


Abstract
The human clearance of omeprazole and lansoprazole is conducted primarily by the hepatic cytochrome P450 (CYP) system. Efficacy data indicate few differences between these two drugs, but they may exhibit discrete drug interaction profiles. To compare the potency and specificity of these drugs as inhibitors of CYP isoforms, we performedin vitro studies with human liver microsomal preparations. Both drugs were potent, competitive inhibitors of CYP2C19, as measured by the conversion of S-mephenytoin to 4-hydroxymephenytoin (ki = 3.1 ± 2.2 μM for omeprazole,Ki = 3.2 ± 1.3 μM for lansoprazole). For omeprazole, the highest concentration at which >70% inhibition of CYP2C19 was observed with no significant inhibitory effect on other isoforms was at least 20 times greater than Ki . Both drugs were competitive inhibitors of CYP2C9-catalyzed conversion of tolbutamide to 4-hydroxytolbutamide (Ki = 40.1 ± 14.8 μM for omeprazole, Ki = 52.1 ± 1.4 μM for lansoprazole) and were noncompetitive inhibitors of CYP3A-catalyzed conversion of dextromethorphan to 3-methoxymorphinan (Ki = 84.4 ± 4.0 μM for omeprazole, Ki = 170.4 ± 7.1 μM for lansoprazole). Lansoprazole was at least 5 times more potent (Ki = 44.7 ± 22.0 μM) than omeprazole (ki = 240.7 ± 102.0 μM) as an inhibitor of CYP2D6-mediated conversion of dextromethorphan to dextrorphan. No inhibition of CYP1A2, assessed by measuring the conversion of phenacetin to acetaminophen, was noted. Our data suggest that whereas the inhibitory profiles of these two drugs are similar, lansoprazole may be the more important in vitro inhibitor of CYP2D6. Since its inhibition is very potent and has a broad “window of selectivity,” omeprazole seems to be a useful, selective inhibitor of CYP2C19.
Omeprazole and lansoprazole are the only two H+,K+-ATPase (proton pump) inhibitors currently approved in the United States. At present, little direct comparative research has been performed that might assist physicians or researchers in distinguishing between these two drugs. In this respect, differences in the ability of these two drugs to cause CYP1-based drug interactions represent an important element in physician’s prescribing and may provide useful tools that researchers might exploit to study human CYP metabolism.
Omeprazole and lansoprazole are substituted benzimidazoles that suppress gastric acid secretion by inhibiting H+,K+-ATPase in the secretory membrane of gastric parietal cells. These drugs are considered the most effective tools to treat reflux esophagitis and Zollinger-Ellison syndrome (1,2). Both drugs exhibit polymorphic metabolism. The poor metabolizer phenotype of both drugs is highly correlated with the poor metabolizer phenotype of S-mephenytoin (3-5). This phenotype is present in 12–23% of Asian populations, 2% in Black Americans (6), and 2–6% of Caucasian populations (7, 8).
Among the proton pump inhibitors, omeprazole is the most extensively characterized both in vivo and in vitro in terms of metabolic pathways and drug–drug interactions. Omeprazole has been reported to inhibit the metabolism of drugs, such as diazepam (9-11), phenytoin (11-13), and clarithromycin (14). High concentrations have been shown to induce CYP1A2 activity (15, 16), but to have little or no effect on the pharmacokinetics of a variety of drugs metabolized by the CYP system, including estradiol, quinidine, lidocaine, erythromycin and prednisolone or metoprolol (17), propranolol (18), orS-warfarin (19). Omeprazole is metabolized in the liver to two principal metabolites: 5-hydroxy-omeprazole and omeprazole sulfone through S-mephenytoin hydroxylase (CYP2C19) and CYP3A, respectively (20). It is clear that 5-hydroxylation plays a major role in the disposition of omeprazole in vivo (4, 21, 22). The mechanism of the documented interactions seems to be by competitive inhibition of CYP2C19, but the ability of omeprazole to inhibit other important specific CYP isoforms in vitro has not been tested thoroughly.
Furthermore, it is clear that omeprazole can inhibit CYP2C19 in vitro, but the quantitative specificity of omeprazole as anin vitro tool to inhibit CYP2C19 has not been documented. This seems important, because useful inhibitory probes for many other clinically important CYP isoforms have been proposed and are available. These include furafylline for CYP1A2 (23), sulfaphenazole for CYP2C9 (24), quinidine for CYP2D6 (25), diethyldithiocarbamate for CYP2E1 (26), and troleandomycin for CYP3A4 (27).
The two major metabolites of lansoprazole found in plasma are 5-hydroxy-lansoprazole and lansoprazole sulfone (28). The metabolism of this drug is different from that of omeprazole in that, whereas sulfoxidation seems to be mediated by CYP3A, hydroxylation seems to be mediated by several isoforms, including both CYP3A and CYP2C19 (29). In a recent study, Pearce et al. (30) suggested that 5-hydroxylation of lansoprazole is mediated by two kinetically distinct enzymes. At pharmacologically relevant concentrations, it is primarily catalyzed by CYP2C19; at higher concentrations, it is highly correlated with CYP3A activity. Based on the limited information about drug–drug interactions involving lansoprazole, it does not seem to have the same profile of drug interactions as omeprazole. In contrast with omeprazole, lansoprazole seems to have little or no inhibitory effect on the metabolism of diazepam (31) or phenytoin (32) in human subjects.
Inhibitory effects of these drugs on metabolism of isoform-specific substrates of CYP have not been described or compared. This seems important because there is a great deal of precedent in the literature for drugs inhibiting CYP isoforms for which they are not substrates. For example, chlorpheniramine (33), halofantrine (34), quinidine (25,35), methadone (36), and fluoxetine (37) all inhibit the metabolism of CYP2D6, but none of them are significantly metabolized by it. For these reasons, we studied the potency and specificity of omeprazole and lansoprazole as inhibitors of the principal CYP isoforms present in human liver microsomes.
Materials and Methods
Chemicals and Reagents.
Phenacetin, acetaminophen, tolbutamide, chlorpropamide, dextromethorphan, chlorzoxazone, G6P, G6PDH, NADP, and the disodium salt of EDTA were purchased from Sigma Chemical Co. (St. Louis, MO).S-mephenytoin, 6-hydroxychlorzoxazone, and 4-hydroxytolbutamide were purchased from Ultrafine Chemicals (Manchester, UK). Dextrophan and 3-methoxymorphinan were purchased from Hoffmann-La Roche, Inc. (Nutley, NJ). Omeprazole was a generous gift from Dr. Tommy Andersson (Clinical Pharmacology, Astra Hässle AB, Mölndal, Sweden), and lansoprazole was a gift from Dr. S.-G. Shin (Clinical Pharmacology Unit, Seoul National University College of Medicine, Seoul, Korea). N-(4-Hydroxyphenyl) butamide was kindly provided by John Strong, Ph.D. (Division of Clinical Pharmacology, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Rockville, MD).
Human Liver Microsomes.
The human liver samples used for preparation of microsomes were medically unsuitable for liver transplantation and frozen at −80°C within 3 hr of the cross-clamp time. The liver donor characteristics, procedure for preparation of microsomal fractions we used, and microsomal CYP contents have been previously described by Harriset al. (38). The microsomes were resuspended to a protein concentration of 5–12 mg/ml in reaction buffer (0.1 M Na+and K+ phosphate:1.0 mM EDTA:5.0 mM MgCl2; pH 7.4) and were stored at −80°C until used. Protein concentrations were determined using the method described by Pollard et al.(39).
Enzyme Assays.
Substrates with high specificity for CYP isoforms were selected: phenacetin O-deethylation for CYP1A2 (40), tolbutamide 4-methylhydroxylation for CYP2C9 (41), S-mephenytoin 4-hydroxylation for CYP2C19 (42, 43), dextromethorphanO-demethylation for CYP2D6 (25, 44), dextromethorphanN-demethylation for CYP3A (45), and chlorzoxazone 6-hydroxylation for CYP2E1 (46). To measure the effects of omeprazole and lansoprazole on the metabolism of these substrates, we incubated human liver microsomes with increasing concentrations of the inhibitor and substrate using incubation conditions, specific to each isoform, that were linear for time and protein concentration. Rates of production of each metabolite were quantified by using the ratio of AUC of the metabolite to AUC of each internal standard.
Instruments used for HPLC were controlled by a Waters (Milford, MA) Millennium 2010 Chromatography Manager and included a Waters model 510 or 600 HPLC pump, Waters 710B or 717 Autosampler, Waters 490 or 484 UV detector, and Spectrovision FD-300 Dual Monochromator Fluorescence Detector (Groton Technology, Inc., Concord, MA).
For each incubation, omeprazole and lansoprazole were dissolved in ethanol (10 mg/ml) and sequentially diluted with water to prepare the concentrations used. The final concentrations of ethanol for each assay were as follows: 0.18% and 0.17% for the CYP2C19 assay; 0.35% and 0.37% for the CYP2C9 assay; and 0.69% and 0.74% for the CYP2D6 and CYP3A assays. Stock solutions of S-mephenytoin for the CYP2C19 assay were dissolved in methanol (10 mg/ml) and sequentially diluted with water, and the methanol concentration in the incubation mixture was 0.13%. For the CYP1A2 and CYP2E1 assays, any alcoholic component was eliminated by dry-down and reconstitution with 0.1 M phosphate buffer before starting the incubation.
S-Mephenytoin Assay (CYP2C19).
After 5 min of preincubation, reactions were started by adding microsomal protein (final concentration = 0.5 mg/ml) to an incubation mixture (final volume = 250 μl) that consisted of 5 mM potassium phosphate buffer, 25 μl of S-mephenytoin, in final concentrations ranging from 5 to 60 μM, and NADPH-regenerating system (0.5 mM NADP, 2.0 mM G6P, 0.4 units/ml G6PDH, 0.1 mM EDTA, and 4.0 mM MgCl2). The range of concentrations of omeprazole and lansoprazole used were from 0.25 to 50 μM. Incubations were terminated after 120 min of incubation by placing the reaction tubes on ice and adding 100 μl of acetonitrile. After adding internal standard (50 μl of 20 μg/ml phenytoin), 3 ml of methylene chloride was added. The organic layer was dried by speed vacuum after centrifuging at 2000 rpm for 5 min in a Beckman J-6M centrifuge (JS4.0 rotor). Dried pellets were reconstituted with 250 μl of mobile phase, and 100 μl was injected into the HPLC apparatus. Phenytoin and 4-hydroxymephenytoin were separated using isocratic elution from a Microsorb C18 column at a flow rate of 0.7 ml/min in a mobile phase of 26% acetonitrile in 0.05 M potassium phosphate (pH 4.0), and detected using a UV wavelength of 211 nm. Standard curves for 4-hydroxy-mephenytoin were linear between 0.1 and 10 μg, and the interday coefficients of variation were between 5.6 and 15.3%. The range of KM and Vmax forS-mephenytoin 4-hydroxylation were from 13.6 to 56.5 μM and from 150 pmol/mg protein/min to 220 pmol/mg protein/min, respectively, in the three microsomal preparations used (HL2, HL4, and HL9).
Dextromethorphan Assay (CYP2D6 and CYP3A).
Dextromethorphan O-demethylation andN-demethylation were evaluated essentially according to the method described by Broly et al. (25). After 30 min of incubation with human hepatic microsomes (final protein concentration = 80 μg/ml) in 80 mM sodium phosphate buffer (pH 7.4) with NADPH-regenerating system (1.3 mM NADP, 3.3 mM G6P, 0.4 units/ml G6PDH, and 3.3 mM MgCl2) and increasing dextromethorphan concentrations ranging from 2.5 to 75 μM. The range of concentrations of omeprazole and lansoprazole used were from 2.5 to 200 μM. Incubations were terminated by placing on ice and adding 20 μl of 60% perchloric acid and internal standard (40 μl of 16 μM levallorphan). The tubes were then centrifuged in an Eppendorf model 5415C microfuge (Brinkman Instruments, Westbury, NY) at 14,000 rpm for 5 min, and 150 μl of supernatant was used for analysis. Dextromethorphan, dextrorphan, 3-methoxymorphinan, and levallorphan were separated on a DuPont Zorbax SB-Phenyl 150 × 4.6 mm column at a flow rate = 0.5 ml/min, using a mobile phase consisting of 5.7 mM monobasic potassium phosphate, 23% acetonitrile, 20% methanol, and 0.114% triethylamine (pH 4.0). Detection was conducted using an excitation wavelength of 200 nm and an emission wavelength of 304 nm. Standard curves for dextrorphan and 3-methoxymorphinan were linear between 40 and 830 ng/ml, and the interday coefficients of variation were between 1.4 and 11.6% and between 2.6 and 8.3%, respectively. The range of KM and Vmaxfor dextromethorphan O-demethylation were from 8.6 to 15.5 μM and from 71 to 480 pmol/mg protein/min in the three microsomal preparations used (HL2, HL6, and HL9). To test the possibility of mechanism-based inhibition, human liver microsomes and regeneration system were preincubated for 0, 15, 30, and 60 min with or without inhibitors (25 μl of 10, 50, and 100 μM) and then incubation was started by adding dextromethorphan.
Tolbutamide Assay (CYP2C9).
The incubation method for tolbutamide hydroxylation was the same as that for the dextromethorphan assay (CYP2D6 and CYP3A), except that the incubation time was 1 hr. Concentrations used for incubations were from 5 to 50 μM for tolbutamide and from 10 to 100 μM for omeprazole and lansoprazole. Chlorpropamide (0.1 mg/ml, 15 μl) was used as an internal standard. After incubation, 1 ml of ethylether was added, protein was precipitated by centrifugation (15,000 rpm for 5 min in the Eppendorf model 5415C microfuge), and the organic phase was dried by speed vacuum. Samples were resuspended in 100 μl of mobile phase, and then 75 μl was used for HPLC analysis. Chromatography was conducted by a modification of the method of Relling et al. (41). Chlorpropamide and tolbutamide were separated on an Alltech Spherisorb ODS-2, 5-μm (250 × 4.6 mm) column with Waters Guard-Pak Novapak C18 guard column, in a mobile phase consisting of 40% acetonitrile in 10 mM ammonium acetate at pH 5.4, using a flow rate of 0.7 ml/min. Detection was by UV absorbance at 230 nm. Standard curves for 4-hydroxytolbutamide were linear between 0.07 and 2.87 μg/ml, and the interday coefficients of variation were between 6.3 and 15.0%. TheKM and Vmax for 4-hydroxylation of tolbutamide were 7.0 μM and 4 nmol/mg protein/min for HL9, and 29 μM and 22 nmol/mg protein/min for HL2.
Phenacetin Assay (CYP1A2).
Phenacetin O-deethylation was measured after 30-min incubations with human hepatic microsomes. The range of concentrations of omeprazole and lansoprazole used were from 5 to 25 μM. After 3 min of preincubation, reactions were started by adding microsomal protein (final protein concentration = 1 mg/ml) to incubation mixtures that consisted of 0.1 M phosphate buffer (pH 7.4), phenacetin (17–140 μM), and NADPH-regenerating system (1.2 mM NADP, 11.1 mM G6P, 1.3 units/ml G6PDH, 1.0 mM EDTA, and 5.0 mM MgCl2). Incubations were terminated by placing on ice and adding 1.0 ml of acetonitrile with internal standard, N-(4-hydroxyphenyl)butamide (2 μg/ml). After removing the precipitated protein by centrifugation at 4,000 rpm for 10 min in a Beckman J-6M centrifuge (JS4.0 rotor), the supernatants were dried by nitrogen speed vacuum and reconstituted in ethanol. Suspensions were again centrifuged at 14,000 rpm for 5 min at 4°C (Eppendorf model 5415C microfuge) after mixing with 1 ml of acetonitrile, and supernatants were dried on speed vacuum a second time. Finally samples were reconstituted in 250 μl of 50% methanol in H2O, and 25 μl was injected into the HPLC apparatus. Acetaminophen and phenacetin were separated on an Alltech Spherisorb ODS-2, 5-μm (250 × 4.6 mm) column with Waters Novapak C18 Guard-Pak in a mobile phase consisting of 35% methanol with 5% acetonitrile and 6 mM ammonium acetate at pH 5.0 at a flow rate of 0.5 ml/min. Detection was by UV absorbance at 243 nm. Standard curves for acetaminophen were linear over the tested range, from 0.2 to 8.0 μg/ml, and the intraday and interday coefficients of variation were within 15%. The range of KM andVmax for phenacetin O-deethylation were 47.5 μM and 9.4 pmol/mg protein/min in HL4, and 198.5 μM and 128.8 pmol/mg protein/min in HL6.
Chlorzoxazone Assay (CYP2E1).
The rate of 6-hydroxychlorzoxazone formation from chlorzoxazone was evaluated through the method described by Kim et al. (46) with slight modification. The range of concentrations of omeprazole and lansoprazole used were from 2.5 to 100 μM at a chlorzoxazone concentration of 20 μM.
Analysis of Data.
Kinetic parameters were determined by linear regression of unweighted data using the Excel (Microsoft Corp., Redmond, WA) statistical package. KM and Vmaxvalues for each reaction were obtained by Lineweaver-Burk plot or Hanes plot. The mechanism of inhibition was decided through graphical analysis of primary Lineweaver-Burk and Dixon plots.Ki values were determined by both Dixon plot and secondary plot using the slope of the primary Lineweaver-Burk plots (“KM/Vmax” ratio as a function of inhibitor concentration). For Ki determination, mean value and standard deviation were obtained from at least two different human liver microsomal preparations.
Results
CYP2C19 Activity.
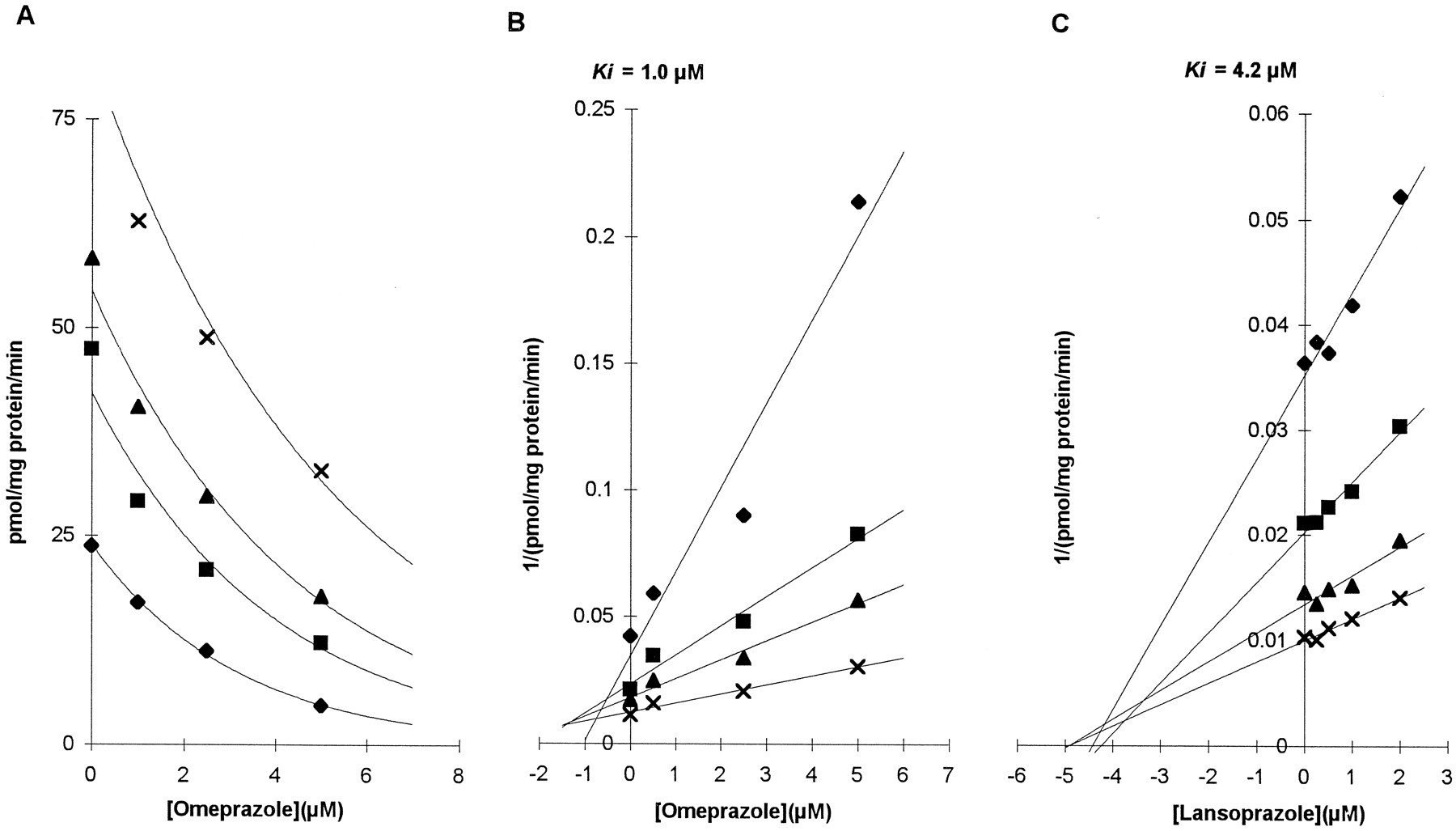
Both omeprazole and lansoprazole showed potent inhibitory effects on the formation of 4-hydroxymephenytoin in two microsomal preparations (HL4 and HL9 for omeprazole and HL2 and HL9 for lansoprazole). The inhibition observed occurred over a broad range of substrate and inhibitor concentrations, but was more prominent at lower concentrations of substrate (fig. 1A). With high inhibitor concentrations >10 μM, metabolite formation was consistently reduced to undetectable levels. Primary Lineweaver-Burk plots revealed common intercepts at the ordinate consistent with pure competitive inhibition. By linear regression using the competitive model of Dixon plots, estimates of the mean Ki for microsomal preparations were 1.0 μM for omeprazole and 3.4 ± 1.1 μM for lansoprazole. Representative graphs of omeprazole and lansoprazole inhibition are presented in fig. 1, B andC.

Effect of omeprazole on CYP2C19-mediated S-mephenytoin 4-hydroxylation.
Data used for graphs are from assays with microsomal preparation HL4 (omeprazole) and HL9 (lansoprazole). S-mephenytoin concentrations used were 10 (⧫), 20 (▪), 30 (▴), and 60 (×) μM for omeprazole, and 5 (⧫), 10 (▪), 20 (▴), and 40 (×) μM for lansoprazole. At concentrations of omeprazole of 10 μM or more, metabolite formation was below the limit of quantitation of the assay. (A) Plot of omeprazole concentration against the rate of production of 4-hydroxymephenytoin from S-mephenytoin substrates. (B) Dixon plot of omeprazole inhibition. (C) Dixon plot of lansoprazole inhibition.
CYP2C9 Activity.
Both of the drugs showed inhibitory effects on tolbutamide 4-methylhydroxylation (fig. 2). The mechanism of inhibition by both drugs seemed to be competitive. Lineweaver-Burk plots showed a common intercept at the y-axis, and the common intercepts of Dixon plots were left of the y-axis. The respective Ki ’s from Dixon plots were 40.1 ± 2.10 μM for omeprazole and 49.1 ± 23.08 μM for lansoprazole (HL2 and HL9).
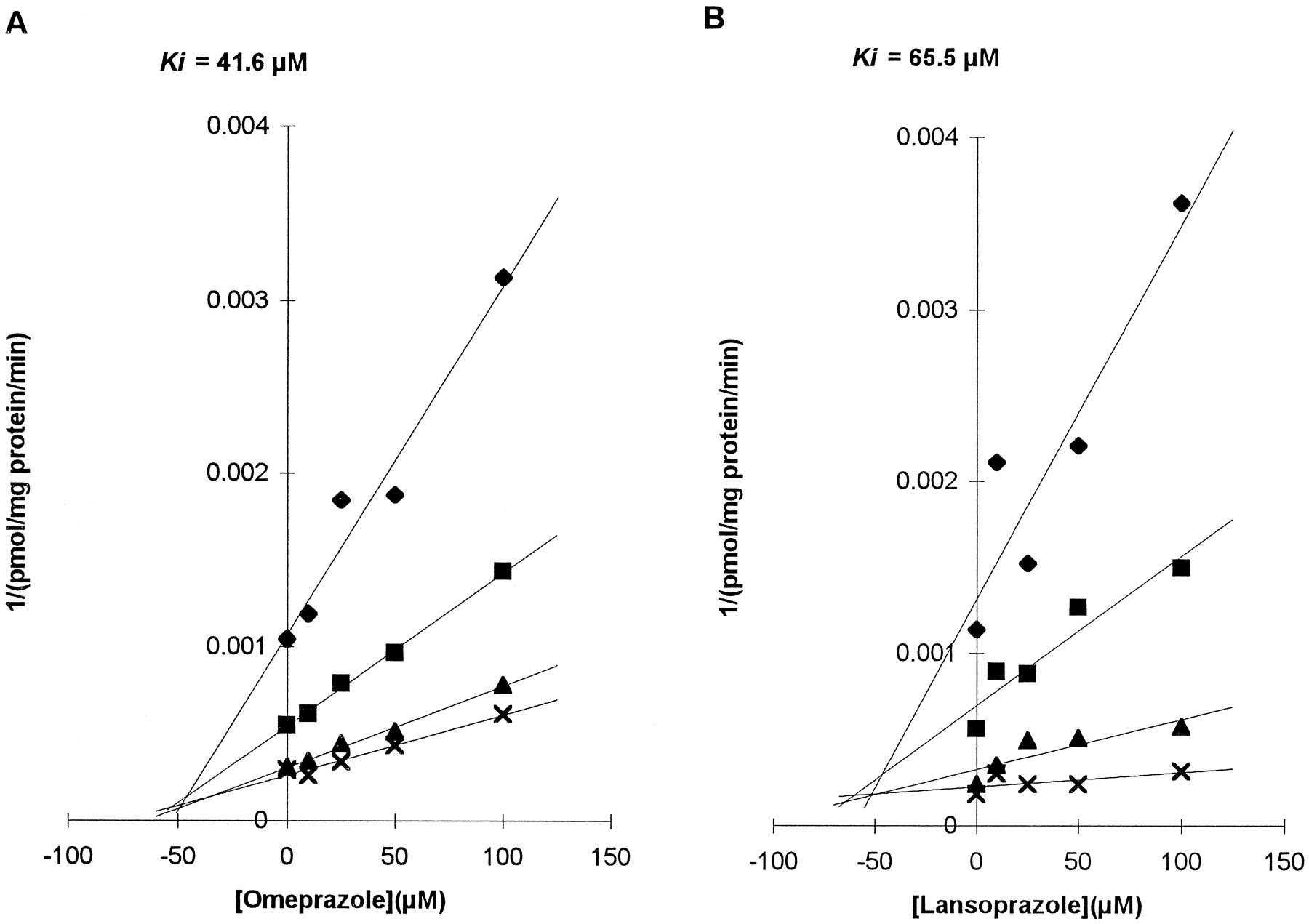
Effects of omeprazole and lansoprazole on CYP2C9-mediated tolbutamide 4-methylhydroxylation.
Dixon plots of omeprazole (A) and lansoprazole (B) are presented. Data are from assays with the same microsomal preparation (HL2). Concentrations of tolbutamide used were 5 (⧫), 10 (▪), 25 (▴), and 50 (×) μM.
Dextromethorphan Assay (CYP2D6 and CYP3A Activity).
The effect of omeprazole and lansoprazole on dextromethorphanO-demethylation to dextrorphan (CYP2D6) andN-demethylation to 3-methoxymorphinan (CYP3A) were investigated using microsomal preparations HL2 and HL9 (and HL6 for lansoprazole). Compared with omeprazole, lansoprazole showed a relatively strong inhibitory effect on dextromethorphanO-demethylation (Ki = 240.7 ± 102.03 μM for omeprazole, Ki = 44.7 ± 22.02 μM for lansoprazole by a secondary Lineweaver-Burk plot). The pattern of inhibition by lansoprazole was compatible with mixed inhibition, and that of omeprazole was fit to pure competitive type by graphical analysis of Lineweaver-Burk, Dixon, and Hanes-Woolf plots. Representative graphs showing the difference in inhibitory potency are presented in fig. 3 (A and B).
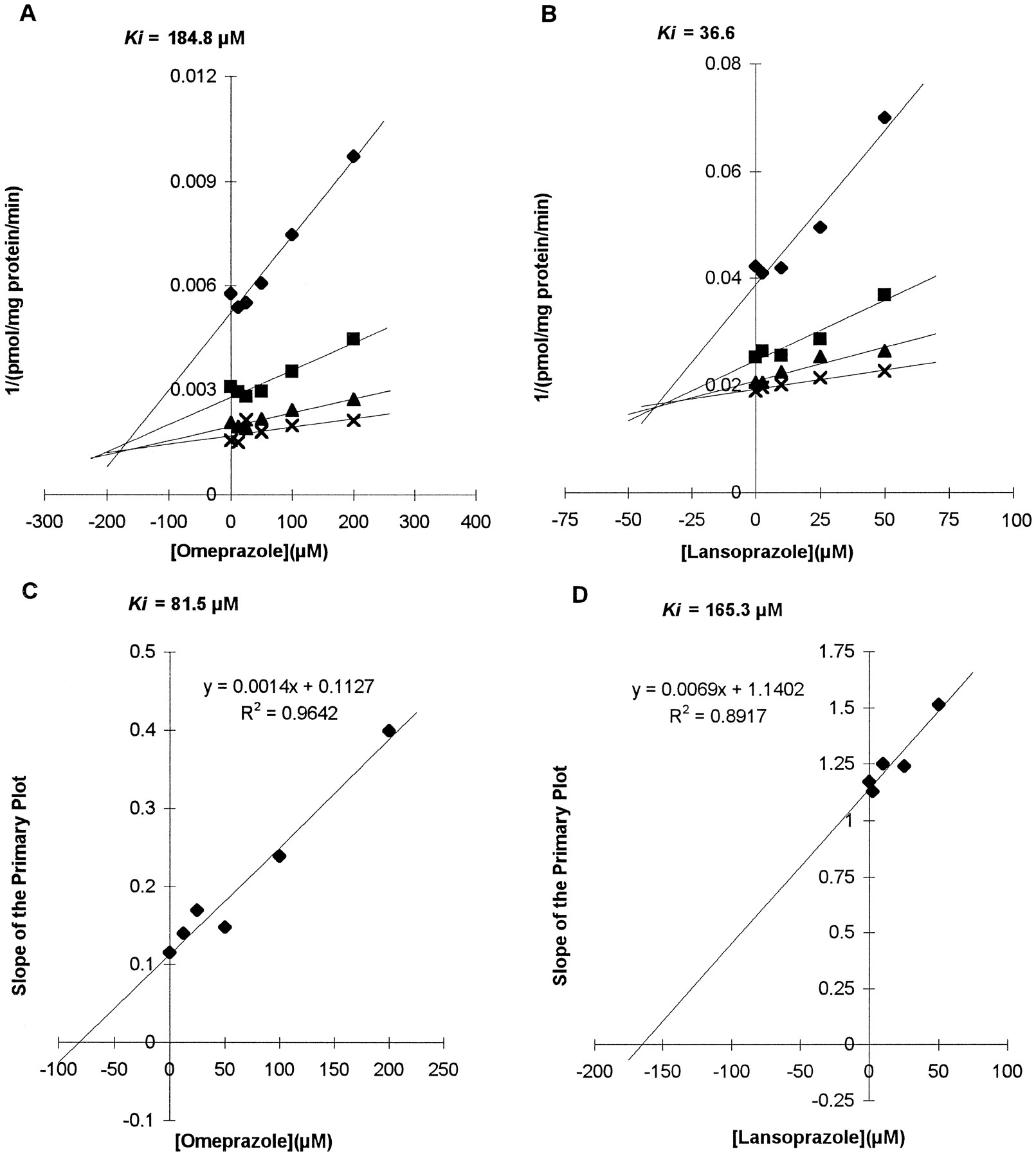
Effects of omeprazole and lansoprazole on CYP2D6-mediated dextromethorphan O-demethylation and CYP3A-mediated dextromethorphan N-demethylation.
(A) Dixon plot obtained from the assay with microsomal preparation HL9 and omeprazole showing effects on CYP2D6. Concentrations of dextromethorphan used were 2.5 (⧫), 7.5 (▪), 22.5 (▴), and 67.5 (×) μM, and the range of concentrations of omeprazole used was 0–200 μM. (B) Dixon plot obtained from the assay with microsomal preparation HL2 and lansoprazole showing effects on CYP2D6. Concentrations of dextromethorphan used were 2.5 (⧫), 10 (▪), 25 (▴), and 50 (×) μM, and the range of concentrations of lansoprazole used was 0–50 μM. (C) Secondary Lineweaver-Burk plot using slopes of the primary plot to get the Ki value of omeprazole for CYP3A (HL2). (D) Secondary Lineweaver-Burk plot of lansoprazole (HL2).
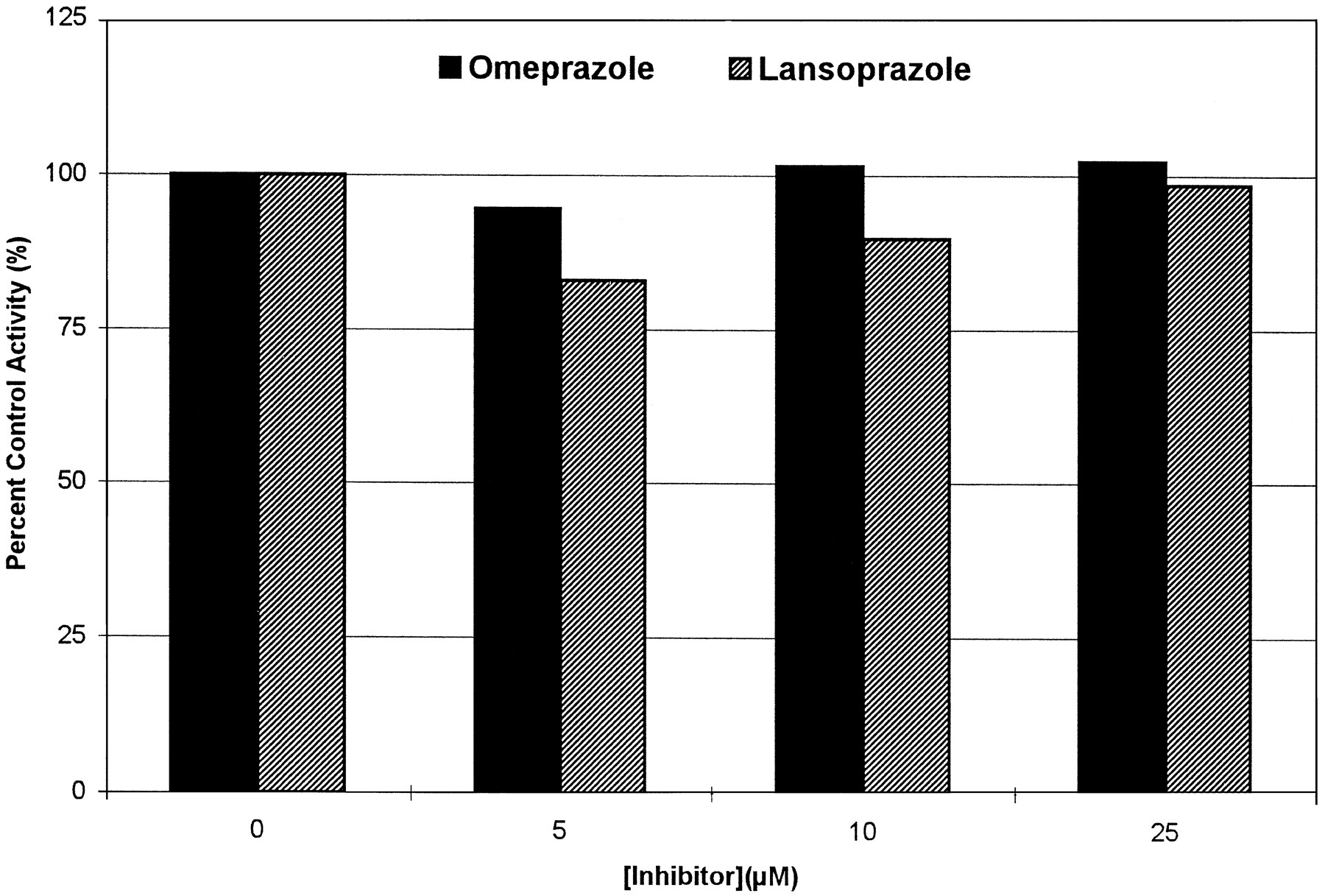
Both drugs showed similar inhibitory effects on the production of the 3-methoxymorphinan metabolite of dextromethorphan, indicating inhibition of the CYP3A isoform. The inhibitory patterns were consistent with a noncompetitive inhibitory mechanism, showing intercepts around x-axis on Lineweaver-Burk plot that imply a common KM with decreasingVmax. Using the noncompetitive model of the secondary Lineweaver-Burk plot, the Ki ’s obtained were 84.4 ± 4.04 μM for omeprazole and 170.4 ± 7.13 μM for lansoprazole (fig. 3, C and D). A preincubation test done to test the possibility of mechanism-based inhibition showed that the rate of 3-methoxymorphinan production after preincubation with omeprazole or lansoprazole was not significantly decreased, and the pattern of inhibition was not changed by preincubation with inhibitors (fig. 4).
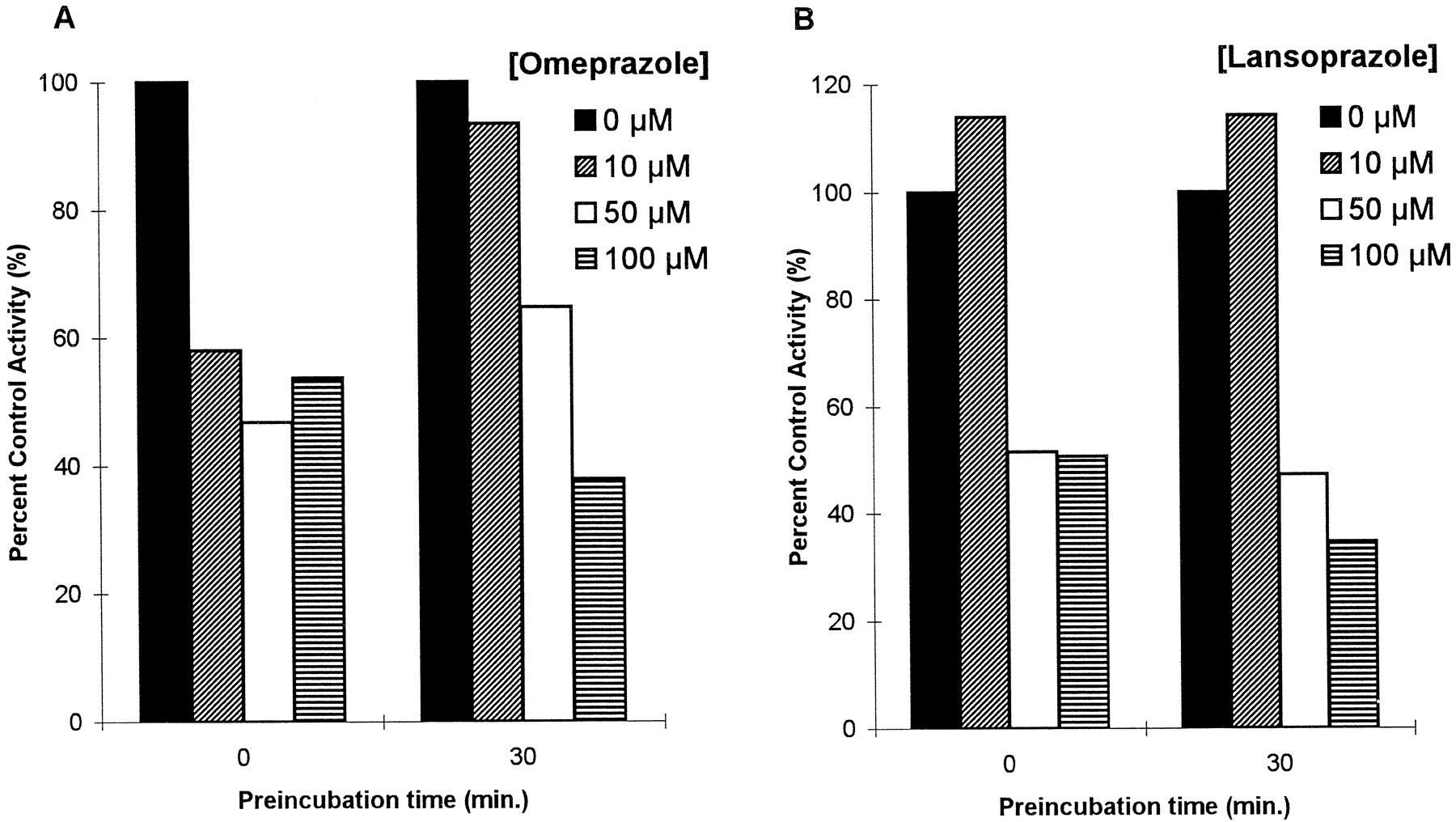
Effects of preincubation with omeprazole or lansoprazole on CYP3A-mediated dextromethorphan N-demethylation.
Data shown on graphs were from HL2. Values are per cent control activities at each preincubation time. (A) Omeprazole. (B) Lansoprazole.
CYP1A2 Activity.
No significant effects of omeprazole or lansoprazole on phenacetinO-deethylation were found (fig. 5). Within what seems to be the pharmacologically relevant range of concentrations (1–5 μM), both drugs showed no detectable effect on the rate of deethylation of phenacetin at substrate concentrations close to theKM for acetaminophen (78 μM).

Effects of omeprazole and lansoprazole on CYP1A2-mediated phenacetin O-deethylation.
Data shown on the graph were mean values of the per cent control activity in two HL assays (HL4 and HL6). The phenacetin concentration used was 70.4 μM.
CYP2E1 Activity.
Formation of 6-hydroxychlorzoxazone after incubations with omeprazole and lansoprazole could not be evaluated because of interfering peaks at the retention times of internal standard or 6-hydroxychlorzoxazone.
Discussion
In this study, we investigated the inhibitory effects of omeprazole and lansoprazole on human CYP isoforms using human liver microsomal preparations. A summary of the kinetic results is presented in table 1. Omeprazole and lansoprazole had inhibitory effects in vitro on CYP isoforms that included not only CYP2C19 and CYP3A, which are the two major isoenzymes involved in their metabolism, but also CYP2C9 and CYP2D6. Of note, the potency of the inhibition of CYP3A and CYP2C9 is such that one would expect only weak clinical interactions to take place with drugs metabolized by these isoforms. Consistent with the similarities in the structure and metabolism of omeprazole and lansoprazole (30), the spectrum and the potency of inhibition of the two drugs were similar, except that lansoprazole showed relatively strong inhibition of CYP2D6 compared with omeprazole.
Inhibition of CYP isoforms by omeprazole and lansoprazole
Both drugs showed strong competitive inhibition ofS-mephenytoin 4-hydroxylation (CYP2C19) with similarKi ’s (3.1 ± 2.18 μM for omeprazole and 3.2 ± 1.29 μM for lansoprazole) that are consistent with the work of VandenBranden et al. (47) and Chiba et al. (48), who reported Ki ’s of 4.1 ± 0.4 and 2.0 μM for omeprazole, respectively. It is possible that inhibitors may be extensively metabolized, and this may result in changes in enzyme kinetics, especially when an inhibitor has high affinity for an enzyme, is highly protein-bound, or has to go through a relatively long period of incubation (as is the case in theS-mephenytoin incubation we use). As a result, the apparent inhibitory effect might be less prominent than expected. In fact, however, it is difficult to quantitate the inhibition due to parent drug precisely, because the effect of the changes in the concentration of the inhibitor or the substrate are not routinely incorporated in enzyme kinetic models as they are in clinical pharmacokinetics. In addition, metabolites of the inhibitor (and even of the substrate) can have effects on the metabolism of the parent compound or the substrate. In fact, for omeprazole, it has been reported that one of omeprazole’s major metabolites, omeprazole sulfone, exhibits a similarly strong inhibition of CYP2C19, with a Ki value of 8 μM (48). Because of these problems in analysis, omeprazole’s effect onS-mephenytoin hydroxylation should be considered as a net effect, not as an effect of parent drug alone.
Several in vivo drug–drug interaction studies are consistent with a significant inhibition of CYP2C19 by omeprazole. The ability of omeprazole to inhibit diazepam clearance in healthy volunteers is well documented (9-11). These results seem reasonable because CYP2C19 plays a major role in the primary metabolism of both omeprazole and diazepam, and has a high inhibitory potency (20, 49,50).
Fewer drugs, or CYP isoforms, have been evaluated for interaction with lansoprazole. Recently, Pearce et al. (30) presented data that strongly suggest that CYP2C19 is the primary metabolic route at pharmacologically relevant concentrations of lansoprazole (1–5 μM). The clinical correlate of these data was presented by Sohn et al. (5), who reported that poor metabolizers ofS-mephenytoin are also poor metabolizers of lansoprazole in Korean healthy volunteers. Data presented herein further suggest that interactions between lansoprazole and CYP2C19 may be important.
Our data suggest that omeprazole and lansoprazole are indistinguishable as inhibitors of CYP2C19. This result seems at odds with clinical data presented by Lefebvre et al. (31), in which lansoprazole seemed to have no inhibitory effect on plasma concentrations of single intravenous doses of diazepam administered to 12 healthy male volunteers. An explanation for this finding was suggested by Andersson (51), who proposed that the affinity of lansoprazole for CYP2C19 may be low in comparison with that of omeprazole. Our data suggest that this is not the case, because the affinities of omeprazole and lansoprazole for CYP2C19 seem similar. It is notable that the mean value of total body clearance of diazepam during the period of placebo (22.5 ml/hr/kg) in this study seems closer to that of slow metabolizers of omeprazole in the data presented by Andersson et al. (10) (17.5 ml/hr/kg in slow metabolizers and 41.0 ml/hr/kg in rapid metabolizers), where an inhibitory effect was noted only in extensive metabolizers of omeprazole. This difference may therefore be related to the population under study, and one might expect that lansoprazole would have an effect on diazepam clearance in populations where CYP2C19 is relatively active. Until more clinical data are available, it seems difficult to say clearly whether lansoprazole has clinically important effects or not on drugs, such as the tricyclic antidepressants, mephenytoin and mephobarbital, that are metabolized by CYP2C19.
Both omeprazole and lansoprazole seem to be weak inhibitors of CYP3A, as measured by N-demethylation of dextromethorphan (Ki = 84.4 ± 4.04 μM for omeprazole,Ki = 170.4 ± 7.13 μM for lansoprazole). These data are consistent with the results of other investigators who have shown competitive inhibition of midazolam 1-hydroxylation withKi ’s in the 40–200 μM range (47) or IC50 values from 0.2 to 1.5 mmol/liter (52). A number of clinical studies involving proton pump inhibitors and drugs that are substrates of CYP3A have been reported. Gustavson et al.(14) reported that omeprazole decreased clarithromycin clearance, and we have shown that clarithromycin inhibits omeprazole sulfonationin vitro (53). No significant interaction between lansoprazole and clarithromycin was noted in a clinical study by Karolet al. (54). Omeprazole did not significantly interfere with cyclosporine metabolism in stabilized renal transplant patients (55), although a possible role of omeprazole in elevation of cyclosporine blood concentration in a single case of liver transplantation has been suggested (56). In contrast, Arranz et al. (57) reported a case of decreased cyclosporine plasma concentration with chronic omeprazole administration.
Our data showing that omeprazole is a weak inhibitor of CYP2D6 are consistent with the results of VandenBranden et al. (47), who showed a mixed type of inhibitory effect (Ki = 302 ± 57 μM) on the 1′-hydroxylation of bufuralol, a specific probe for this CYP isoform, and with several clinical studies showing that omeprazole has no clinically important inhibitory effect on drugs metabolized through CYP2D6. These include metoprolol (17) and propranolol (18). Our data indicate that lansoprazole is a relatively potent inhibitor of CYP2D6, with a Ki forO-demethylation of dextromethorphan of 44.7 ± 22.02 μM. Further studies in this area seem important, given the large number of important drugs metabolized by CYP2D6 and the possibility that lansoprazole might affect their metabolism, particularly in poor metabolizers of CYP2C19.
This study is the first to demonstrate any interaction between proton pump inhibitors and CYP2C9. When tolbutamide 4-methylhydroxylation was used as a probe for this enzyme, both omeprazole (Ki = 40.1 ± 14.8 μM) and lansoprazole (Ki = 52.1 ± 1.4 μM) were shown to be competitive inhibitors. These data suggest that inhibition of this isoform might contribute to interactions with drugs metabolized by CYP2C9, such as the active S-enantiomer of warfarin (58) and phenytoin (59), but these Ki values are considerably higher than the serum concentrations one would expect, even in poor metabolizers and potent clinical inhibition seems unlikely.
It has been suggested that lansoprazole has less significant drug interactions than other proton pump inhibitors. This suggestion is based on the observations that lansoprazole has no significant effect on diazepam (31), phenytoin (32), or R-warfarin metabolism (60). Most of these clinical studies, however, were performed before the report of Sohn et al. (5) that showed clearly that lansoprazole exhibits polymorphic metabolism. These data seem surprising in light of the similar affinities of lansoprazole and omeprazole for CYP isoforms that we have shown. In addition, when Simonet al. (61) compared inhibitory effects of omeprazole, lansoprazole, and pantoprazole, they noted that lansoprazole had the greatest inhibitory effect on O-dealkylation of 7-ethoxycoumarin, N-demethylation of ethylmorphine, and hydroxylation of lonazolac.
Although more clinical studies are necessary to clarify the ability of lansoprazole to interact with other drugs, our data suggest that lansoprazole has an important an effect as omeprazole on CYP isoforms, and that it may be a clinically more important inhibitor of CYP2D6 in some populations.
This study clearly demonstrates that both omeprazole and lansoprazole can serve as useful, potent, and selective in vitroinhibitors of CYP2C19. As such, these drugs represent a valuable addition to the battery of CYP isoform-specific inhibitors that can be used to characterize the hepatic metabolism of drugs in vitro (62). Omeprazole seems to be a particularly selective inhibitor of CYP2C19, because it has little interaction with CYP2D6. Although it has been suspected for some time that omeprazole might serve such a purpose (8, 63), the selectivity of omeprazole as an inhibitor of CYP2C19 has not been described until now. In our study, omeprazole exhibited a strong competitive inhibition ofS-mephenytoin 4-hydroxylation (CYP2C19,Ki = 1.0 μM from Dixon plot), but relatively weak inhibition of other isoforms with Ki ’s >40 μM.
Most chemical inhibitors seem to lose their selectivity at higher concentrations. For example, quinidine, a well-known inhibitor of CYP2D6, also inhibits nifedipine dehydrogenation (CYP3A4) at high concentrations (64) and ketoconazole—a useful and selective inhibitor of CYP3A4—inhibits bufuralol 1′-hydroxylation (CYP2D6) and tolbutamide methylhydroxylation (CYP2C9) at high concentrations (27). For this reason, Rodrigues (65) suggested that inhibitors should be characterized by their “window of selectivity.” Thus, when doing inhibitory studies, it is important to choose low inhibitor concentrations (within the window of selectivity) at substrate concentrations near KM to inhibit selectively the high-affinity activity (64). Compared with other inhibitors, the “window of selectivity” of omeprazole for CYP2C19 seems broad. The range of concentration in which >70% inhibition of CYP2C19 was observed with no significant inhibitory effect on other isoforms was from 5 to 25 μM, a concentration that is at least 20-fold greater than the Ki (fig. 6). Several other CYP isoforms such as CYP1A1, CYP2B6, and CYP2C8 have not been included as part of this work. The possibility remains that they may be inhibited by omeprazole or lansoprazole, but the potency of the inhibition by omeprazole and the lack of documented involvement of these CYP isoforms in the metabolism of proton pump inhibitors make this possibility seem unlikely. We, therefore, believe that omeprazole at low concentrations (up to 10 μM) can be used as an isoform-selective inhibitor to test the involvement of CYP2C19 in oxidative reactions in human liver microsomes.

Effects of omeprazole on CYP-mediated reactions in human liver microsomes.
Data shown on the graph are from data at approximately their respectiveKM of the reaction; 70 μM for phenacetinO-deethylation (CYP1A2), 50 μM for tolbutamide 4-methylhydroxylation (CYP2C9), 30 μM for S-mephenytoin 4-hydroxylation (CYP2C19), 22.5 μM for dextromethorphanO-demethylation (CYP2D6), and 67.5 μM for dextromethorphanN-demethylation (CYP3A). Activities are expressed as a percentage of control activity.
Footnotes
-
Send reprint requests to: Dr. Jae-Wook Ko, Division of Clinical Pharmacology, Georgetown University Medical Center, 3900 Reservoir Road, NW, Washington, D.C. 20007.
-
This work was supported in part by Clinical Pharmacology Training Grant T32-GM 08386 from the National Institute of General Medical Sciences (Bethesda, MD), and by a fellowship award to Dr. Ko from the World Health Organization (WPRO 0630/95).
- Abbreviations used are::
- CYP
- cytochrome P450
- G6P
- glucose 6-phosphate
- G6PDH
- glucose 6-phosphate dehydrogenase
- AUC
- area under the curve
- HL
- human liver
- Received December 11, 1996.
- Accepted March 25, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}