


Abstract
Both ritonavir and indinavir were readily metabolized by human intestinal microsomes. Comparison of the patterns of metabolites in incubations with enterocyte microsomes and expressed cytochrome P450 (CYP) isozymes and immunoinhibition and chemical inhibition studies showed the essential role of the CYP3A subfamily in the metabolism of both protease inhibitors by the small intestine. Ritonavir was similarly biotransformed by microsomes containing expressed CYP3A4 or CYP3A5 isozymes (KM = 0.05–0.07 μM,Vmax = 1–1.4 nmol/min/nmol CYP). In contrast, both the patterns of metabolites and the enzyme kinetic parameters for the metabolism of indinavir by expressed CYP3A5 (KM = 0.21 μM,Vmax = 0.24 nmol/min/nmol CYP) and CYP3A4 (KM = 0.04 μM,Vmax = 0.68 nmol/min/nmol CYP) were different. The biotransformation of both indinavir and ritonavir in human enterocyte microsomes was characterized by very lowKM values (0.2–0.4 μM for indinavir and <0.1 μM for ritonavir). The Vmaxfor indinavir metabolism was greater in enterocyte (163 ± 35 pmol/min/mg protein) than in liver (68 ± 44 pmol/min/mg protein) microsomes. The metabolism of ritonavir in liver and enterocyte microsomes was associated with inactivation of CYP3A. The initialVmax for ritonavir metabolism by enterocyte microsomes was 89 ± 59 pmol/min/mg protein. The apparent inactivation rate constants for intestinal CYP3A and expressed CYP3A4 were 0.078 and 0.135 min−1, respectively. Metabolic inactivation of CYP3A by ritonavir explains the improved bioavailability and pharmacokinetics of ritonavir and the sustained elevation of blood levels of other, concomitantly administered, substrates of CYP3A.
Indinavir and ritonavir (fig.1) represent a new class of anti-HIV1 agents that selectively inhibit the HIV type 1 protease. The HIV protease cleaves viral precursor polyproteins (Darke et al., 1988). This process is essential for the maturation of infectious virions (Gottlinger et al., 1989). Numerous protease inhibitors have been discovered. Most of them display low oral bioavailability and rapid elimination. Indinavir is one of the first protease inhibitors with enhanced oral bioavailability, varying from 10 to 70% in different species (Vacca et al., 1994; Lin et al., 1996). Its average half-life in humans is 1.8 hr. Improved pharmacokinetic properties (half-life, 3.1–5.7 hr) and high oral bioavailability in humans have been recently reported for ritonavir (Kempf et al., 1995; Hsu et al., 1997). It has been shown that the protease inhibitors indinavir, ritonavir, and saquinavir are metabolized primarily by isozymes of the CYP3A subfamily and to a lesser extent by CYP2D6 (Kumar et al., 1996; Chibaet al., 1996, 1997; Fitzsimmons and Collins, 1997). CYP3A is a major subfamily of oxidative enzymes in the small intestine and accounts for 70% of the total intestinal CYP content (Watkins et al., 1987). CYP3A4 and CYP3A5 are two CYP3A isozymes that are commonly expressed in the gastrointestinal tract. The small intestine has been shown to be an important site for presystemic metabolism of cyclosporine (Hebert et al., 1992), midazolam (Thummelet al., 1996), rifabutin (Iatsimirskaia et al., 1997), and possibly other CYP3A substrates, including saquinavir (Fitzsimmons and Collins, 1997). Biotransformation of protease inhibitors by CYP3A enzymes in the intestine may account for low and variable bioavailability. Recently, Chiba et al. (1997)showed that the small intestine is capable of metabolizing indinavir. However, the contribution of the gut to the first-pass metabolism of indinavir, estimated from in vitro metabolic data (Vmax/KM ) and intestinal mucosal blood flow, was suggested to be minor. The metabolism of ritonavir by intestinal enzymes has not been examined.

Chemical structures and labeling positions (*) of [14C]indinavir and [14C]ritonavir and characteristic fragments produced by CID-MS of the molecular ions.
a, present in CID spectra of the daughter ion atm/z 296; b, present in CID spectra of some metabolites.
The coadministration of ritonavir significantly increased the plasma levels of other drugs metabolized by CYP3A, including saquinavir, for which a 63–83-fold increase in AUC was reported (Kempf et al., 1997). Ritonavir also potently inhibited the CYP-mediated metabolism of indinavir, saquinavir, and nelfinavir in human liver microsomes; however, none of these protease inhibitors altered the metabolism of ritonavir in vivo or in vitro. The objectives of the present study were to 1) determine and compare enzyme kinetic parameters for indinavir and ritonavir in incubations with human intestinal microsomes, 2) estimate the relative contributions of specific CYP enzymes (CYP3A4/3A5 and CYP2D6) to the biotransformation of indinavir and ritonavir by liver and intestine, and 3) provide anin vitro explanation for the sustained inhibition by ritonavir of the elimination of other, concurrently administered, CYP3A substrates.
Materials and Methods
Materials.
Unlabeled indinavir as the sulfate salt (>98% purity by HPLC) and [14C]indinavir (7.84 mCi/mmol) were kindly supplied by Merck Research Laboratories (West Point, PA). Unlabeled ritonavir (>98% purity by HPLC) and [14C]ritonavir (39.3 mCi/mmol) were generously provided by Abbott Laboratories (Abbott Park, IL). The radiolabeled compounds were repurified by HPLC to yield >99% purity. Ketoconazole was received from Janssen Life Science Products (Beerse, Belgium). 5-Methoxypsoralen was from Aldrich Chemical Co. (Milwaukee, WI). HPLC-grade ACN, methanol, and o-phosphoric acid were obtained from Fisher Scientific Products (Fair Lawn, NJ), and TEA was from J. T. Baker Inc. (Phillipsburg, NJ). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
Human liver samples were received from the operating rooms (The Ohio State University Hospitals) or the International Institute for the Advancement of Medicine (Exton, PA). Samples of human small intestine were all from the operating rooms of The Ohio State University Hospitals, after Roux-en-Y operations (Mason et al., 1980). The collection of specimens for research was approved by The Ohio State University Biomedical Sciences Human Subjects Review Committee.
Intestinal and liver microsomes were prepared as described previously (Iatsimirskaia et al., 1997). Microsomal protein was determined by the method of Lowry et al. (1951). Mouse MAB3A4, rabbit polyclonal antibody against CYP2D6, and microsomes containing expressed human CYP (CYP3A4, CYP3A5, and CYP2D6) and reductase were purchased from Gentest (Woburn, MA).
In Vitro Incubations .
Typically, microsomal incubations were conducted in a volume of 0.5–3 ml, in duplicate (duplicates were within 15% of their mean). Enterocyte or liver microsomal protein (0.05–2 mg/ml) or recombinant CYP (2–20 pmol/ml), suspended in 50 mM potassium phosphate buffer, pH 7.4, was preincubated for 2 min at 37°C in a shaking bath with 0.04–20 μM radiolabeled ritonavir or 0.1–20 μM indinavir (unlabeled or labeled) added from stock solutions in methanol; the reaction was initiated by the addition of NADPH (2 mM final concentration). Incubations with no NADPH served as controls. The reaction was terminated at 15 min (ritonavir) or 2–40 min (indinavir) by the addition of 4 volumes of ACN, and the samples were centrifuged at 1500g for 10 min. The supernatant was evaporated to dryness under nitrogen at 40°C, the residue was resuspended in 100–120 μl of mobile phase, and 80 μl was analyzed by HPLC for the total metabolism of ritonavir (method 2) or indinavir (method 6). Recovery of the label after precipitation with ACN was ≥98% for both indinavir and ritonavir. For estimation of enzyme kinetic parameters, conditions were maintained to ensure ≤20% disappearance of substrate.
Because of the low specific activity of [14C]indinavir, unlabeled drug was used to determine KM andVmax values for enterocyte and liver microsomes. All other quantitative studies used [14C]indinavir. The samples with unlabeled indinavir were incubated for 2–40 min in triplicate, and the reaction was stopped by vigorous mixing with 1-chlorobutane (4 volumes) containing 5-methoxypsoralen (50 ng/ml) as an internal standard. After centrifugation (1500g for 10 min), the 1-chlorobutane extract was evaporated to dryness under nitrogen, the residue was resuspended in 100 μl of mobile phase, and 80 μl was analyzed by HPLC as described below (method 6). The retention times of the internal standard and indinavir were 14.6 and 16.1 min, respectively. The extraction efficiency was 80% for indinavir and 93% for the internal standard. Incubations in the absence of NADPH were used to construct calibration curves, for which the correlation coefficients were routinely >0.999. Concentrations of indinavir were determined using drug/internal standard peak area ratios. The instrumental detection limit for unlabeled indinavir was 30 nM. The parametersKM and Vmaxwere determined using Eadie-Hofstee linearization.
Time-Course Studies.
For time-course studies, [14C]indinavir (5 μM) or [14C]ritonavir (5 μM) was incubated with enterocyte (0.4 or 2 mg/ml protein) or liver (0.05 mg/ml protein) microsomes, in the presence of NADPH (2 mM), for up to 1 hr. [14C]Ritonavir (2 μM) was also incubated for up to 1 hr with microsomes containing expressed CYP3A4 (50 pmol/ml). The reaction was terminated at specific times by the addition of ACN, as described above, and the samples were analyzed by HPLC using method 1 or 2 for ritonavir and method 6 for indinavir (see HPLC Analysis). To examine the effect of preincubation on the rate of ritonavir metabolism, liver or enterocyte microsomes were also preincubated with [14C]ritonavir for up to 10 min at 37°C before the addition of NADPH.
Profiles and Isolation of Metabolites.
Profiles of metabolites were studied by incubation of [14C]indinavir (5 μM) or [14C]ritonavir (2 or 5 μM) with enterocyte or liver microsomal protein (2 mg/ml for indinavir and 0.4 mg/ml for ritonavir) or expressed CYP (100 and 25 pmol CYP/ml for indinavir and ritonavir, respectively), in a volume of 1 ml, for 15 min (ritonavir) or 30 min (indinavir). The parent drug and metabolites were separated by HPLC using conditions described below (method 1 for ritonavir and method 4 for indinavir).
Indinavir (10 μM) was incubated with human liver microsomal protein (2 mg/ml, 80 ml) in the presence of NADPH (1 mM) for 1 hr; the mixture was combined with an equal volume of 4 M potassium/sodium phosphate buffer, pH 7, and extracted with 3 volumes of ethyl acetate (with shaking, for 10 min). After centrifugation at 1500g for 10 min, the upper layer was evaporated to dryness in a vacuum evaporator and the residue was redissolved in 6 ml of methanol. The methanol extract was evaporated under nitrogen at 40°C, resuspended in 100 μl of mobile phase, filtered through a 0.2-μm filter, and injected into the HPLC system to separate metabolites using method 4. Fractions of the eluate corresponding to the major radioactive peaks (see fig. 4) were collected, and each was extracted twice with 6 ml of ethyl acetate. After centrifugation, the combined organic layers were dried under nitrogen at 40°C, and the residue was redissolved in 100 μl of mobile phase and injected into the HPLC system for final purification (method 5).

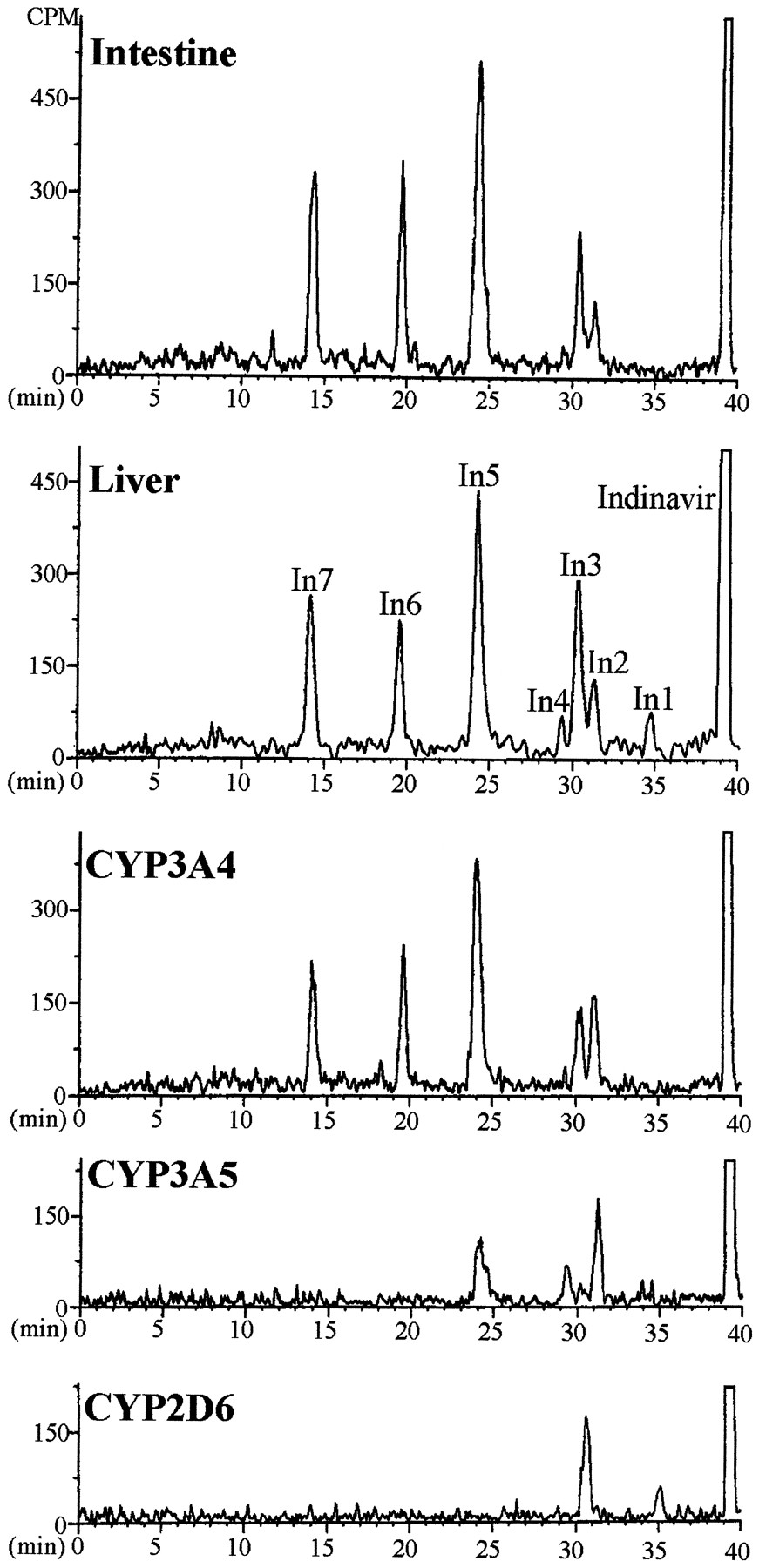
Patterns of metabolites of [14C]indinavir (5 μM) in incubations with human liver and intestinal microsomes (2 mg/ml protein) and recombinant CYP enzymes (100 pmol CYP/ml).
The major peaks were identified as products of oxidation of the 1) indanyl (one site) and 1,1-dimethylethylaminocarbonylpiperazinyl (two sites) groups (In1 and In2), 2) piperidinyl (one site) and 1,1-dimethylethylaminocarbonylpiperazinyl (two sites) groups (In3), 3) phenylmethyl (one site) and 1,1-dimethylethylaminocarbonylpiperazinyl (two sites) groups (In4), and 4) 1,1-dimethylethylaminocarbonyl (one site) and indanyl (one site) moieties (In6).
Ritonavir (5 μM) was incubated with liver microsomes (2 mg/ml protein, with 1 mM NADPH, in a final volume of 70 ml) for 30 min and extracted twice with an equal volume of 1-chlorobutane. The organic layers were pooled and evaporated to dryness under nitrogen at 40°C, and the residue was redissolved in 150 μl of mobile phase and injected into the HPLC system to separate metabolites (method 1). Fractions corresponding to the major radioactive peaks (see fig. 5) were collected, and each was mixed with an equal volume of 2 M potassium/sodium phosphate buffer, pH 7, and extracted with 1-chlorobutane as described above. Final purification was achieved by rechromatography of each metabolite using method 3 (see HPLC Analysis).
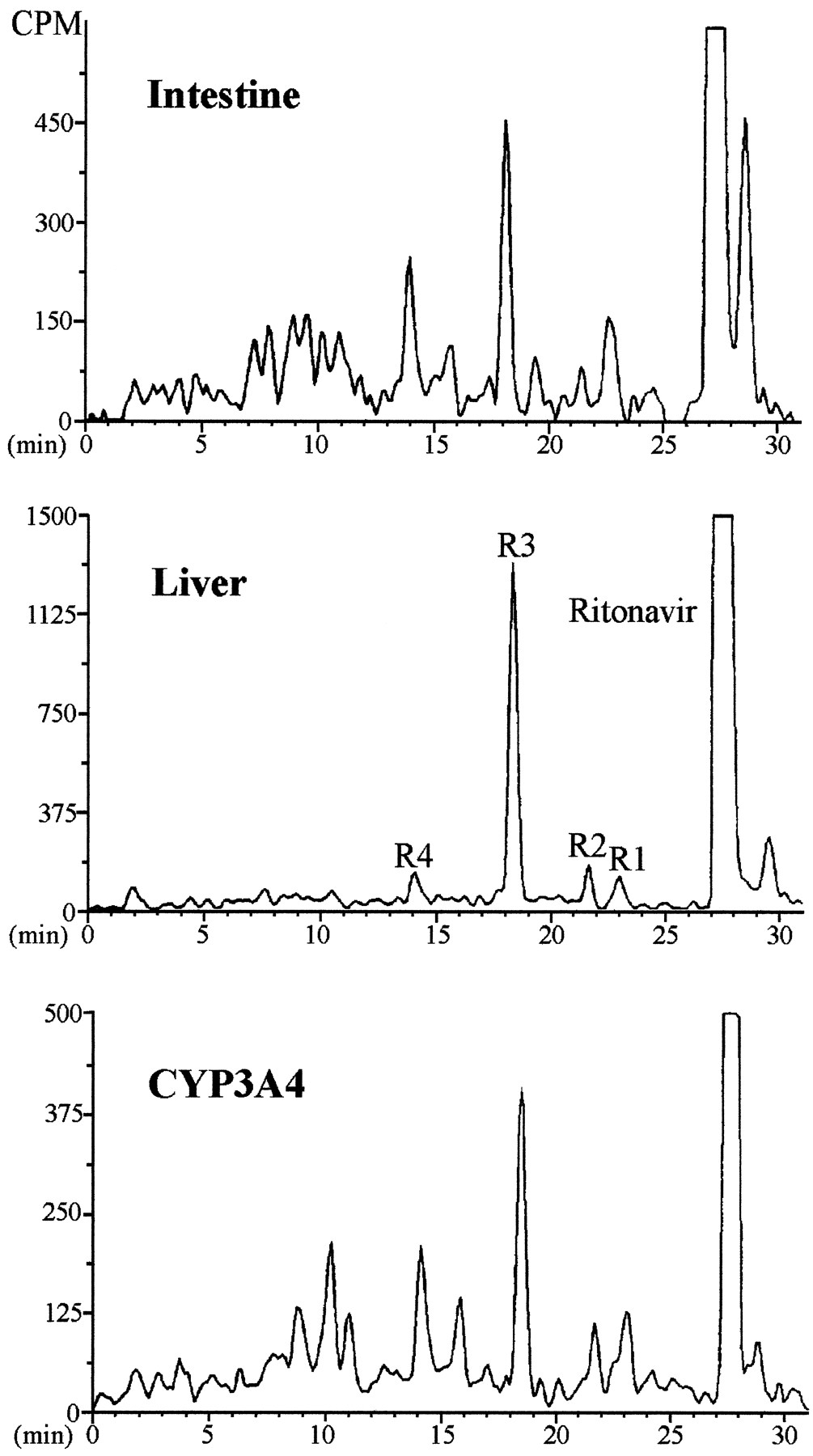
Patterns of metabolites of [14C]ritonavir (5 μM) in incubations with human enterocyte and liver microsomes (0.4 mg/ml protein) and recombinant CYP3A4 (25 pmol CYP/ml).
The major peaks were identified as products of 1)N-demethylation (R1), 2) oxidation of the methylthiazolyl moiety (R2), 3) hydroxylation of the isopropyl side chain (R3), and 4) cleavage of the 2-(1-methylethyl)thiazolylmethyl group (R4).
Inhibition by Ketoconazole.
Inhibition of metabolism by ketoconazole was studied by incubating enterocyte or liver microsomal protein (0.2–0.6 mg/ml) with 1 μM [14C]indinavir (20 min) or 0.5 μM [14C]ritonavir (15 min) in the presence of different concentrations of ketoconazole (0.05–20 μM). The samples were analyzed for the formation of total metabolites using methods 2 (ritonavir) and 6 (indinavir).
Immunoinhibition Studies.
Typical (by HPLC profiles) liver (LMS 9) and enterocyte (EMS 10 and EMS 13) microsomes were used for immunoinhibition studies. Microsomes were preincubated with MAB3A4 (0.5–5 mg IgG/mg microsomal protein) or antibody against CYP2D6 (0.2–2 mg IgG/mg microsomal protein) at room temperature for 15 min (MAB3A4) or 30 min (anti-CYP2D6), followed by the addition of [14C]indinavir (1 μM) or [14C]ritonavir (0.5 μM). The reaction was initiated by the addition of NADPH, and the samples were incubated for 15 min (ritonavir) or 20 min (indinavir) at 37°C and analyzed for total metabolism of indinavir (method 6) or ritonavir (method 2).
Coincubation of Indinavir and Ritonavir.
Ritonavir (0.05 μM) and [14C]indinavir (5 μM) were coincubated with human enterocyte microsomes (0.4–0.5 mg/ml protein), in 1-ml aliquots, in the presence of NADPH for up to 1 hr. The reaction was stopped at 10, 20, 30, 40, 50, or 60 min by the addition of ACN, and samples were analyzed for total metabolism of indinavir (by the formation of products, using method 6) or ritonavir (by the disappearance of the drug, using method 2), as described below (see HPLC Analysis).
Inactivation of CYP by Ritonavir.
First, enterocyte microsomes (0.5 mg/ml protein) were preincubated with ritonavir (0.075 μM) and NADPH for 20 min (under these conditions, ritonavir is completely metabolized), [14C]indinavir (5 μM) was added, and the mixture was incubated for 20 min. Two controls consisted of preincubation of microsomes for 20 min in the absence of ritonavir, addition of [14C]indinavir (alone or together with ritonavir), and incubation for an additional 20 min. The reactions were stopped by mixing with ACN, and total radiolabeled metabolites were measured by method 6.
The nature of the inhibition of indinavir metabolism by ritonavir was further examined by coincubating 100 μM [14C]indinavir and 0.075 μM ritonavir with enterocyte microsomes (in triplicate) for 15 min. Incubations containing only [14C]indinavir served as controls. The reaction was stopped by the addition of ACN, and the samples were analyzed for total metabolism of indinavir (method 6).
Second, the effect of ritonavir on the activity of hepatic CYP was studied by incubating [14C]ritonavir (5 μM) with liver microsomes (0.5 mg/ml protein, in a volume of 5 ml) and NADPH, in triplicate, for 30 min at 37°C (∼20% conversion into metabolites); the incubation mixtures were placed in ice for 1 min and ultracentrifuged at 100,000g for 30 min at 4°C. The supernatant containing metabolites was collected to study the effect of the metabolites on the metabolism of ritonavir (see below). The pellets were resuspended in 10 ml of 0.05 M phosphate buffer, and the centrifugation was repeated. The removal of radioactivity from the incubation with ritonavir after two washes was approximately 90%. The microsomal pellet from each incubation was homogenized in 5 ml of cold phosphate buffer and incubated with ritonavir (5 μM) and NADPH (2 mM), in 0.5-ml aliquots, for 5, 10, or 20 min; the disappearance of ritonavir was measured by method 2. Liver microsomes incubated without ritonavir and treated similarly served as controls.
The effect of metabolites on the metabolism of ritonavir was studied by incubating fresh liver microsomal protein (0.5 mg/ml) with the supernatant from the first ultracentrifugation and [14C]ritonavir (adjusted to a final concentration of 5 μM), at 37°C, for 5, 10, or 20 min. After termination of the reaction, the samples were analyzed for the disappearance of ritonavir (method 2). In a separate experiment, unlabeled ritonavir (5 μM) was incubated with enterocyte microsomal protein (2 mg/ml, 1.5 ml) in the presence of NADPH for 15 min at 37°C (20% conversion into metabolites). The reaction was stopped by the addition of ACN, and the samples were processed as described above forin vitro incubations. The residue was resuspended in 120 μl of a mixture of methanol/0.01% TFA (1:1), and 80 μl was injected into the HPLC system. Ritonavir metabolites were separated from the parent compound using a linear gradient of 25–75% ACN and an aqueous phase of 0.01% TFA in distilled water, with UV detection at 210 nm. The eluate (15 ml), containing a mixture of metabolites, was collected into an incubation tube and evaporated to dryness under nitrogen. The residue was redissolved in 20 μl of 50% methanol, mixed with fresh enterocyte microsomes (2 mg/ml, 1 ml), and incubated with [14C]ritonavir (5 μM) for 15 min, and the samples were analyzed by method 2. Eluates from incubations without ritonavir that were treated as described above served as the controls.
Third, the effect of free radical-trapping agents on the inactivation of CYP was studied in incubations of liver microsomes (1 mg/ml protein, 0.3 ml, in duplicate) with ritonavir (5 μM), in the presence or absence of reduced glutathione (1 mM), ascorbic acid (2 mM),N-acetylcysteine (5 mM), or superoxide dismutase (300 units/ml), for 5, 10, or 20 min. The samples were analyzed for the disappearance of ritonavir using method 2.
Estimation of kin.
The time-course experiments under conditions of S ≫KM
(5 μM ritonavir and 2 mg/ml protein for enterocyte microsomes or 2 μM ritonavir and 50 pmol/ml CYP3A4) were used to calculate kin, assuming the following scheme of enzyme inactivation:
The observed relationship between total product concentration (P) and time (t) was linear in double-reciprocal plots (correlation coefficient of >0.99 for both enterocyte and CYP3A4 microsomes) and, therefore, could be expressed by the equation
In view of the nonlinearity of the metabolism of ritonavir, 15-min incubations with enterocyte and liver microsomes were used for kinetic studies, to increase the sensitivity of the assay. Initial velocities for each substrate concentration were estimated based on the assumption that enzyme inactivation is proportional to product formation (Gray and Tam, 1991).
HPLC Analysis. General Procedure.
All samples were analyzed using an HPLC system (model 1090; Hewlett-Packard, Palo Alto, CA) equipped with a UV diode-array detector and a Radiomatic A-500 series flow scintillation analyzer, with a solid scintillation flow cell (Packard, Meriden, CT). The mobile phase flow rate was 1.5 ml/min.
Ritonavir.
For isolation of metabolites and analysis of profiles, the conditions previously described (Kumar et al., 1996) were used, with minor modifications. Separation of metabolites (detected by radioactivity or absorption at 210 nm) used a Hypersil 5C18 column (250 × 4.6 mm; Phenomenex, Torrance, CA) and a 40-min linear gradient of 25–67% ACN with an aqueous phase containing 0.1% TFA, adjusted to pH 4.8 with ammonium acetate (method 1). For determination of total metabolism (formation of metabolites and/or disappearance of the parent drug), samples were analyzed with a 15-min gradient of 25–75% ACN, with on-line detection of radioactivity (method 2). The retention time of ritonavir under these conditions was 12.9 min. Final purification of the major metabolites, which were monitored by UV absorption at 210 nm, was achieved with a 40-min linear gradient of 25–67% ACN with an aqueous phase containing 10 mM potassium phosphate buffer, pH 7 (method 3).
Indinavir.
Analysis of profiles and collection of metabolites used a Capcell Pak 5-μm C18 UG-120A column (250 × 4.6 mm; Phenomenex) and a mobile phase gradient of 20–31% ACN over 0–33 min and then 31–80% ACN over 33–48 min, with an aqueous phase of 0.2% TEA adjusted to pH 7 with o-phosphoric acid (method 4). Final purification of the metabolites used a 42-min linear gradient of 20–34% ACN with an aqueous phase containing 0.02% TEA, pH 7 (adjusted with o-phosphoric acid) (method 5). For analysis of total metabolism, a Hypersil 5C18 column (250 × 4.6 mm; Phenomenex) and an 18-min linear gradient of 25–45% ACN with an aqueous phase of 0.3% TEA, pH 6, with UV detection at 240 nm or on-line detection of radioactivity, were used (method 6). The retention time of indinavir under these conditions was 16.1 min.
Identification of Metabolites by MS.
MS data were obtained using a VG Quattro I triple-quadrupole mass spectrometer (Micromass, Beverly, MA) equipped with a triaxial ESI probe. Samples, after HPLC purification, were dissolved in a solution of isopropanol/water/acetic acid (75:25:0.1). A sheath liquid composed of the same solvents was infused at a flow rate of 5 μl/min, to maintain a stable spray. Samples were infused manually at a flow rate of ∼10 μl/min. Argon was used as the collision gas to obtain CID spectra. The spectra were acquired in the positive-ion mode. The cone voltage setting of 35 V was used to obtain ESI or CID spectra. To pinpoint the location of parent compound modifications, a cone voltage of 75 V was used to induce in-source fragmentation. All full-scan ESI spectra were obtained at unit resolution, whereas CID spectra were obtained at lower resolutions to improve detectability.
CID-MS of the protonated molecule produced a number of characteristic fragments for indinavir [m/z 614 ([M+H]+), 513, 465, 421, 338, and 133] and ritonavir [m/z 721 ([M+H]+), 551, 426, 296, 268, 197, 171, 140, and 98], which were consistent with the proposed fragmentation patterns shown in fig. 1. The following diagnostic ions were used for structural identification of indinavir metabolites: In1 and In2, m/z 662 (614+48) ([M+H]+), 569 (521+48), 354 (338+16), 149 (133+16), and 131 (149−18); In3, m/z662 (614+48) ([M+H]+), 553 (521+32), 338, 133, and 107 (91+16); In4, m/z 662 (614+48) ([M+H]+), 569 (521+48), 354 (338+16), 133, and 107 (91+16); In6, m/z 646 (614+32) ([M+H]+), 529 (513+16), 481 (465+16), 437 (421+16), 354 (338+16), and 149 (133+16). Assignment of structures to ritonavir metabolites was based on the following characteristic fragments: R1, m/z 707 (721−14) ([M+H]+), 426, 282 (296−14), 254 (268−14), 157 (171−14), 140, and 98; R2, m/z 737 (721+16) ([M+H]+), 442 (426+16), 296, and 114 (98+16);R3, m/z 737 ([M+H]+), 551, 426, 312 (296+16), 284 (268+16), 213 (197+16), 187 (171+16), 98, and 59 (43+16); R4, m/z 582 (721−139) ([M+H]+), 525, 426, and 98.
Results
Time Course of the Metabolism of Indinavir and Ritonavir.
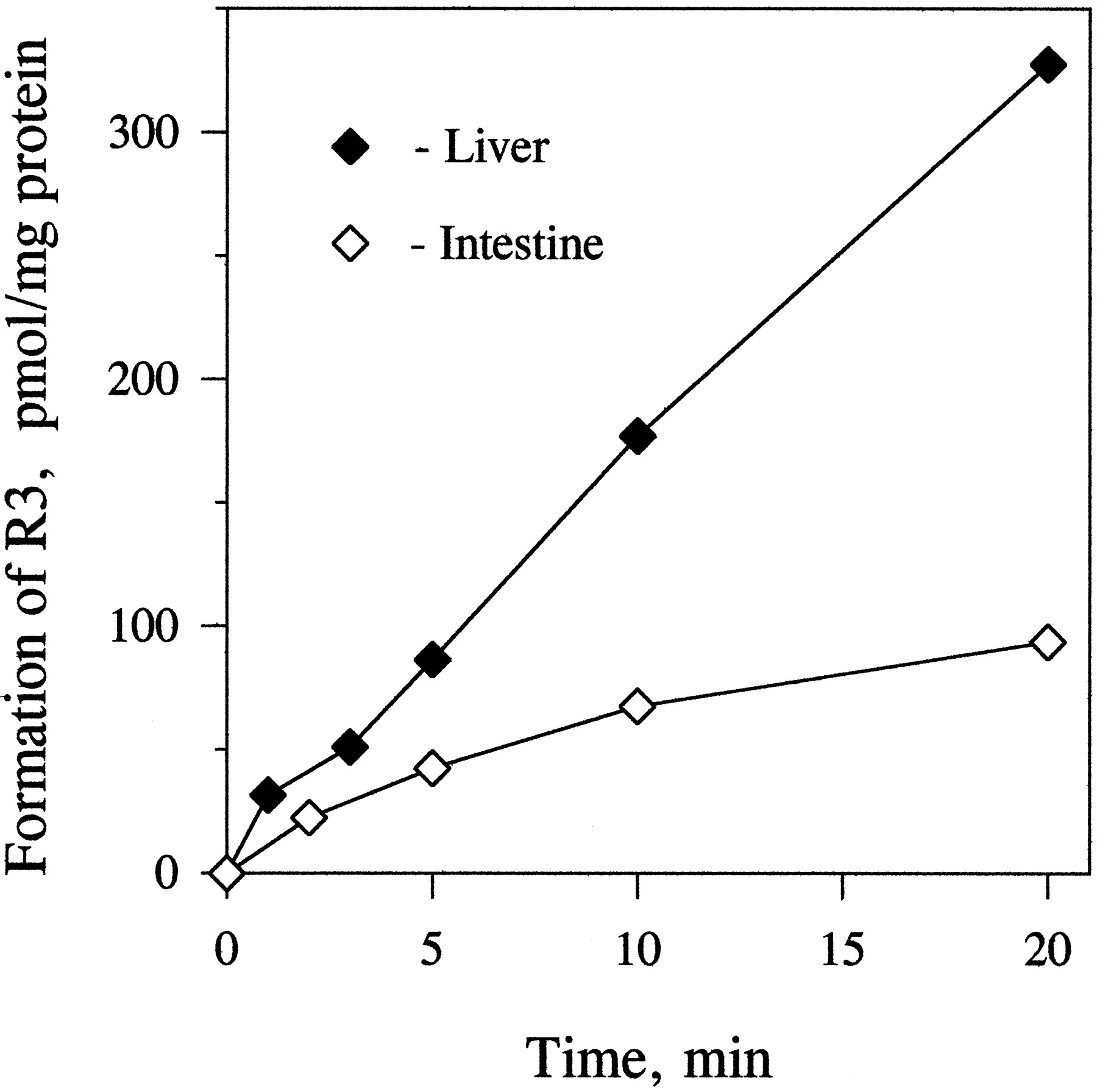
The biotransformation of both protease inhibitors in human enterocyte microsomes was NADPH dependent. The rate of metabolism of indinavir was linear during 1-hr incubations (fig. 2). In contrast to indinavir, the time course of the metabolism of ritonavir in incubations with enterocyte and liver microsomes and those containing expressed CYP3A4 was nonlinear (fig. 2). The time courses of metabolism of ritonavir in enterocyte microsomes at two different protein concentrations (0.4 and 2 mg/ml) were the same, when expressed per milligram of microsomal protein. Preincubation of ritonavir with enterocyte or liver microsomes for 10 min without NADPH had no effect on the time course of its metabolism initiated by addition of NADPH. The time course of the formation of ritonavir metabolite R3was linear for 20 min in liver microsomes but not in enterocyte microsomes (fig. 3).

Time course of the in vitrometabolism of indinavir and ritonavir.
Enterocyte microsomes (EMS) (2 mg/ml) were incubated with 5 μM [14C]indinavir or [14C]ritonavir in the presence of NADPH (2 mM). [14C]Ritonavir (2 μM) was also incubated with microsomes containing expressed CYP3A4 (50 pmol/ml). Samples were analyzed for the formation of total metabolites as described inMaterials and Methods. Each data point is the mean of two determinations. The time-course data for ritonavir metabolism were fitted to eq. 2 using SigmaPlot software (Jandel Scientific).

Time course of the formation of metabolite R3 in incubations of ritonavir with liver and intestinal microsomes.
Ritonavir (5 μM) was incubated with human liver (0.05 mg/ml) or enterocyte (0.4 mg/ml) microsomes for up to 20 min, and the samples were analyzed for the formation of R3 as described inMaterials and Methods. Each data point is the mean of two determinations.
Metabolism of Indinavir.
The profiles of metabolites in enterocyte and liver microsomes and expressed CYP3A4 were basically the same (fig.4). A minor difference involved metabolite In1, which was produced by liver but not enterocyte microsomes or expressed CYP3A4. In1 was one of two products formed by CYP2D6. The patterns of metabolites of indinavir in incubations with microsomes containing expressed CYP3A5 or CYP3A4 were different. Five products were formed in incubations with CYP3A4, whereas only three metabolites appeared in incubations with CYP3A5. Metabolites produced in incubations of indinavir with human liver microsomes resulted from oxidation of the 1) indanyl (one oxidation site) and 1,1-dimethylethylaminocarbonylpiperazinyl (two oxidation sites) groups (In1 and In2), 2) piperidinyl (one oxidation site) and 1,1-dimethylethylaminocarbonylpiperazinyl (two oxidation sites) groups (In3), 3) phenylmethyl (one oxidation site) and 1,1-dimethylethylaminocarbonylpiperazinyl (two oxidation sites) groups (In4), and 4) 1,1-dimethylethylaminocarbonyl (one oxidation site) and indanyl (one oxidation site) moieties (In6).
The enzyme kinetic parameters for indinavir metabolism by CYP3A4 and CYP3A5 also differed from each other (table1). The apparentKM values were similar in enterocyte and liver microsomes (0.25 ± 0.07 and 0.21 ± 0.13 μM, respectively), whereas the Vmax values were >2-fold higher in enterocyte than liver microsomes (163 ± 35 and 68 ± 44 pmol/min/mg, respectively).
Kinetic parameters for indinavir metabolism by individual liver (LMS) and enterocyte (EMS) microsomes and expressed CYP3A4/CYP3A5 enzymes
Metabolism of Ritonavir.
The profile of ritonavir metabolites in enterocyte microsomes differed from that in liver microsomes (fig. 5). Enterocyte microsomes biotransformed ritonavir to many products, of which only four (R1–R4) were detectable in incubations with liver microsomes. The major metabolites of ritonavir isolated from incubations with human liver microsomes were identified as products of 1) N-demethylation (R1), 2) oxidation of the methylthiazolyl moiety (the exact site, on the methylene carbon, nitrogen, or sulfur of the thiazolyl ring, is still unknown) (R2), 3) hydroxylation of the isopropyl side chain (R3), and 4) cleavage of the 2-(1-methylethyl)thiazolylmethyl group (R4). The ritonavir metabolites R2, R3, and R4 had been previously isolated and identified in incubations with human liver microsomes (Kumar et al., 1996), whereas the demethylated product (R1) was new.
The KM andVmax values for the total metabolism of ritonavir in enterocyte microsomes were 0.063 ± 0.045 μM and 89.4 ± 59.3 pmol/min/mg protein, respectively. Both the patterns of metabolites and the kinetic parameters for metabolism of ritonavir by recombinant CYP3A enzymes were similar to those in enterocyte microsomes (fig. 5, table 2). Expressed CYP3A4 and CYP3A5 metabolized ritonavir with very similar profiles of metabolites and kinetic parameters (table 2). In liver microsomes, theKM for the formation of the major metabolite (R3) was 0.92 ± 0.07 μM and theVmax was 20.1 ± 2.2 pmol/min/mg protein. CYP2D6 catalyzed the formation of only one metabolite of ritonavir, R3 (not shown), with aKM of 1.0 μM and aVmax of 0.93 pmol/min/pmol CYP.
Kinetic parameters for ritonavir metabolism by individual liver (LMS) and enterocyte (EMS) microsomes and expressed CYP enzymes
Inhibition by Ketoconazole.
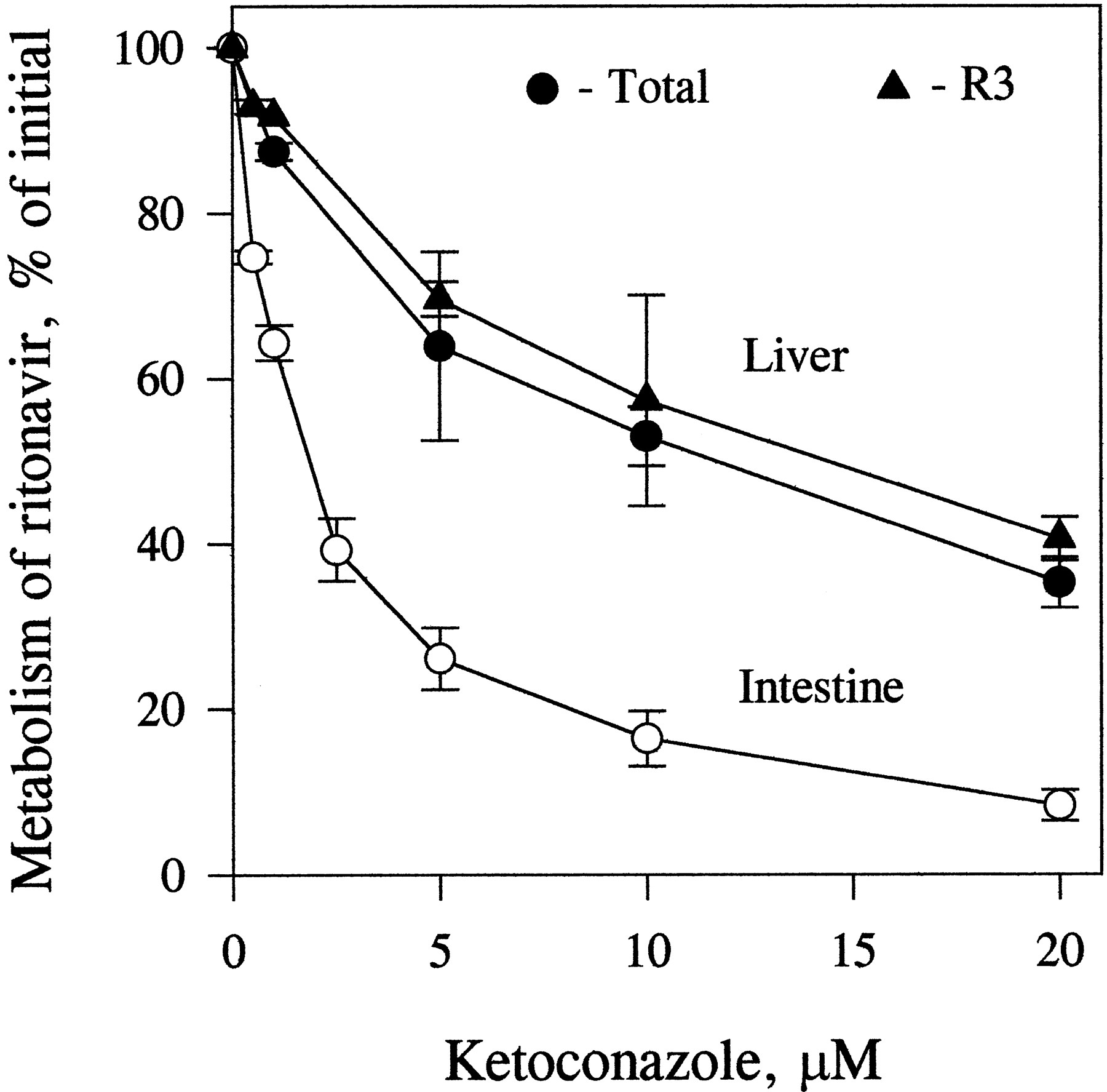
Ketoconazole inhibited indinavir metabolism similarly in enterocyte and liver microsomes, in a concentration-dependent fashion (>90% at 1 μM). Ketoconazole also inhibited the metabolism of ritonavir in both enterocyte and liver microsomes (fig. 6). However, the inhibition was more marked in enterocyte microsomes than in liver microsomes (75 and 35%, respectively, at 5 μM).

Inhibition of the metabolism of ritonavir by ketoconazole in incubations with liver and enterocyte microsomes.
Liver (0.4 mg/ml) and enterocyte (0.2 mg/ml) microsomes were incubated with [14C]ritonavir (0.5 μM) in the presence of different concentrations of ketoconazole for 15 min at 37°C. The percentage of control activity was calculated for the formation of total metabolites and R3 (liver microsomes). Data points are means ± SD for three determinations.
Immunoinhibition.
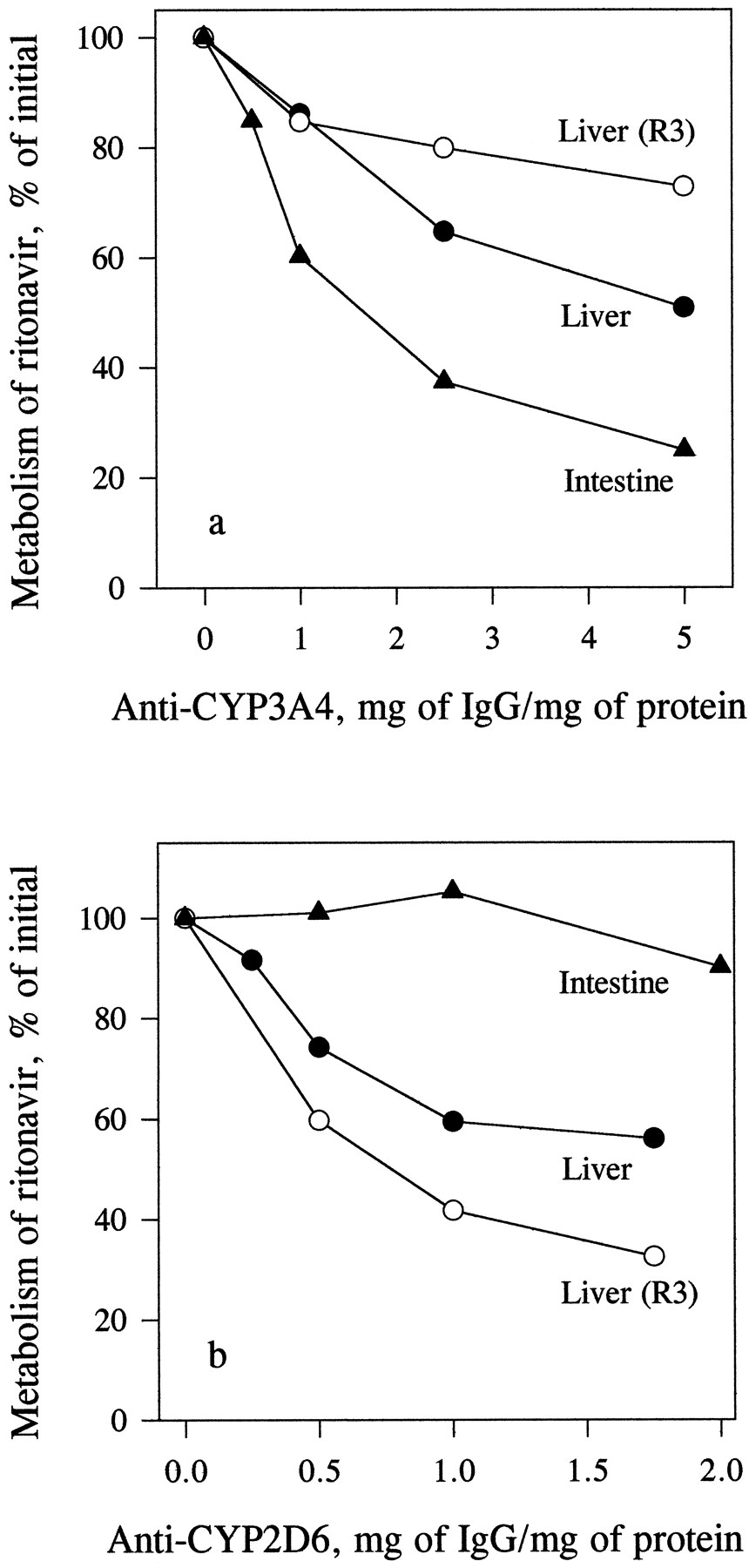
MAB3A4 (5 mg/mg protein) inhibited the metabolism of indinavir in human enterocyte microsomes by 100% and that in liver microsomes by 78%. The inhibition of the metabolism of ritonavir by MAB3A4 (5 mg/mg protein) in enterocyte microsomes was 75%; in liver microsomes, the inhibition of total metabolism was 49%, whereas formation ofR3 was inhibited by only 27% (fig.7a). Preincubation with polyclonal antibody against CYP2D6 (2 mg/mg protein) did not significantly affect the metabolism of ritonavir in enterocyte microsomes but inhibited total metabolism of ritonavir in liver microsomes by 44% and the formation of R3 by 67% (fig.7b).

Inhibition of the metabolism of ritonavir in liver and enterocyte microsomes by antibodies to CYP3A4 (a) and CYP2D6 (b).
Liver (0.4 mg/ml) and enterocyte (0.2 mg/ml) microsomes, preincubated with varying concentrations of MAB3A4 for 15 min or polyclonal anti-CYP2D6 antibody for 30 min at room temperature, were incubated with [14C]ritonavir (0.5 μM) for 15 min, and the samples were analyzed for total metabolism of ritonavir as described inMaterials and Methods. Each data point is the mean of two determinations.
Coincubation of Indinavir and Ritonavir.
When the two protease inhibitors, ritonavir (0.05 μM) and indinavir (5 μM), were together incubated with human enterocyte microsomes for up to 1 hr, the rate of indinavir metabolism was decreased by about 35% at all times (except for 10-min incubations, where the metabolism was reduced by 10%), compared with control incubations without ritonavir (fig. 8). In these incubations, 75% of ritonavir was metabolized in the first 10 min.

Metabolism of indinavir in human enterocyte microsomes in the presence of ritonavir.
[14C]Indinavir (5 μM) and ritonavir (0.05 μM) were simultaneously incubated with enterocyte microsomes (0.5 mg/ml) for up to 1 hr, and the samples were analyzed by HPLC for total metabolism of indinavir, as described in Materials and Methods. Each data point is the mean of two determinations.
Metabolic Inactivation of CYP by Ritonavir.
Ritonavir at a concentration 0.075 μM inhibited the metabolism of indinavir (5 μM) in enterocyte microsomes by 62%. The inhibition of indinavir metabolism by ritonavir was significantly greater (96%) when enterocyte microsomes were preincubated with ritonavir, in the presence of NADPH, before the addition of indinavir (fig.9). During the 20-min preincubation, ritonavir was completely metabolized.
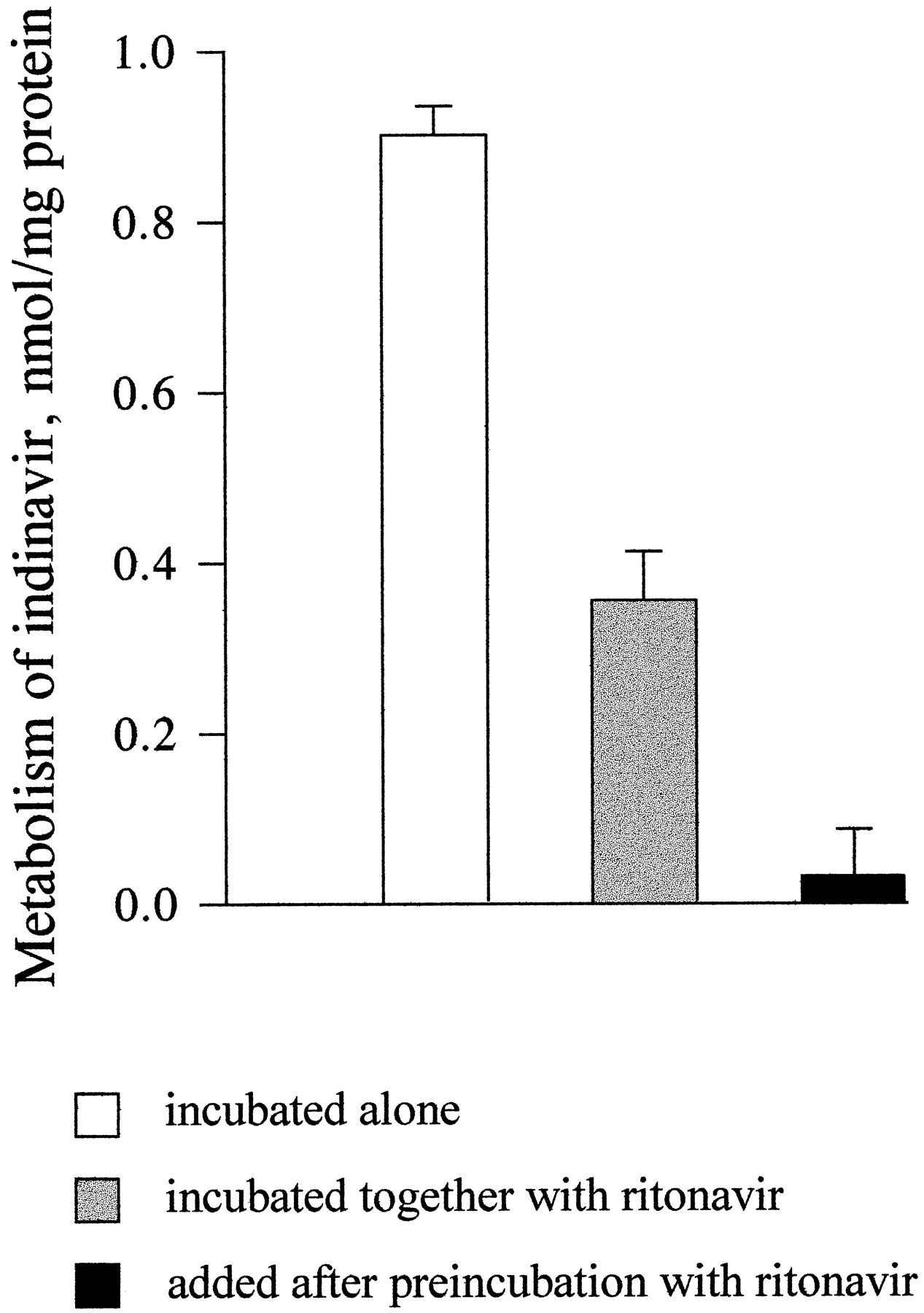
Metabolism of indinavir by intestinal microsomes preincubated with ritonavir.
[14C]Indinavir (5 μM) was added to human enterocyte microsomes (0.5 mg/ml) alone, together with ritonavir (0.075 μM), or after a 20-min preincubation with 0.075 μM ritonavir, and the mixtures were incubated for 20 min. The samples were analyzed for the formation of total metabolites of indinavir as described inMaterials and Methods. Data are the mean ± SD of three determinations.
The metabolism of ritonavir by enterocyte microsomes was suppressed by >90% when indinavir (100 μM) and ritonavir (0.075 μM) were coincubated. Under these conditions, the presence of ritonavir had no effect on the rate of indinavir metabolism (139.0 ± 14.2 and 130.4 ± 4.6 pmol/min/mg in the presence and absence of ritonavir, respectively).
The mixture of ritonavir metabolites produced in a 15-min incubation of the drug with human enterocyte microsomes and separated from the parent compound by HPLC had no effect on the metabolism of ritonavir by enterocyte microsomes. The supernatant, containing metabolites of ritonavir, obtained by ultracentrifugation after a 30-min incubation of ritonavir with human liver microsomes failed to alter the metabolism of the drug by fresh liver microsomes. In contrast, the rate of metabolism of ritonavir by liver microsomal protein that had been previously incubated with the drug and repeatedly washed was significantly reduced, compared with similarly treated control microsomes (fig.10). The addition of reduced glutathione, ascorbic acid, N-acetylcysteine, or superoxide dismutase failed to protect liver CYPs from inactivation by ritonavir.

Metabolism of ritonavir by liver microsomal protein preincubated with (○) or without (•) ritonavir and repeatedly washed in phosphate buffer, pH 7.
Human liver microsomes (0.5 mg/ml) in a volume of 5 ml were preincubated with [14C]ritonavir (5 μM) and NADPH (2 mM) in triplicate for 30 min at 37°C. Similar incubations without ritonavir served as controls. The incubation mixtures were placed in ice for 1 min and centrifuged for 30 min at 100,000g at 4°C. Each pellet was resuspended in 10 ml of ice-cold phosphate buffer, and the centrifugation was repeated. The microsomal pellet from each incubation was homogenized in 5 ml of fresh phosphate buffer and incubated with [14C]ritonavir (5 μM) and NADPH, in 0.5-ml aliquots, for up to 20 min. The samples were analyzed for the disappearance of ritonavir as described in Materials and Methods. Data are the mean ± SD.
The kin values determined for CYP3A4 and enterocyte microsomes (EMS 15) from time-course studies were 0.135 and 0.078 min−1, respectively. The partition ratio for inactivation of CYP3A4 was 10.
Discussion
This study demonstrated that both indinavir and ritonavir are readily metabolized by CYP3A enzymes in the small intestine. The metabolite patterns for indinavir were similar in enterocyte, liver, and CYP3A4 microsomes, with the exception of In1, which was formed only by liver or CYP2D6 microsomes. These results, together with the immunoinhibition data, suggest a minimal contribution of intestinal CYP2D6 to the metabolism of indinavir. The kinetic parameters for indinavir were different in incubations with CYP3A4 and CYP3A5; theKM and Vmaxvalues for CYP3A5 were 5 times and one third of the values for CYP3A4, respectively. Moreover, CYP3A5 failed to produce some of the metabolites (In3, In6, and In7) formed by CYP3A4. Marked interpatient heterogeneity (>10-fold) in the expression of both CYP3A4 and CYP3A5 in the intestine (Lown et al., 1994) may account for significant interindividual variability in the metabolism of indinavir in humans. The metabolism of indinavir by intestinal, hepatic, and recombinant CYP3A enzymes was characterized by low KM values (0.04–0.2 μM). TheKM values estimated in the present study were 5–10-fold lower and the Vmax values were 3–10-fold higher than those previously reported (Chiba et al., 1997). This discrepancy in the kinetic parameters can be explained by the different sources of microsomes and/or a lack of standardization of the procedure for preparation of human intestinal microsomes. An additional factor contributing to the variability of parameters is the difficulty of maintaining Michaelis-Menten conditions (i.e. constant substrate concentrations) for compounds with low KM values (<1 μM).
Chiba et al. (1997) estimated the intestinal first-pass metabolism of indinavir based on in vitro data and intestinal blood flow. However, available models of organ clearance are not applicable for estimation of the intestinal extraction ratio or the contribution of the intestine to bioavailability, because drug reaches the circulation after passage through the epithelial lining of the intestine, a process that is not limited by blood flow. Several variables, including the rates of biotransformation and absorption, determine the extent of first-pass metabolism in the wall of the gut. The concentration of indinavir in enterocytes during absorption appears to be ≫KM . Under these conditions, indinavir is metabolized at a velocity comparable toVmax. The concentration of CYP is highest in the duodenum and decreases gradually toward the ileum (Kolarset al., 1992). Therefore, the exact mass of enzyme to which the drug is exposed during absorption is unknown. We estimated the quantity of microsomal protein in a 3-m segment of the small intestine, based on the recovery from a 4-cm segment of jejunum, to be approximately 9 g. The amount of indinavir that can be biotransformed by 9 g of intestinal microsomal protein atVmax (163 pmol/min/mg protein) during the absorption phase (approximately 60 min) is 88 μmol or 63 mg of indinavir sulfate. Therefore, the contribution of the small intestine to the first-pass metabolism of indinavir depends on the dose. Recently, we demonstrated (Koudriakova et al., 1996) that the oral bioavailability of rifabutin, which is primarily biotransformed by enterocytes in rats, is dose dependent (>90% at the 25 mg/kg dose and <50% at the 1 mg/kg dose).
A single oral dose of 400 mg of indinavir given to healthy volunteers (Balani et al., 1996) produces a mean maximal plasma concentration of 4.7 μM, which is much greater than theKM , even adjusting for binding of indinavir to plasma proteins (70%). Similarly to calculations for the intestine, the amount of indinavir sulfate that could be metabolized by the liver (Vmax of 68 pmol/min/mg protein), scaled up for the whole organ (Houston, 1994), during a 1-hr absorption phase can be estimated to be approximately 205 mg. The hepatic intrinsic clearance (CLint), expressed using the Michaelis-Menten kinetic parameters (Wilkinson, 1987) asCLint =Vmax/(KM +S), is dose dependent for S ≥KM . The hepatic extraction ratio (E) expressed, according to the “well-stirred” model (Pang and Rowland, 1977), as E =CLint/(Q +CLint), where Q is hepatic blood flow, also depends on the dose. Thus, the first-pass metabolism of indinavir in the liver would be expected to be dose dependent. A greater than dose-proportional increase in plasma concentrations of indinavir in the 200-1000-mg dose range has been reported (Merck & Co., 1996).
The profiles of ritonavir metabolites in incubations with enterocyte and liver microsomes differed mainly in the 5–10-fold increase in the proportion of metabolite R3 in liver microsomes. As reported earlier (Kumar et al., 1996) and confirmed in our study, the formation of R3 is catalyzed by both CYP3A4 and CYP2D6 enzymes. The comparison of the KM values for metabolism in enterocyte, liver, and cDNA-expressed CYP microsomes, together with immunoinhibition studies, indicated that CYP2D6 contributes significantly to the overall metabolism of ritonavir in the liver but not in the intestine. The metabolism of ritonavir in enterocyte microsomes was substantially inhibited by both ketoconazole (a potent inhibitor of CYP3A) and MAB3A4, indicating the essential role of CYP3A in the intestinal metabolism of the drug. In contrast to indinavir, there was no significant difference in the metabolism of ritonavir by CYP3A4 and CYP3A5.
The metabolism of ritonavir by enterocyte microsomes and expressed CYP3A enzymes was also characterized by very low apparentKM values (< 0.1 μM). However, in contrast to indinavir, the time courses of its biotransformation by enterocyte and liver microsomes and expressed CYP3A4 were not linear. Detailed investigation of reasons for this nonlinearity, including nonspecific binding of ritonavir to microsomal proteins or inhibition by products, showed that this decrease in the velocity of the reaction was directly related to the metabolism of ritonavir. The metabolites of ritonavir added to incubations had no effect on the rate of the metabolism of ritonavir by intact microsomes. The velocity of the reaction rapidly declined over time, even when enterocyte or liver microsomes were incubated with a large excess of ritonavir (<5% substrate disappearance in 1 hr). This ruled out the possibility of competitive inhibition by products as a reason for the nonlinearity. The loss of CYP activity appears to follow pseudo-first order kinetics. Moreover, the enzyme activity of microsomal protein that had been preincubated with ritonavir and repeatedly washed was significantly decreased. Collectively, these data indicate that the metabolism of ritonavir is accompanied by inactivation of CYP. The formation of metabolite R3, which is primarily catalyzed by CYP2D6 in the liver, was proportional to time in incubations with liver microsomes. This indicates a selective inactivation of CYP3A without a comparable effect on CYP2D6. The kin (0.135 min−1), which is 3-fold lower than that for gestodene, one of the most powerful mechanism-based enzyme inactivators (Guengerich, 1990), and the partition ratio of 10 indicate that ritonavir is a potent inactivator of CYP3A4. Many compounds are mechanism-based inactivators of CYP, including phencyclidine (Crowley and Hollenberg, 1995), halothane (Saton et al., 1985), parathion (Butler and Murray, 1997), chloramphenicol (Kraner et al., 1994), and methoxsalen (Tinel et al., 1987; Mayset al., 1990). L-754,394, an investigational protease inhibitor exhibiting nonlinear pharmacokinetics, was reported to be a potent inactivator of CYP3A4 and, to a lesser extent, CYP2D6 (Chibaet al., 1995; Lin et al., 1995).
Ritonavir potently inhibits the metabolism of indinavir and other HIV protease inhibitors (Kempf et al., 1997). We investigated the nature of this inhibition in vitro. When ritonavir and indinavir were incubated together, the inhibitory effect of ritonavir lasted long after the latter had disappeared. The suppression of indinavir metabolism was substantially greater when ritonavir was completely metabolized by preincubation with enterocyte microsomes before the addition of indinavir. Moreover, when the metabolism of ritonavir was inhibited by coincubation with a very high concentration of indinavir (100 μM), no inhibition of indinavir metabolism occurred. This shows that the potent inhibitory effect of ritonavir is associated with its metabolism. These data, together with a lack of protection by free radical-trapping agents, indicate that the sustained inhibition by ritonavir of its own metabolism and that of indinavir is related to formation of reactive intermediates that inactivate CYP3A. Comparison of ritonavir with some of its structural analogs (Kempfet al., 1997) showed that the potent inhibitory properties of ritonavir required the presence of both the 5-thiazolyl and 2-(1-methylethyl)thiazolyl groups. We theorize that inactivation of CYP3A by ritonavir occurs via the formation of a chemically reactive intermediate, possibly a fragment containing the 2-(1-methylethyl)thiazolyl group. The product of cleavage of the 2-(1-methylethyl)thiazolylmethyl portion of ritonavir (R4) was detected in incubations with enterocyte, liver, and expressed CYP3A4 and CYP3A5 microsomes.
It is difficult to extrapolate in vitro kinetic parameters for ritonavir to in vivo conditions, because of time- and dose-dependent inactivation of hepatic and intestinal enzymes by the drug in vivo. Nevertheless, some extrapolations are appropriate. For example, the pharmacokinetics of ritonavir in humans would be expected to be nonlinear, based on our in vitrodata. Indeed, the kinetics of the drug in HIV-positive subjects were reported to be dose dependent (Hsu et al., 1997). Inactivation of intestinal and hepatic CYP3A during initial transit of the oral dose of ritonavir increases its bioavailability and diminishes the rate of its systemic elimination. This explains the high bioavailability and longer half-life of ritonavir, compared with other protease inhibitors, such as saquinavir and indinavir. Ritonavir is also biotransformed by CYP2D6, which, in contrast to CYP3A, is not inactivated and likely becomes the predominant CYP that metabolizes the drug in vivo. The hydroxyisopropyl derivative (R3), formed by CYP2D6, was the only metabolite of ritonavir detected in human plasma and the major metabolite excreted in feces and urine (Denissen et al., 1997). Finally, inactivation of CYP3A by ritonavir also explains the pronounced and sustained elevation of the plasma levels of other protease inhibitors that are substrates of CYP3A.
Acknowledgments
We thank Dr. Richard Granneman (Abbott Laboratories) for valuable comments during preparation of this manuscript.
Footnotes
-
Send reprint requests to: Tatiana Koudriakova, Department of Pharmacology, The Ohio State University, 5084 Graves Hall, 333 W. 10th Ave., Columbus, OH 43210.
- Abbreviations used are::
- HIV
- human immunodeficiency virus
- CYP
- cytochrome P450
- ACN
- acetonitrile
- TEA
- triethylamine
- TFA
- trifluoroacetic acid
- MAB3A4
- monoclonal antibody to cytochrome P4503A4
- kin
- inactivation rate constant
- ESI
- electrospray ionization
- CID
- collision-induced dissociation
- Received October 31, 1997.
- Accepted February 26, 1998.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}