1-mediated DDI between pitavastatin and these drugs occurs in the clinical stage. However, we should pay attention to the OATP1B1-mediated DDI for pitavastatin with coadministration of cyclosporin A, rifampicin, rifamycin SV, clarithromycin, and indinavir because their R values exceeded 2.5 (Table 2), although the degree of the inhibition could be overestimated. We should notice that these drugs may also cause DDI with compounds that are mainly eliminated from liver via OATP1B1, including other statins. The previous clinical studies have shown that plasma concentration of pitavastatin was increased by cyclosporin A (Hasunuma et al., 2003), but not gemfibrozil and fenofibrate (Mathew et al., 2004). This evidence was consistent with our prediction (Table 2).
Conversely, gemfibrozil caused an increase in plasma concentration of cerivastatin (Backman et al., 2002). One of the major mechanisms of DDI between cerivastatin and gemfibrozil was considered to be the inhibition of CYP2C8-mediated metabolism of cerivastatin by gemfibrozil-1-O-glucuronide, which is thought to be concentrated in hepatocytes (Shitara et al., 2004). Gemfibrozil is metabolized into M3 and its glucuronide in the liver. Therefore, to investigate whether gemfibrozil, gemfibrozil-M3, and gemfibrozil-1-O-glucuronide could affect the transcellular transport by the inhibition of efflux transporter in liver, we checked their inhibitory effects on transcellular transport clearance (PSnet) and efflux clearance (PSapical) in double-transfected cells. As a result, gemfibrozil and its metabolites could not inhibit the efflux transporters and affect transcellular transport, whereas cyclosporin A strongly inhibited both PSnet and PSapical in all kinds of double-transfected cells (Figs. 4 and 5). This result suggested that the DDI between cyclosporin A and pitavastatin may be caused not only by the inhibition of OATP1B1-mediated uptake, but also by the inhibition of efflux transport mediated by MRP2, MDR1, and BCRP.
In conclusion, we have confirmed the major contribution of OATP1B1 to the hepatic uptake of pitavastatin in human hepatocytes. In addition, focusing on OATP1B1, inhibitory effects of various drugs on pitavastatin uptake were determined by OATP1B1-expressing cells, and its clinical relevance was discussed by considering the R values. Our results suggested that OATP1B1-mediated DDI between pitavastatin and some drugs indicated above may be clinically relevant and should be taken notice of during coadministration of inhibitors of OATP1B1.
Acknowledgments
We thank Dr. Piet Borst (The Netherlands Cancer Institutes, Amsterdam, The Netherlands) for providing the MDCKII cells expressing MRP2 and MDR1, Dr. Yoshihiro Miwa (University of Tsukuba, Japan) for providing pEB6CAGMCS/SRZeo vector, and Ying Tian and Miyuki Kambara for the construction of OATP2B1-expressing cells. We also thank Kowa Co. Ltd. (Tokyo, Japan) for providing radiolabeled pitavastatin and unlabeled pitavastatin, and Sankyo Co., Ltd. (Tokyo, Japan) and Chemtech Labo. Inc. (Tokyo, Japan) for providing gemfibrozil-1-O-glucuronide.
Footnotes
-
This work was supported by Health and Labour Sciences Research Grants from the Ministry of Health, Labour and Welfare for the Research on Advanced Medical Technology and a Grant-in Aid for Young Scientists (B) (15790087) from the Ministry of Education, Culture, Sports, Science and Technology.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009290.
-
ABBREVIATIONS: HEK, human embryonic kidney; MDCK, Madin-Darby canine kidney; OATP, organic anion-transporting polypeptide; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; Km, Michaelis constant; Vmax, maximum transport velocity; Ki, inhibition constant; E217βG, estradiol 17β-d-glucuronide; E1S, estrone-3-sulfate; DDI, drug-drug interaction.
- Received January 6, 2006.
- Accepted March 29, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}