


Abstract
Disposition and biotransformation of the new antipsychotic agent olanzapine (OLZ) were studied in six male healthy volunteers after a single oral dose of 12.5 mg containing 100 μCi of [14C]OLZ. Biological fluids were analyzed for total radioactivity, the parent compound (GC/MS), and metabolites (electrospray LC/MS and LC/MS/MS). Mean radiocarbon recovery was ∼87%, with 30% appearing in the feces and 57% excreted in the urine. Approximately half of the radiocarbon was excreted within 3 days, whereas >70% of the dose was recovered within 7 days of dosing. Circulating radioactivity was mostly restricted to the plasma compartment of blood. Mean peak plasma concentration of OLZ was 11 ng/ml, whereas that of radioactivity was 39 ng eq/ml. Mean plasma terminal elimination half-lives were 27 and 59 hr, respectively, for OLZ and total radioactivity. With the help of NMR and MS data, a major metabolite of OLZ in humans was characterized as a novel tertiaryN-glucuronide in which the glucuronic acid moiety is attached to the nitrogen at position 10 of the benzodiazepine ring. Another N-glucuronide was detected in urine and identified as the quaternary N-linked 4′-N-glucuronide. Oxidative metabolism on the allylic methyl group resulted in 2-hydroxymethyl and 2-carboxylic acid derivatives of OLZ. The methyl piperazine moiety was also subject to oxidative attack, giving rise to the N-oxide and N-desmethyl metabolites. Other metabolites, including the N-desmethyl-2-carboxy derivative, resulted from metabolic reactions at both the 4′ nitrogen and 2-methyl groups. The 10-N-glucuronide and OLZ were the two most abundant urinary components, accounting for ∼13% and 7% of the dose, respectively. In fecal extracts, the only significant radioactive HPLC peaks were due to 10-N-glucuronide and OLZ representing, respectively, ∼8% and 2% of the administered dose. Semiquantitative data obtained from plasma samples from subjects given [14C]OLZ suggest that the main circulating metabolite is 10-N-glucuronide. Thus, OLZ was extensively metabolized in humans via N-glucuronidation, allylic hydroxylation,N-oxidation, N-dealkylation and a combination thereof. The 10-N-glucuronidation pathway was the most important pathway both in terms of contribution to drug-related circulating species and as an excretory product in feces and urine.
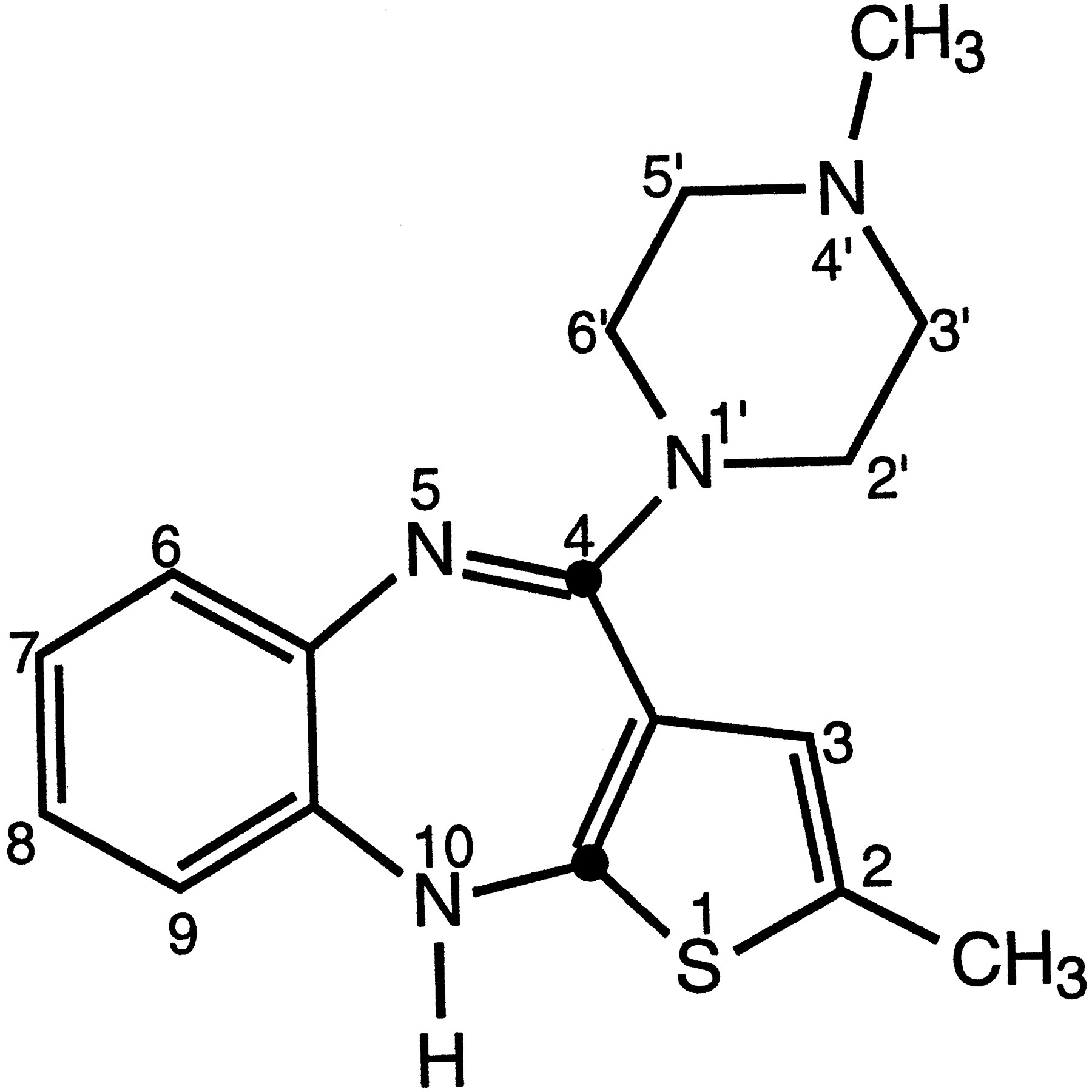
OLZ1, (fig.1) is a new antipsychotic drug with a thienobenzodiazepinyl structure. Biochemical studies have shown that OLZ has a broad pharmacological profile, with activity at dopamine (D1/D2/D3/D4), serotonin (5-HT2A/2C), muscarinic (especially M1), histamine (H1), and adrenergic (α1) receptors (1, 2).

Chemical structure of OLZ.
The position of 14C atoms is indicated by the filled circles.
Anticholinergic activity and 5-HT2 receptor antagonism, as shown by the atypical neuroleptic clozapine, are thought to reduce the extrapyramidal effects exhibited by classical D2-antagonist antipsychotic agents (3, 4). Significant 5-HT2 receptor antagonism has been proposed to be responsible for the treatment of the negative symptoms of schizophrenia, as well as improved activity of atypical antipsychotic agents (5).
In clinical studies in patients suffering from schizophrenia or schizophreniform disorder, OLZ was effective in the treatment of both positive and negative symptoms of schizophrenia, with a low incidence of extrapyramidal side effects (6-8). Antipsychotic efficacy of OLZ was demonstrated in the dose range of 5–20 mg/day.
The purpose of the present study was to evaluate the disposition and biotransformation of OLZ in healthy volunteers as part of the clinical development of the compound. The study was conducted in six healthy volunteers after administration of a single oral dose of 12.5 mg containing 100 μCi of [14C]OLZ.
Materials and Methods
Reference Compounds and Other Materials.
The following compounds were synthesized at Lilly Research Laboratories as described elsewhere (9): OLZ (2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-B][1,5]benzodiazepine), [4,10a-14C2]OLZ ([14C]OLZ, radiochemical purity, 99.5%; specific activity, 22.9 μCi/mg), 4′-N-desmethyl OLZ (N-desmethyl OLZ, 2-methyl-4-(1-piperazinyl)-10H-thieno[2,3-B][1,5]benzodiazepine), 4′-N-oxide OLZ (N-oxide OLZ, 4-(2-methyl-10H-thieno[2,3-B][benzodiazepin-4-yl)-1-methylpiperazine-1-oxide), 2-hydroxymethyl OLZ (4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-B][1,5]benzodiazepine-2-methanol), OLZ 4′-N-glucuronide (4′-N-glucuronide, 1-[4-(2-methyl-10H-thieno[2,3-B][1,5]benzodiazepin-4-yl)-1-methylpiperazinyl]-1-deoxy-β-d-glucopyranosiduronic acid), 2-carboxymethyl OLZ (methyl 4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-B][1,5]benzodiazepine-2-carboxylate), and 4′-N-desmethyl-2-hydroxymethyl OLZ (4-(1-piperazinyl)-10H-thieno[2,3-B][1,5]benzodiazepine-2-methanol). NEE-154 Glusulase was purchased from DuPont (Wilmington, DE). HPLC grade acetonitrile and methylene chloride were bought from Mallinckrodt (Paris, KY). Scintisol was purchased from Isolab, Inc. (Akron, OH). PITC was supplied by Aldrich Chemical Co. (Milwaukee, WI).
OLZ 10-N-glucuronide (10-N-glucuronide) was synthesized as follows. To a silylated amber vial, 100 mg (0.32 mmol) of OLZ and 4 ml of acetone were added, and the vial was vortexed until the OLZ dissolved completely. A solution of glucuronic acid (124 mg, 0.64 mmol) in 0.1 M phosphate buffer (pH 7.2) (4 ml) was added, and the mixture was stirred at room temperature for 3 days. Mixtures from several parallel reactions were combined, and the pH of the resulting mixture was adjusted to ∼10 with 25% ammonium hydroxide solution. The solution was extracted with ethyl acetate (100 ml), and the organic layer that contained unreacted OLZ was discarded. The aqueous layer was lyophilized to dryness, and the residue dissolved in water. Aliquots (200 μl) of the aqueous fraction were separated on an analytical Inertsil C18 HPLC column. The initial mobile phase was 0.05 M ammonium acetate/acetonitrile (90:10), and a linear gradient of 2.5% acetonitrile/min was started 2 min after injection and continued for 20 min. Flow rate was maintained at 1.0 ml/min. The column effluent was collected at the retention volumes of two closely eluting peaks that corresponded to the two “isomers” of the 10-N-glucuronide. Isolated material was further purified by fractionation on the same HPLC system using isocratic elution (0.05 M ammonium acetate/acetonitrile, 77:23). Purified material was analyzed by proton NMR and electrospray MS.
The 2-carboxylic acid derivative of OLZ (2-carboxy OLZ) was prepared by de-esterification of 2-carboxymethyl OLZ. Approximately 2 mg of 2-carboxymethyl OLZ was placed in a siliconized tube and dissolved in methylene chloride (2 ml). The solution was flushed with nitrogen and treated with boron tribromide (2 ml of a 25% in solution in methylene chloride). The tube was capped immediately and allowed to stand at room temperature for 2 hr. Approximately 50% of the ester was converted to the corresponding acid. Electrospray MS analysis gave m/z343 (MH+).
2-Carboxy-4′-N-desmethyl OLZ (N-desmethyl-2-carboxy OLZ) was prepared by oxidizing 4′-N-desmethyl-2-hydroxymethyl OLZ using chromium trioxide (10). Approximately 9% of the starting material was converted to the desired acid. Electrospray MS yielded m/z 329 (MH+).
Subjects, Dosing, and Sample Collection.
Six healthy male volunteers (23–42 years old, weighing 68.5–89.8 kg) participated in the study. All subjects were smokers. Each subject was given a single dose of 12.5 mg OLZ containing ∼100 μCi of radioactivity. Volunteers fasted from midnight the evening before, until 5 hr after drug administration at 8 a.m. The dose was formulated as a capsule and was administered orally along with ∼250 ml water. No other medications were administered to the volunteers during the trial. Serial blood samples (10 ml) were obtained at 0, 20, 40, and 60 min, and 2, 2.5, 3, 3,5, 4, 4.5, 5, 6, 8, 12, 16, 24, 36, 48, 72, 96, 120, and 144 hr postdose. Both whole blood and plasma were analyzed for radioactivity, as were breath samples collected at the same time as blood samples. Fecal samples were collected at 24-hr intervals for 21 days. Urine samples were collected at 0–4, 4–8, 8–16, and 16–24 hr after drug administration, and daily thereafter for 21 days. All samples were kept at approximately −70°C until analyzed.
Analysis of Radioactivity.
The radioactivity in plasma (0.25 ml) and urine (1.0 ml) samples was determined by liquid scintillation counting after addition of Scintisol. Breath samples were obtained by having the subject blow into a vial containing a trapping solution and were counted directly after addition of Permafluor V. Whole blood and fecal samples were oxidized before counting. Approximately 0.5 ml of whole blood was pipetted into sample oxidizer combustion cones, weighed, air-dried, and combusted in a sample oxidizer. Fecal samples (∼1 g) were prepared for radioactivity determination by making a 1:1 water/feces homogenate. Aliquots of the homogenate were placed in sample oxidizer combustion cones, weighed, air-dried, and combusted in a sample oxidizer. Blood and fecal samples prepared in this manner were then counted after addition of Permafluor V.
Analysis of Unchanged OLZ.
A GC/MS assay was used to quantitate OLZ in plasma. Briefly, the method consisted of liquid-liquid extraction of OLZ and the internal standard (2-ethyl homolog of OLZ) from 1 ml of plasma. The extraction was a three-step process consisting of a basic extraction into ethyl acetate, a backextraction into acid, and finally extraction into methylene chloride. The methylene chloride was removed using a Speed Vac system, and the resulting dried extract was treated with heptafluorobutyric anhydride. The instrument used was a Finnigan 4500 GC/MS. Derivatized materials were dissolved in 25 μl of ethyl acetate and separated by capillary GC on a 30-m DB-1701 fused silica column. Analysis was conducted in the methane negative-ion chemical ionization mode using selected ion monitoring. The ions at m/z 370 (OLZ) andm/z 384 (internal standard) were monitored, and the ratio of responses (m/z 370:384) thus obtained was related to a standard curve. The limit of quantitation was 0.1 ng/ml.
Pharmacokinetics.
Noncompartmental analysis was used to determine the pharmacokinetics of OLZ and radioactivity. Cmax andtmax were assessed by visual inspection. Terminal elimination half-life was calculated using the relationship 0.693/k, where k is the elimination rate constant. The AUC0 to t was calculated up to the last time point t by the trapezoidal rule. The AUC0 to twas extrapolated to infinite time using k. The apparent systemic clearance (CLs/F) and apparent volume of distribution (V/F) were found as Dose/AUC0 to inf and CLs/k, respectively.
In Vitro Protein Binding.
Plasma was spiked with [14C]OLZ and incubated for 1 hr at 37°C. After ultracentrifugation (100,000 rpm at 37°C for 4 hr), the amount of OLZ in the supernatant was determined by liquid scintillation spectroscopy. The fraction of OLZ bound to protein was calculated from the radioactivity concentrations in the spiked sample and the supernatant.
Metabolite Isolation. Urinary Metabolites.
Urine samples from individual subjects (∼400 ml) were made alkaline with a solution of ammonium hydroxide (47 ml, 0.1 M) and extracted with ethyl acetate (1800 ml). The ethyl acetate extract was evaporated to dryness under reduced pressure in a 40°C water bath. The residue was reconstituted in ∼2 ml of acetonitrile/water (1:1) for HPLC and LC/MS/MS analysis. The remaining aqueous fraction was lyophilized and the residue reconstituted in 0.1 M ammonium acetate/1% TEA in acetonitrile (4:1), and fractions containing metabolites were collected from the HPLC system for MS analysis. Metabolites were also isolated from urine using column chromatography. Approximately 400 g of Amberlite XAD-2 resin was packed in a 2.5 × 30 cm glass column. The column was conditioned by passing methanol (2 liters) followed by purified water (5 liters). The remaining urine samples from the volunteers were individually pooled and passed slowly through the column. The column was washed with water and the radioactivity eluted with methanol. Methanolic extracts were concentrated in vacuo at 40°C using a rotary evaporator, and the resulting residue was reconstituted in 50 ml methanol/water (50:50).
Plasma Metabolites.
Samples for LC/MS/MS analysis were prepared by solvent (ethyl acetate) extraction as described for urine samples. For metabolite profiling using HPLC with radiochemical detection, the remaining plasma samples from three volunteers were processed as follows. To 10 ml plasma from subject 4 (0.33–144 hr postdose), acetonitrile (15 ml) was added, and the solution was vortex-mixed for 30 sec. The mixture was allowed to stand for 5 min and then centrifuged for 15 min at 1,000g.The supernatant was decanted into a siliconized vial and evaporated to dryness in a water bath (40°C) under a stream of nitrogen. The residue was dissolved in water (∼400 μl) for HPLC analysis. Similarly, samples were prepared from plasma aliquots obtained from subject 3 (7 ml, 0.33–72 hr postdose) and subject 1 (37 ml, 0.66–144 hr postdose), and subjected to HPLC and LC/MS analysis.
Fecal Metabolites.
An aliquot (10 g) of an aqueous homogenate of fecal sample was placed in an amber, siliconized glass vial and extracted with two 20-ml portions of methanol. The sample was centrifuged for 10 min at 833g. After removal of the supernatant, the remaining fecal material was transferred to an Alundum thimble (20 × 80 mm, course porosity) and placed in a Soxhlet extraction tube. Fecal material was extracted for 4 hr using methanol (100 ml). The methanol extract was combined with the previous methanol extracts, and placed in a −20°C freezer for ∼2 hr. The combined methanol extract was then centrifuged for 10 min at 833g. The methanol layer was decanted into a siliconized vial and evaporated to dryness in a 40°C water bath under a nitrogen stream. Acetonitrile/water (10 ml) was added to the residue, the resulting mixture was vortexed for several minutes, and centrifuged for 10 min at 1,300g. Liquid extract was decanted into a siliconized tube and evaporated to near dryness under a stream of nitrogen at 40°C. The remaining residue was dissolved in 2 ml of acetonitrile/water (30:70).
Hydrolysis and Derivatization Reactions. Enzyme Hydrolysis of OLZ 4′-N-Glucuronide.
Aliquots (∼3.4 mg 14C-equivalents) of a sample of 4′-N-glucuronide isolated from urine were pipetted into three siliconized glass tubes, and 100 μl acetate buffer (0.2 M, pH 5.0) was added to each tube. To each tube was added either 15 μl of buffer, 15 μl Glusulase (containing 2,025 units β-glucuronidase and 150 units sulfatase), or 2 mg of β-saccharolactone and 15 μl Glusulase. Tubes were capped and incubated in a water bath at 37°C for 16 hr.
Attempted Enzyme Hydrolysis of OLZ 10-N-Glucuronide.
To examine the susceptibility of 10-N-glucuronide to enzyme hydrolysis, a sample of the conjugate (isolated from urine and/or chemically synthesized) was incubated with β-glucuronidase from either Helix pomatia or Escherichia coli as described previously.
Chemical Hydrolysis of OLZ 10-N-Glucuronide.
The susceptibility of 10-N-glucuronide to acid/base hydrolysis was evaluated in plasma spiked with 10-N-glucuronide (8 ng/ml) using either HCl (0.1, 1, 2, 3, or 6 N) or NaOH (0.1 or 1.0 N). To siliconized glass tubes, 1-ml aliquots of spiked plasma and 1 ml of the HCl or NaOH solution were added. Tubes were capped, vortexed for 10 sec, and placed in a water bath for 1 hr at 50°C, following which the samples were neutralized with an equimolar amount of NaOH or HCl. Samples were then extracted and analyzed for OLZ by HPLC with electrochemical detection as described previously (11).
Derivatization of the Secondary Amine of OLZ and Its Glucuronide.
To obtain information on the site of attachment of the glucuronic acid moiety to the OLZ molecule, OLZ and the two N-glucuronides of OLZ were treated with PITC as follows. OLZ (1 mg), 10-N-glucuronide (∼0.1 mg), and 4′-N-glucuronide (∼0.1 mg) were added to separate siliconized tubes. To each tube, 1 ml of pyridine/acetonitrile (1:1) was added, and the tubes were vortexed until the samples were completely dissolved. To each tube, neat PITC (500 μl) was added, and samples were allowed to stand at room temperature for 1 hr. Tubes were placed in a 50°C water bath, and the solvent was evaporated under a stream of nitrogen. The remaining residue was reconstituted in acetonitrile (1 ml), and aliquots were analyzed by LC/MS and LC/MS/MS.
Metabolite Identification. HPLC.
The HPLC system consisted of a Beckman model 126 pump, NEC PC8300 controller, Waters model 712 WISP autosampler, Applied Biosystem model 785A UV detector, and a Berthold model LB 507A radiodetector equipped with a 150-μl yttrium solid cell. For profiling plasma metabolites, samples were analyzed on an Inertsil C18 column (5 μm particle size, 0.46 × 25 cm) using a gradient of 0.05 M ammonium acetate and acetonitrile. Solvent composition was maintained 90/10 (0.05 M ammonium acetate/acetonitrile for 2 min) and programmed at 2.5%/min until a mobile phase composition of 60% acetonitrile was achieved. The mobile phase was maintained at that composition for 8 min. The column was maintained at room temperature, and the flow rate was 1.0 ml/min. The level of each metabolite in plasma, urine, and fecal extracts was estimated by injecting an aliquot of an extract into the column, collecting the peak of interest as it eluted from the column, and measuring the amount of radioactivity in the sample by liquid scintillation spectroscopy. Total radioactivity in the injected sample was estimated by passing an aliquot through the HPLC autoinjector and collecting the whole sample before the sample passed through the column. Scintisol (15 ml) was added to each sample and the level of radioactivity determined. For HPLC separation of urinary and fecal components, aliquots (150 μl) of the ethyl acetate or aqueous urine extracts and fecal extracts were injected onto a Hypersil C18 column (5 μm particle size, 0.46 × 25 cm), with an initial mobile phase of 0.1 M ammonium acetate/1% TEA in acetonitrile (9:1). Two minutes after the injection, a linear gradient of 2.5% acetonitrile/TEA/min was used until a solvent composition of 40:60 0.1 M ammonium acetate/1% TEA in acetonitrile was attained. The run was continued for an additional 8 min at this solvent composition. Metabolites were isolated from the aqueous fraction of urine and fecal extracts by collecting radioactive peaks as they eluted from the HPLC column. Samples were injected successively to obtain sufficient quantities of each metabolite for MS identification. Samples were lyophilized to remove the mobile phase and were reconstituted in ∼500 μl acetonitrile/water (1:1). Each urine extract obtained using an XAD-2 column as described previously was separated on a semipreparative (5 μm particle size, 1 × 25 cm) Hypersil C18 column using the same solvent and program described previously for the Hypersil analytical column. Flow rate was 3.5 ml/min, and injection volume was 500 μl. Collected fractions were further purified using an analytical Hypersil C18 column.
LC/MS and LC/MS/MS.
Extracts were analyzed for metabolites and parent compound by LC/MS and LC/MS/MS on either a Finnigan MAT TSQ700 or a Sciex API III mass spectrometer. For ionspray full-scan analysis, the Sciex API III was programmed to scan from m/z 200 to 800 every 1.66 sec. For MS/MS experiments, argon/nitrogen (90:10) was used as the target gas at a thickness of 250 × 1012 atoms/cm2, with the collision energy set at 30 eV. Positive-ion, full-scan mass spectra of isolated or synthetic metabolites and product ion spectra obtained by CID of the corresponding MH+ precursor ions were recorded either by direct infusion or LC introduction. For direct infusion experiments, the orifice potential was maintained at 70 V to analyze urine extracts or 140 V for fecal extracts. Samples were infused into the ion source at a rate of 20 μl/min by a syringe pump directly coupled to the fused silica from the ionspray interface. LC/MS was performed by injecting aliquots of extracts onto an Inertsil ODS-2 column coupled to the mass spectrometer via a splitting tee. Samples were injected at an initial mobile phase of 10% acetonitrile in 0.025 M ammonium acetate that was maintained for 2 min. Then, the proportion of acetonitrile was increased to 60% in a linear fashion at a rate of 2.5%/min and was maintained at that level for an additional 8 min. The mobile phase (flow rate 1 ml/min) was delivered by a pair of Shimadzu model LC-10AD pumps controlled by a model SCL-10A gradient controller. Column effluent was split such that the flow rate to the ion source was 20 μl/min, the remaining being diverted to a UV detector (Shimadzu model SPD-10AV UV detector). With the Finnigan MAT TSQ700 MS/MS system, the electrospray LC interface was used. For analysis by direct infusion, the sample was infused at a rate of 2 μl/min. For LC/MS and LC/MS/MS analysis, a Waters model 600 pump and a model 600 MS system controller were used. Analytes were separated on an Inertsil C18 column using the same conditions as those described previously for the LC/MS analysis using the Sciex API III mass spectrometer. The flow rate was 1 ml/min, and the effluent was split such that equal volumes were delivered into the ion source and a Raytest Ramona model 5LS radiodetector. MS spectra were obtained by scanning from m/z 200 to 600 every second. For MS/MS analysis, collision gas (argon) pressure was maintained at 2.0 m torr, and the collision offset voltage was −20 eV.
NMR Spectroscopy.
Proton NMR spectrum of the synthesized 10-N-glucuronide was recorded in d6-dimethylsulfoxide on a Bruker AMX spectrometer operating at 500 MHz. Chemical shifts are reported in ppm (δ) relative to tetramethylsilane.
Results
Radiocarbon Excretion.
After a single oral dose of [14C]OLZ to six male subjects, radioactivity was predominantly excreted in the urine (fig.2). Recovery of radiocarbon was 74.8 ± 18.8% (mean ± SD), with 22.6 ± 13.8% appearing in the feces and 52.2 ± 8.9% excreted in the urine. Fecal recovery for subjects 3 and 4 (fig. 2) was noticeably lower than for the other subjects, perhaps due to failure by these two subjects to collect complete samples. If values for these two subjects are omitted from the calculation, urinary and fecal recovery were 56.6 ± 6.2% and 30.2 ± 9.1%, respectively, for a total recovery of 86.8 ± 3.2%. Greater than 70% of the radiocarbon was recovered within 7 days of dosing. No radioactivity was detectable in breath samples.

Recovery of radioactivity in urine and feces.
Samples were collected for 21 days after a single dose of [14C]OLZ.
Plasma OLZ and Blood Radioactivity Concentrations.
OLZ concentrations were measured by a validated GC/MS assay with a limit of quantitation of 0.1 ng/ml. Mean plasma concentrationvs. time plots of OLZ and radioactivity (fig.3) show that the level of OLZ is much lower than radioactivity at all time points. The amount of radiocarbon in red blood cells was calculated using the concentrations of radioactivity obtained for plasma and whole blood, and the hematocrit was found on the day of dosing. Little or no radioactivity was detectable in red blood cells.

Mean plasma concentrations of OLZ and radioactivity after a single oral dose of [14C]OLZ to six normal subjects.
Error bars represent SEMs.
In Vitro Plasma Protein Binding.
OLZ was highly protein-bound, with a mean binding of ∼93% in plasma from normal subjects. Binding was concentration-independent over the range tested (7–1,100 ng/ml). Using purified proteins, the drug was found to bind to a higher extent to albumin (90%) than α1-acid glycoprotein (77%).
Pharmacokinetics.
Noncompartmental plasma pharmacokinetic parameters for OLZ and radioactivity are shown in tables 1 and2. Maximum plasma concentration of OLZ averaged 10.5 ng/ml (range: 9.6–12.5 ng/ml) and was reached 3–8 hr after dosing. The average maximum concentration of radioactivity in plasma was 39.0 ng eq/ml (range: 26.4–65.8 ng equiv/ml) and occurred between 4 and 6 hr postdose. The mean elimination half-lives of OLZ and radioactivity were, respectively, 26.5 ± 1.6 and 58.7 ± 7.1 hr. The mean apparent systemic clearance (CLs/F) and volume of distribution (V/F) of OLZ were 0.571 ± 0.059 liter/kg/hr and 21.9 ± 3.2 liter/kg, respectively. The ratio of plasma OLZ AUC0 to t to radioactivity was 0.14.
Calculated pharmacokinetic parameters of OLZ after the administration of a single dose of [14C]OLZ
Calculated pharmacokinetic parameters of radioactivity after the administration of a single dose of [14C]OLZ
Metabolism Study. Identification of Metabolites.
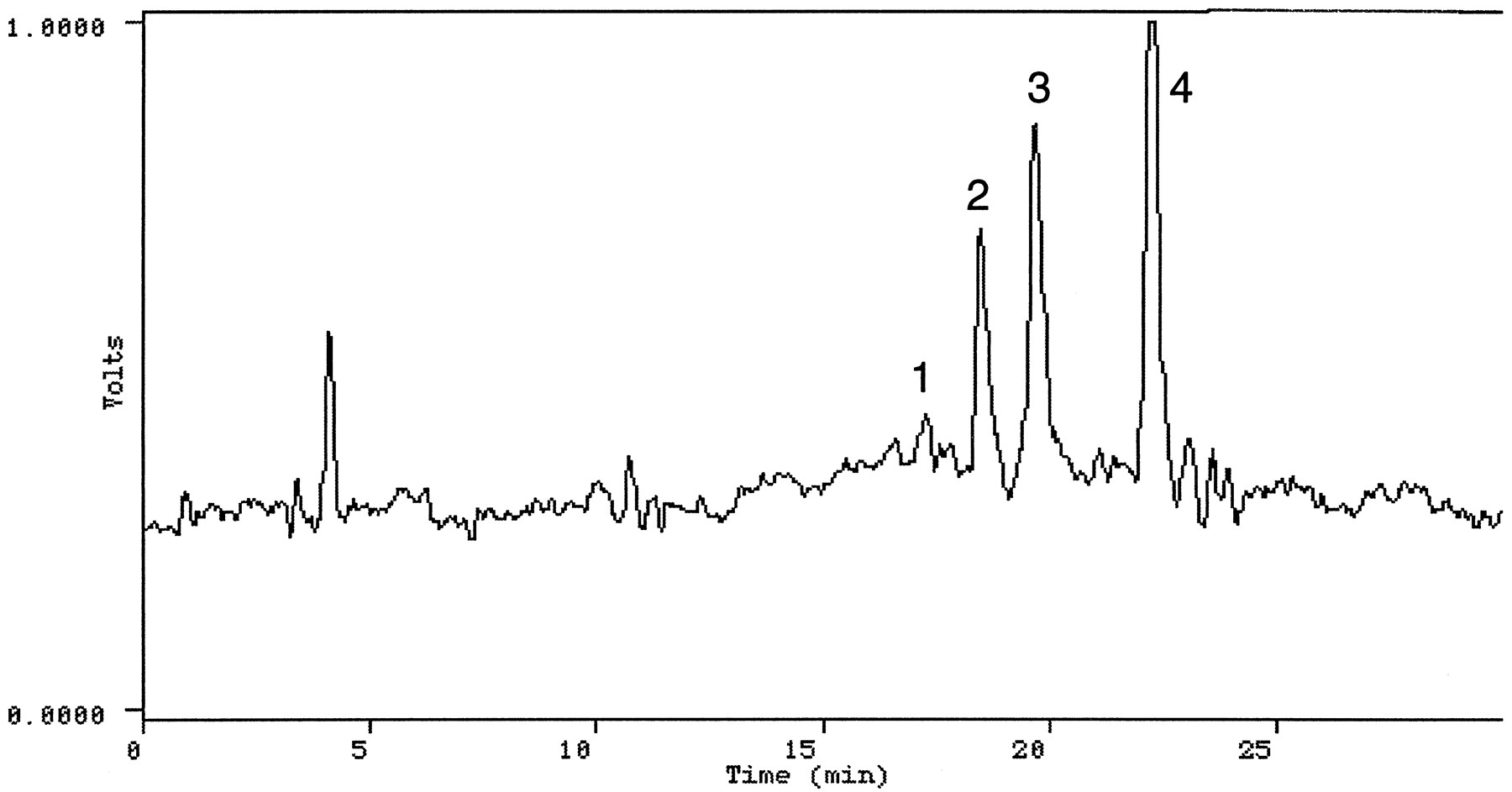
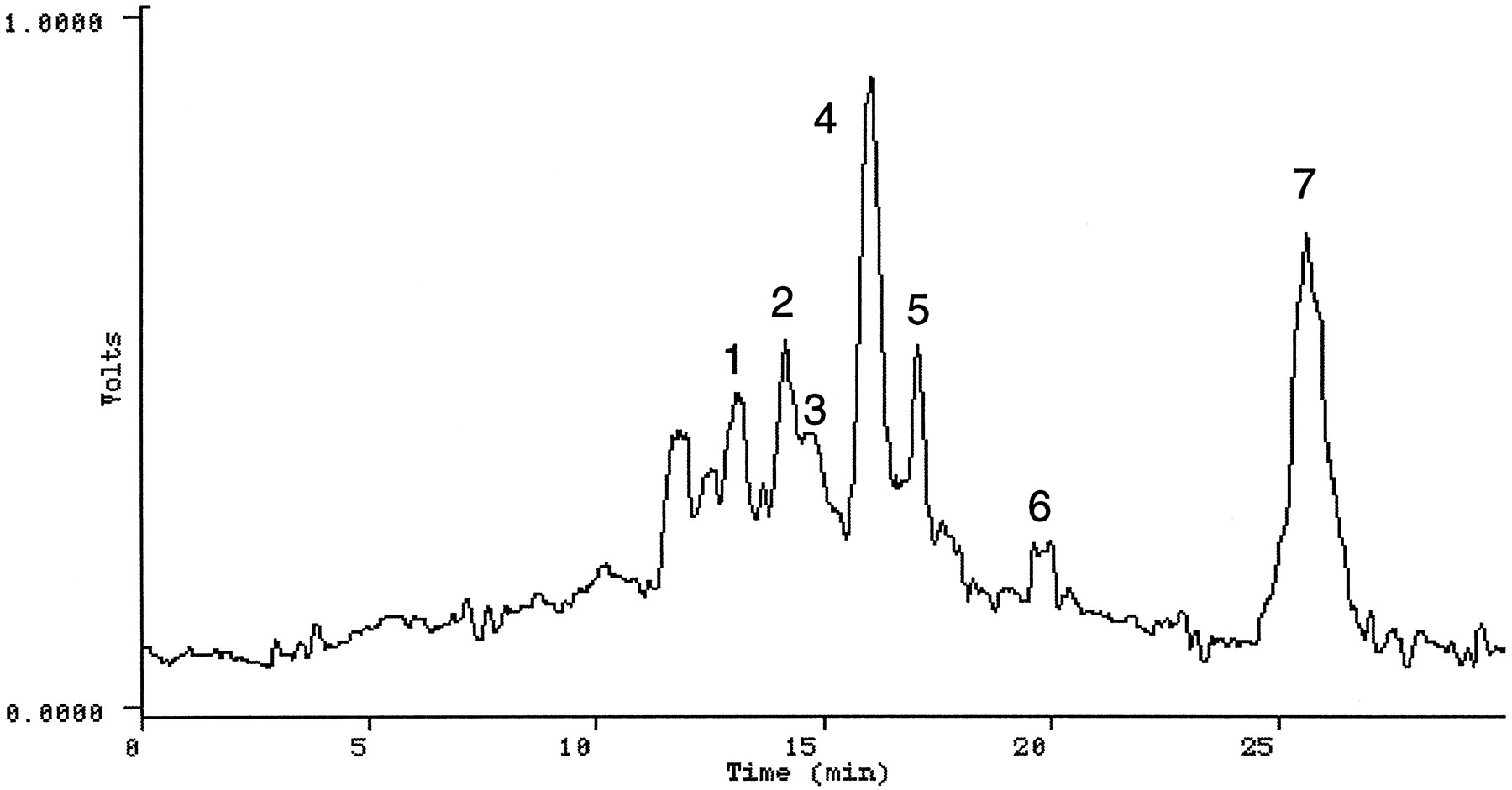
Urine—The HPLC radiochromatogram shown in fig. 4 was obtained from the aqueous fraction of the extract of a urine sample (subject 1, 8–16 hr; 8–16 hr urine from all subjects registered a high concentration of radioactivity). Seven radioactive components were detected and isolated by HPLC for analysis by direct infusion electrospray MS and MS/MS. The HPLC profile obtained from the ethyl acetate extract (subject 4, 8–16 hr) showed at least 5 peaks (fig.5). The extract was directly analyzed by electrospray LC/MS and LC/MS/MS. Figure 6 shows the HPLC separation of radiolabeled components in a urine extract obtained using an XAD-2 column. The following compounds were identified by analyzing the various urinary extracts.

HPLC radioprofile obtained from the aqueous portion of an ethyl acetate extract of a urine sample (subject 1, 8–16 hr) from a normal subject given [14C]OLZ.
Peaks were identified as: 1, 2-carboxy OLZ; 2,10-N-glucuronide; 3, N-oxide OLZ.

HPLC radioprofile of the ethyl acetate extract of a urine sample (subject 4, 8–16 hr) from a subject given [14C]OLZ.
Peaks were identified as: 1, 2-hydroxymethyl OLZ; 2, N-oxide OLZ; 3, N-desmethyl OLZ; 4, OLZ.

HPLC radioprofile obtained from the XAD-2 extract of pooled urine samples from three subjects given a single dose of [14C]OLZ.
Peaks were identified as: 1, N-desmethyl-2-carboxy OLZ;2, 2-carboxy OLZ; 3, N-oxide-2-carboxy OLZ glucuronide; 4, 10-N-glucuronide; 5,4′-N-glucuronide + 2-carboxy OLZ glucuronide; 6, N-oxide OLZ + 2-hydroxymethyl OLZ; 7, OLZ.
10-N-Glucuronide—Electrospray MS analysis of the large peak eluting at ∼16 min (fig. 4) exhibited an MH+ atm/z 489. When the same peak was isolated and rechromatographed on an analytical Hypersil C18 column with a mobile phase of 0.1 M ammonium acetate/1% TEA in acetonitrile (4:1), the peak resolved into two components having a ratio of 3:1. Both components yielded a protonated molecular ion of m/z 489 and nearly identical product ion spectra (shown in fig. 7for one of the “isomers”). In both spectra, the ion m/z313 (protonated OLZ), which could arise as a result of loss of dehydroglucuronic acid (176 Da) as a neutral, was present. Neutral loss scan of 176 Da yielded a peak corresponding to m/z 489, as did a neutral loss scan of 57 Da (loss of CH2⋕CH—NH—CH3 from the methyl piperazine moiety of OLZ). These data suggested that these two metabolites were glucuronic acid conjugates of OLZ, with the glucuronic acid moiety attached to a nitrogen other than the methyl piperazine nitrogen.
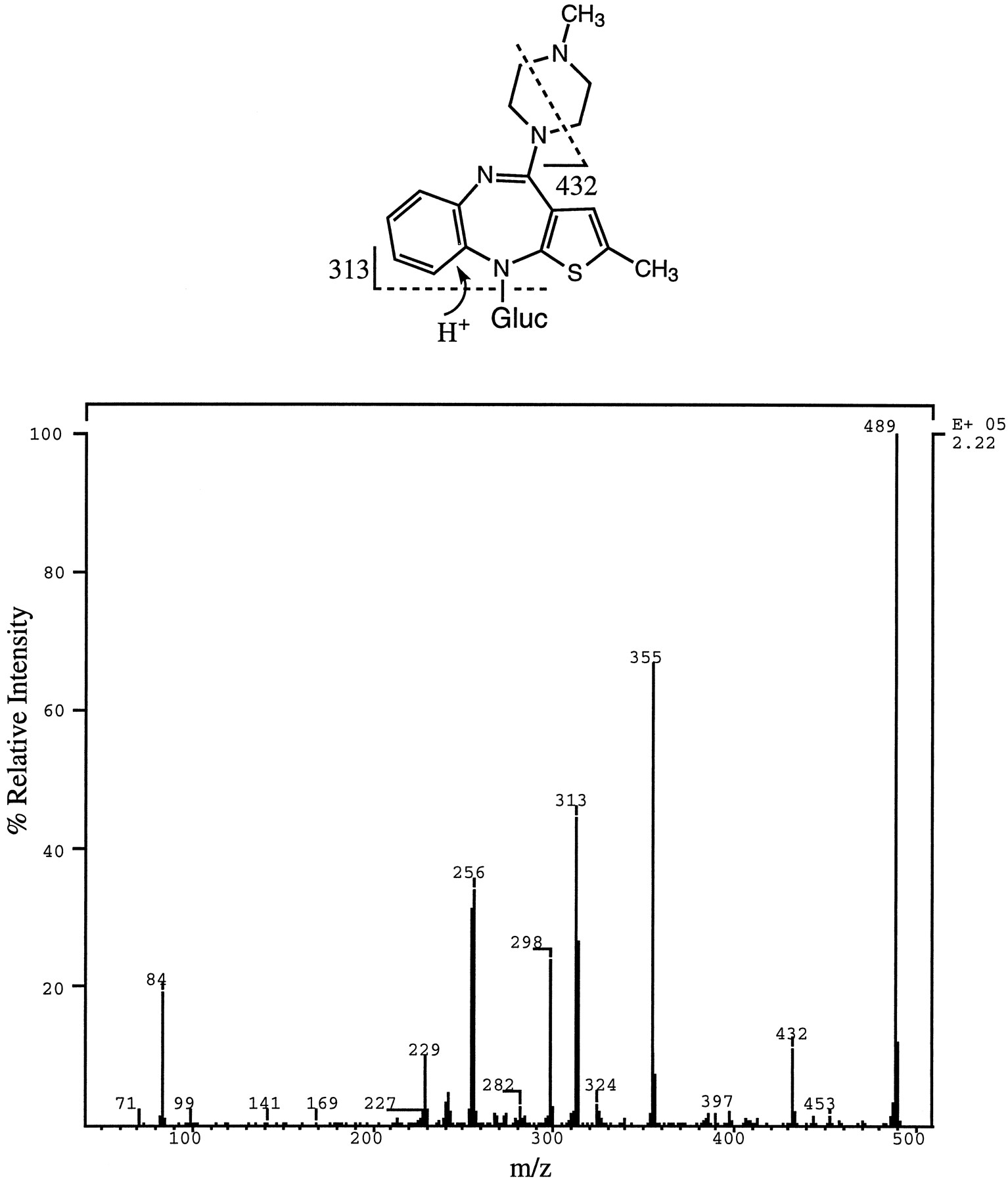
Product ion spectrum of m/z 489 (MH+) from the early eluting “isomer” of 10-N-glucuronide.
Possible structural assignment for two of the fragment ions is shown. Gluc, glucuronic acid.
Acid hydrolysis with either 3 or 6 N HCl solution resulted in the complete disappearance of the peak due to the conjugate and appearance of a new peak at the retention volume of OLZ. Treatment of a sample of the conjugate with 0.1 N HCl solution caused a 6% hydrolysis of the conjugate, whereas 1 and 2 N HCl solutions resulted in 60–75% hydrolysis of the abduct. OLZ remained stable under these conditions. There was no detectable hydrolysis of the conjugate after treatment of a sample of the conjugate with 0.1 or 1.0 N NaOH solution or upon treatment with β-glucuronidase from either H. pomatia orE. coli.
OLZ and another glucuronide conjugate of OLZ (4′-N-glucuronide) both have free secondary amines at position 10, and both formed the corresponding thiourea derivatives (data not shown). However, a sample prepared by isolating the peak with retention time at ∼16 min (fig. 4) failed to form such a derivative, indicating that, in the isolated metabolite, the secondary nitrogen at position 10 was substituted and that the metabolite might be glucuronidated at that position.
For proof of structure, the 10-N-glucuronide of OLZ was chemically synthesized. Conjugation of the secondary amine of OLZ with glucuronic acid was successful, albeit at a slow rate. The reaction also produced several isomeric glucuronide conjugates. The conjugate of interest was purified by HPLC, and its structure was determined using MS and NMR. The MS data and NMR spectrum were consistent with the proposed structure. The NMR spectrum of 10-N-glucuronide showed the following peak assignments: 2.19 d (s, 3H, 4′-CH3); 2.30 d (s, 3H, 2-CH3); 4.43 d (d, 1H, anomeric proton); 6.39 d (s, 1H, H-3); 6.85 d (dd, 1H, H-9 or H-6); 6.91 d (m, 1H, H-7 or H-8); 6.97 d (m, 1H, H-7 or H-8); 7.25 d (d, 1H, H-6 or H-9). Configuration at the anomeric proton of the glucuronyl moiety of the conjugate was determined to be β-glycosidic from the coupling constant of the H-1′ and H-2′ protons that was 9 Hz. When an aliquot of the synthetic standard was analyzed by HPLC, two peaks with identical retention times to the corresponding peaks obtained from the isolated metabolite were detected. The product ion spectra (precursor m/z 489) of the synthetic conjugates were also nearly identical to the corresponding spectra obtained on the conjugates isolated from urine. Although the NMR spectrum of the conjugate was obtained from a sample that contained the two “isomers,” there was only a single signal for the anomeric proton, as well as the aromatic protons of the conjugate.
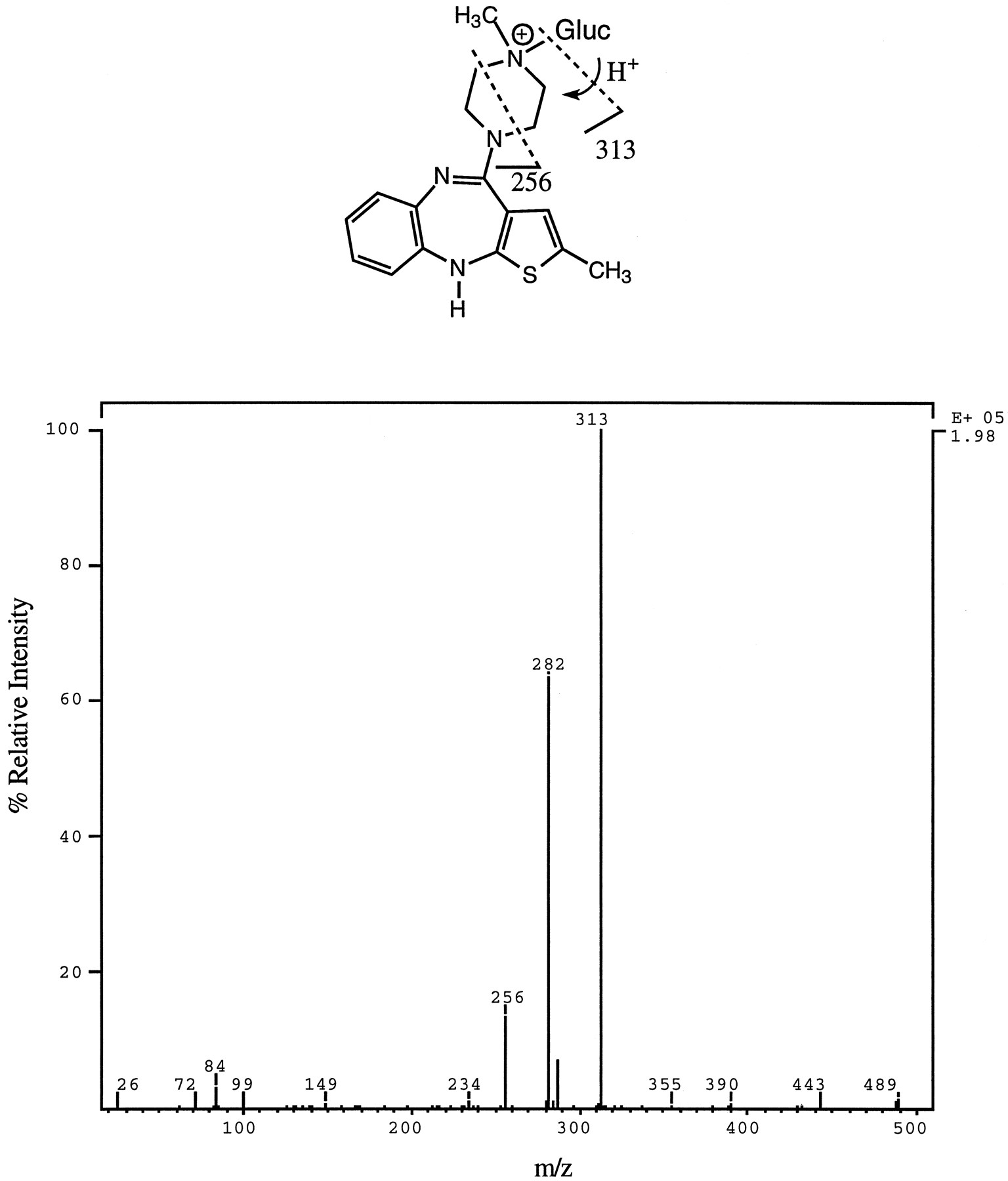
4′-N-Glucuronide—This metabolite eluted as a single peak (peak 5, fig. 6) and yielded a molecular ion ofm/z 489, which, upon CID, afforded the product ion spectrum shown in fig. 8. The fragment ion at m/z 313 (MH-176)+ was the base peak in the spectrum and indicated that the metabolite was a glucuronide adduct of OLZ. Two important differences between the product ion spectrum of 10-N-glucuronide (fig. 7) and 4′-N-glucuronide were that, in the spectrum of the latter, the relative intensity of them/z 313 fragment was much higher and that the fragment ion at m/z 432 was absent. The ion at m/z 432 results from loss of CH2⋕CH—NH—CH3 from the methyl piperazinyl portion of OLZ. The 4′-N-glucuronide, in contrast to the 10-N-glucuronide, afforded a thiourea derivative when treated with PITC (data not shown). From these data and the fact that the isolated metabolite had an identical HPLC retention time and MS/MS spectrum to that obtained from an authentic standard of 4′-N-glucuronide, it was concluded that the conjugate was the quaternary 4′-N-glucuronide. Unlike the 10-N-glucuronide, the 4′-N-glucuronide was susceptible to β-glucuronidase hydrolysis using the enzyme fromH. pomatia. The 4′-N-glucuronide was resistant to hydrolysis by 3 N HCl, and its enzyme hydrolysis was inhibited by β-saccharolactone.
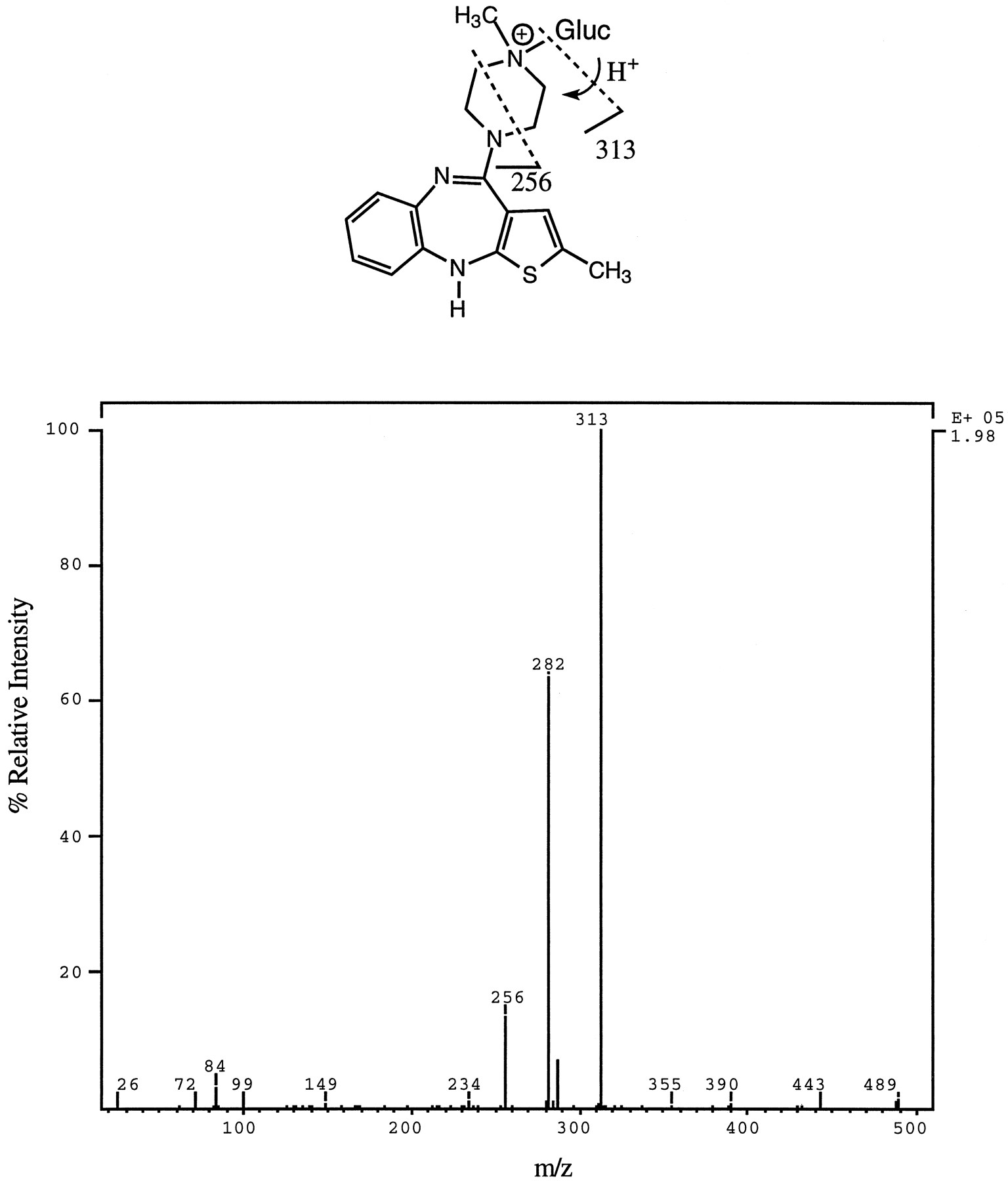
Product ion spectrum of m/z 489 (M+) of a metabolite isolated from urine (peak 4, fig. 6) and characterized as the 4′-N-glucuronide conjugate of OLZ.
Possible structural assignment for two diagnostic fragment ions is shown. The ion at m/z 282 is likely due to the combined loss of CH3NH2 and glucuronic acid. Gluc, glucuronic acid.
N-Desmethyl-2-carboxy OLZ—This metabolite eluted aspeak 1 in fig. 6, and its retention volume matched that of a standard prepared by oxidizing 4′-N-desmethyl-2-hydroxymethyl OLZ with chromium trioxide. The positive-ion spectrum of this metabolite yielded an MH+ion of m/z 329. The product-ion spectrum (fig.9) showed diagnostic losses of 17 Da (loss of NH3 from the piperazine ring) and 43 Da (loss of CH2⋕N—CH3 from the piperazine ring) to givem/z 312 and 286, respectively. The presence of an ion atm/z 70 (CH2⋕CH—NH—CH2—CH2+) also indicated that this compound was a demethylated analog of OLZ. MS analysis of the isolated metabolite after diazomethane derivatization afforded a protonated molecular ion of m/z 343, providing additional evidence for an N-desmethyl-2-carboxy OLZ structure for this metabolite.

Product ion spectrum of m/z 329 (MH+) of a metabolite isolated from urine (peak 1, fig. 6) and identified as N-desmethyl-2-carboxy OLZ.
Possible structural assignment for two of the fragment ions is shown. The fragment ion at m/z 312 is likely to be due to the loss of ammonia from the piperazine ring.
2-Carboxy OLZ—The isolated metabolite (peak 2, fig. 6) had the same retention time as a sample of authentic 2-carboxy OLZ. Product ion spectrum (precursor m/z 343) produced fragment ions atm/z 312 (loss of CH3NH2), 286 (loss of CH2⋕CH—NH—CH3), 243 (loss of the methyl piperazinyl moiety), and 84 [CH2⋕CH—N(CH3)—CH2—CH2+]. The product ion spectrum was identical to that obtained from the authentic standard.
2-Hydroxymethyl OLZ—The metabolite eluting as peak 1 in fig. 5 had the same HPLC retention time as the synthetic standard of 2-hydroxymethyl OLZ. CID on m/z 329 (MH+) yielded ions at m/z 298 (loss of CH3NH2), 272 (loss of CH2⋕CH—NH—CH3), 242 (loss of CH2⋕CH—NH—CH3 and H2CO), 229 (loss of the methyl piperazinyl moiety), and 84 [CH2⋕CH—N(CH3)—CH2—CH2+]. The product ion spectrum was nearly identical to that obtained from the synthetic standard.
N-Oxide OLZ—The metabolite eluting as peak 2 in fig. 5 had the same HPLC retention time as an authentic standard ofN-oxide OLZ. The CID spectrum of m/z 329 (MH+) produced characteristic ions at m/z 282 [loss of CH3—NH2(O)], 229 (scission of theN-oxide-N-methyl piperazine ring), and 213 (loss of the N-oxide N-methyl piperazinyl moiety). This product ion spectrum was nearly identical to that obtained from an authentic sample of N-oxide OLZ.
N-Desmethyl OLZ—This metabolite appeared as peak 3 in fig. 5 and had the same HPLC retention time as a sample of an authentic standard of N-desmethyl OLZ. The product spectrum of m/z 299 (MH+) showed diagnostic ions atm/z 282 (loss of NH3 from the piperazinyl moiety), 256 (loss of CH2⋕CH—NH2), 230 (loss of CH2⋕CH—NH—CH⋕CH2), 213 (loss of the piperazine ring), 198 (loss of the piperazine ring + CH4), and 70 (CH2⋕CH—NH—CH2—CH2+). The spectrum was consistent with that obtained from a synthetic standard of N-desmethyl OLZ.
Putative N-Oxide-2-carboxy OLZ Glucuronide—This putative metabolite was isolated using XAD-2 column chromatography (peak 3, fig. 6). MS analysis of the extract showed an apparent MH+ ion at m/z 535, the CID of which afforded a spectrum with a base peak at m/z 359. The peak atm/z 359, possibly resulting from neutral loss of 176 Da, is indicative of a glucuronide structure. Thus, on the basis of molecular weight and MS/MS fragment ions, the metabolite seems to be the glucuronide conjugate of N-oxide-2-carboxy OLZ.N-Oxide-2-carboxy OLZ was identified as a urinary metabolite of OLZ in rhesus monkeys (12).
Putative 2-Carboxy OLZ Glucuronide—This putative metabolite was isolated using XAD-2 column chromatography (peak 5, fig. 6), and upon MS analysis yielded an apparent protonated molecular ion ofm/z 519. CID of m/z 519 produced fragment ions atm/z 312, 286, and 84 (fig. 10), all of which were observed in the product ion spectrum of authentic 2-carboxy OLZ. On the basis of its MS and MS/MS spectrum, this metabolite was tentatively identified as the glucuronide adduct of 2-carboxy OLZ. However, the aglycone could not be released from the conjugate upon β-glucuronidase, acid, or base treatment.

Product ion spectrum of m/z 519 (apparent protonated molecular ion) of a metabolite present in the XAD-2 extract of urine and tentatively assigned the structure of 2-carboxy OLZ glucuronide.
Possible structural assignment for the major fragment ions is shown. Gluc, glucuronic acid.
OLZ—The parent compound was excreted in urine and is shown aspeak 4 in fig. 5. The product ion spectrum, as well as HPLC retention time obtained from the isolated material, were identical to those obtained from a reference standard of OLZ.
Plasma.
Plasma samples that remained after aliquots had been taken for the assay of OLZ were analyzed by HPLC with radiochemical detection. The plasma from individual time points for each volunteer was pooled, and the proteins were precipitated before analysis. Radioprofiles obtained for subjects 1 and 4 (fig. 11) were dominated by a single radioactive peak having the same HPLC retention volume as the 10-N-glucuronide. The 10-N-glucuronide peak accounted for ∼47% of the total radioactivity in the sample from subject 4. Although individual peaks were not isolated, mass spectral evidence was obtained that supported the presence of 10-N-glucuronide in these samples. The glucuronide was identified by multiple reaction monitoring-MS in which the transition of m/z 489 (MH+) to m/z 355, 313, 256, and 84 was monitored. There were no detectable radioactive components in the plasma sample from subject 3. OLZ was detected in all three subjects using the more sensitive GC/MS assay.

HPLC radiochromatograms obtained from extracts of a plasma sample from subject 4 (10 ml of plasma, from 0.33 to 144 hr postdose).
Feces.
Fecal samples from subjects 1, 3, and 4, collected during the first 120 hr after drug administration, were used for metabolite characterization. Aqueous homogenates of feces were initially extracted with methanol using an Eberbach shaker and then with Soxhlet extraction; on average, 62% (N = 8, coefficient of variation = 9%) of the radioactivity was extracted. A typical HPLC radiochromatogram obtained from a fecal extract is shown in fig.12. At least two radioactive components were present, and were isolated and analyzed by direct infusion electrospray MS. The major component that eluted as peak 1 in fig. 12 exhibited a protonated molecular ion of m/z 489, the MS/MS of which produced fragment ions at m/z 84, 255, and 354—ions also generated by the 10-N-glucuronide isolated from urine. The combination of HPLC retention time, MS/MS spectrum, and generation of OLZ after treatment with acid indicated that the major radiolabeled compound in feces was 10-N-glucuronide. The radioactive component eluting as peak 2 in fig. 12 was identified as OLZ by comparing its LC/MS/MS properties to those obtained from an authentic standard of OLZ. In some fecal extracts, a peak with the same retention time as 2-carboxy OLZ was present, but the amount of material isolated was insufficient for a conclusive MS identification of this metabolite.
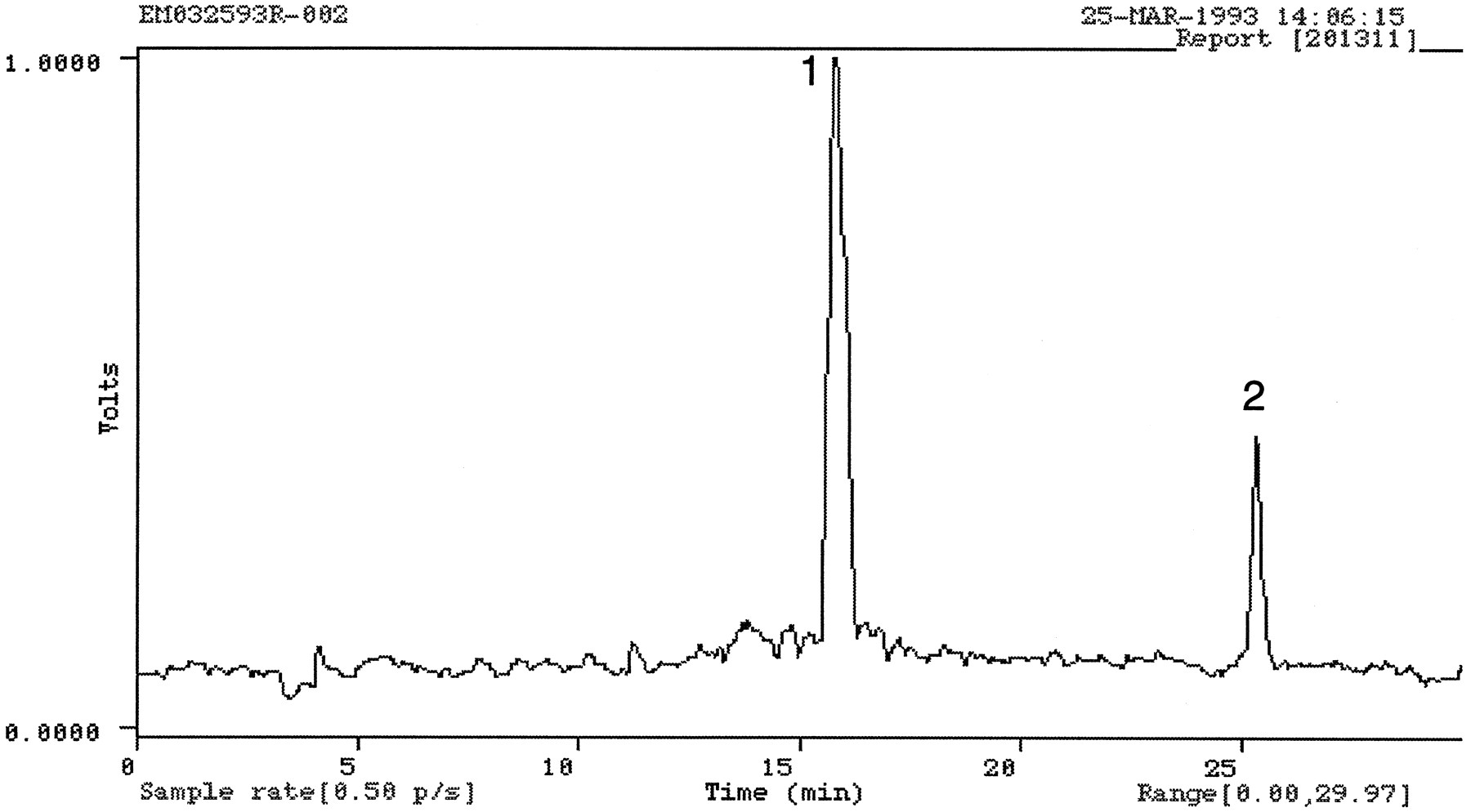
A typical HPLC radiochromatogram obtained from a fecal extract (subject 1, 48–72 hr).
Peaks 1 and 2 were identified as 10-N-glucuronide and OLZ, respectively.
Estimation of the Level of Metabolites.
Based on the percentage of radioactivity extracted into ethyl acetate (26%) and that remaining in the aqueous fraction (74%), the amount of OLZ and its metabolites in a single urine sample was estimated (table3). 10-N-Glucuronide accounted for ∼29% of the radioactivity and was the major radiolabeled component in this sample. The second most abundant species was OLZ itself, respresenting ∼18% of the radiocarbon.
Estimation of OLZ and its metabolites in a urine sample3-a
Combining data from the urine extracts obtained by XAD-2 resin and those from liquid-liquid extraction, an approximate estimate of the various metabolites is presented in table 4. Together with the parent compound excreted in urine, the identified metabolites accounted for ∼70% of the radioactivity or ∼40% of the dose.
Estimation of metabolite recovery in urine4-a
Similarly, the amounts of 10-N-glucuronide and OLZ were estimated in fecal samples from three subjects. On average, the 10-N-glucuronide conjugate accounted for ∼40% of the radioactivity, and OLZ represented ∼8% of the extracted radiocarbon. Because ∼30% of the dose was excreted in the feces, it is estimated that 8% of the dose was eliminated in feces as 10-N-glucuronide and another 2% as OLZ.
Discussion
After oral administration of OLZ to healthy subjects, at least 65% of the dose was absorbed as demonstrated by the radioactivity excreted in urine and metabolites detected in fecal samples (assuming systemic formation of fecal metabolites). Approximately 87% of the dose was recovered in the urine and feces mostly within 7 days of dosing. Renal excretion was the primary mode of radiocarbon elimination. Little or no radiocarbon was detected in red blood cells, suggesting that OLZ and/or its metabolites are mostly restricted to the plasma compartment of blood after a single dose. The pharmacokinetics of OLZ was characterized by a relatively long half-life (27 hr) and a large volume of distribution (22 liters/kg). All of the subjects in the present study were smokers. The half-life of OLZ in these subjects was similar to the median half-life (31 hr) observed in several subsequent studies with OLZ.2
At the time of maximum plasma concentration, the ratio of OLZ to radioactivity was 28%, indicating that OLZ is extensively metabolized in humans. Several lines of evidence suggest that a major metabolite of OLZ in humans is a tertiary N-glucuronide in which the glucuronic acid moiety is attached to the nitrogen at position 10 of the molecule (fig. 13). The ion at m/z 313 (MH-176)+ is present in the product ion spectrum obtained from the isomeric conjugate, 4′-N-glucuronide (fig. 8), as well as the spectrum from 10-N-glucuronide (fig. 7); however, the ratio of m/z 313 to 489 was much higher in the spectrum of the 4′-N-glucuronide conjugate. This observation reflects the ease with which the glycosidic bond is broken in the quaternary 4′-N-glucuronide to give protonated OLZ (m/z 313), compared with the glycosidic linkage in the tertiary 10-N-glucuronide. Additional evidence for a 10-N-glucuronide structure was obtained from a derivatization experiment with PITC. Although the 4′-N-glucuronide formed a thiourea analog, the 10-N-glucuronide failed to form this derivative, as would be expected for a compound lacking a free secondary amine. Moreover, the susceptibility of the 10-N-glucuronide conjugate to chemical hydrolysis was consistent with its tertiary N-glucuronide structure. The 10-N-glucuronide was hydrolyzed to the aglycone in the presence of 3 or 6 N HCl, whereas essentially no hydrolysis of the conjugate was achieved with either 0.1 N HCl or 1 N NaOH solutions. Finally, the 10-N-glucuronide isolated from urine had the same retention time and product ion spectrum to that obtained from a standard prepared by chemical synthesis.

Metabolic pathways of OLZ in humans.
Compound in brackets has not been identified. Gluc, glucuronic acid; bold arrow, a major pathway.
With optimal HPLC conditions, the 10-N-glucuronide conjugate eluted as two distinct peaks, which raises the possibility that there are two isomers of the 10-N-glucuronide. Because in the NMR spectrum of the conjugate a single signal was observed for the anomeric proton, it is unlikely that the two peaks correspond to the α- and β-anomers of the conjugate. The two peaks also yielded product ion spectra that were nearly identical to each other. On standing, one “isomer” tended to be converted to the other. However, from the present data, it is not clear what kind of isomeric relationship the two conjugates might have, other than to state that the conjugates are not regioisomers.
In the urine, at least 13 radioactive HPLC peaks were detected, of which 10 have been identified as OLZ and its metabolites. Two major urinary components were identified as the 4′-N- and 10-N-glucuronides of OLZ. Oxidative metabolism on the allylic methyl group resulted in 2-hydroxymethyl and 2-carboxy derivatives of OLZ. The methyl piperazine moiety was also subject to oxidative attack, giving rise to the N-oxide andN-desmethyl metabolites. Other metabolites, including theN-desmethyl-2-carboxy and possiblyN-oxide-2-carboxy derivatives resulted from metabolic reactions at both the 4′ nitrogen and 2-methyl groups. The precursor ofN-desmethyl-2-carboxy OLZ,N-desmethyl-2-hydroxymethyl OLZ (MR = 314, fig. 13) was not positively identified in the present study; however, a plasma component with an apparent protonated molecular ion at m/z 315 was detected by LC/MS. As shown in table 4, the major urinary component accounting for ∼13% of the dose was 10-N-glucuronide. The parent compound was the second most abundant species in urine. The compounds identified in urine accounted for ∼70% of the urinary radiocarbon or 40% of the administered dose.
In fecal extracts, the only significant radioactive HPLC peaks were due to 10-N-glucuronide and OLZ itself, representing ∼8 and 2% of the administered dose, respectively. If allowances are made for the extraction efficiency (62%), the corresponding amounts would be 12% and 2.4%. It is estimated, therefore, that a majority (perhaps 21–25%) of an acute dose of OLZ is eliminated via the 10-N-glucuronidation pathway by way of urine and feces.
Semiquantitative data obtained from plasma samples of subjects administered [14C]OLZ suggest that the main circulating metabolite is 10-N-glucuronide. Data obtained from the quantitative determination of OLZ and two of its metabolites in plasma samples from patients on multiple dosing show that OLZ itself is the principal circulating entity, whereas levels of 10-N-glucuronide are ∼44% those of OLZ.3 This result, coupled with the information obtained from the present mass balance study, tends to indicate that, whereas 10-N-glucuronide is a major circulating metabolite, the parent compound is the largest single component in plasma, at least under conditions of chronic dosing.
In addition to 10-N-glucuronide, N-desmethyl,N-oxide, and 2-hydroxymethyl OLZ were identified, using LC/MS/MS techniques (data not shown), in plasma from patients administered multiple doses of OLZ. In vitro studies using human liver microsomal preparations indicate that the formation ofN-desmethyl and 2-hydroxymethyl is catalyzed, respectively, by the cytochrome P450 isozymes CYP1A2 and CYP2D6 (13). TheN-oxide metabolite was found to be a product of flavin-containing monooxygenase.
Although similarities in the metabolic fate of OLZ in animals [mouse, rat, and dog (12, 14)] and humans include the 2-alkyl hydroxylation,N-dealkylation and N-oxidation reactions, two significant differences can be noted. First, direct glucuronidation (producing mainly 10-N-glucuronide) and, to a lesser extent, 4′-N-glucuronide, is an important pathway in humans, with apparently no evidence for metabolites arising from aromatic hydroxylation in any of the biological fluids studied. Second, in mice, rats, and dogs, the major metabolites result from oxidative attack on the benzene ring of OLZ. Thus, the major metabolite in dog urine was 7-hydroxy-N-oxide OLZ, whereas in the mouse it was the glucuronide of 7-hydroxy OLZ (12). Likewise, in rats, the major urinary metabolites were 2-hydroxymethyl OLZ, the glucuronide of 7-hydroxy OLZ and possibly the glucuronide of 7-hydroxy-N-desmethyl OLZ. The 10-N-glucuronide conjugate was absent in animal samples, with the exception of a trace amount in dog urine.
It is also instructive to compare the human biotransformation of OLZ with that of clozapine, a drug with a similar structure to OLZ. OLZ and clozapine form common metabolites as a result of modifications at the methyl piperazine ring (i.e. N-oxide,N-desmethyl, and 4′-N-glucuronide). But, the similarity in their metabolic fate ends at the level of these metabolites. As discussed previously, the metabolic disposition of OLZ is dominated by direct glucuronidation. Also, the presence of the allylic methyl group in OLZ makes OLZ metabolically quite distinct from clozapine. Several OLZ metabolites (including 2-hydroxymethyl, 2-carboxy, and N-desmethyl-2-carboxy OLZ) owe their formation to oxidation at the 2-CH3 group. On the other hand, clozapine is known to undergo aromatic oxidation at positions 2 and 7 of the two benzene rings (15-17). In addition, clozapine forms metabolites that result from the replacement of the chlorine attached to the benzene ring by methylthio and hydroxyl groups. Thus, replacement of the chlorobenzene ring with a methyl thiophene moiety seems to make a considerable effect on the metabolic disposition of this class of compounds.
In summary, OLZ was well absorbed and extensively metabolized and unchanged OLZ; its metabolites were excreted mainly via the urine. The metabolism of OLZ was characterized by the presence of several pathways and apparent absence of a single dominant oxidative pathway.
Acknowledgments
We thank Douglas O’Bannon for synthesizing [14C]OLZ, Larry Spangle for the NMR data of 10-N-glucuronide, and Ronald Franklin for helpful discussions during the course of these studies.
Footnotes
-
Send reprint requests to: Dr. Kelem Kassahun, Department of Drug Metabolism, Lilly Research Laboratories, Eli Lilly and Company, Lilly Corporate Center, Mail Drop 0825, Indianapolis, IN 46285.
-
↵2 R. Bergstrom et al., unpublished data.
-
↵3 E. Mattiuz and K. Kassahun, unpublished data.
- Abbreviations used are::
- OLZ
- olanzapine
- 5-HT
- serotonin
- PITC
- phenyl isothiocyanate
- AUC
- area under the plasma concentration-time curve
- TEA
- triethylamine
- CID
- collision-induced dissociation
- Received May 23, 1996.
- Accepted October 17, 1996.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}