


Implications for Drug-Drug Interactions and Pharmacological Activity of the Main Metabolite
Abstract
The metabolism of valspodar (PSC 833; PSC), which is developed as a multidrug resistance-reversing agent, was investigated to assess the potential for drug-drug interactions and the pharmacological activity of major metabolites. The primary metabolites of PSC produced by human liver microsomes were monohydroxylated, as revealed by LC/MS. The major site of hydroxylation was at amino acid 9, resulting in M9, as determined by cochromatography with synthetic M9. Dihydroxylated and N-demethylated metabolites were also detected. PSC metabolism in two human livers exhibitedKM values of 1.3–2.8 μM. The intrinsic clearance was 9–36 ml/min/kg of body weight. PSC biotransformation was cytochrome P450 (CYP or P450) 3A dependent, based on chemical inhibition and on metabolism by Chinese hamster ovary cells expressing CYP3A. Ketoconazole was a competitive inhibitor (Ki = 0.01–0.04 μM). The inhibition by 27 compounds, including four antineoplastic agents, corresponded to the inhibitory potentials of these compounds toward CYP3A. For vinblastine, paclitaxel, doxorubicin, and etoposide, the IC50 values were 5, 12, 20, and 150 μM, respectively. M9 was also an inhibitor, with a lower apparent affinity for CYP3A (IC50 = 21 μM), compared with that of PSC. M9 was also less active as a multidrug resistance-reversing agent. M9 demonstrated low potency in sensitizing resistant cells to paclitaxel and was a poor inhibitor of rhodamine-123 efflux from paclitaxel-resistant cells. In addition, compared with PSC, a higher concentration of M9 was needed to compete with the photoaffinity labeling of P-glycoprotein. Conversely, PSC inhibited only reactions catalyzed by CYP3A, including cyclosporine A metabolism (IC50 = 6.5 μM) andp-hydroxyphenyl-C3′-paclitaxel formation (Ki = 1.2 μM). Thus, PSC behaves in a manner very similar to that of other cyclosporines, and a comparable drug-drug interaction profile is expected.
The development of MDR1 by the overexpression of the multidrug transporter Pgp is one of the major impediments to cancer chemotherapy (Pastan and Gottesman, 1988). Pgp is a member of a large multigene family of ATP-binding cassette membrane transporters and transports basic or neutral amphiphilic compounds, including some anticancer drugs, i.e. the epipodophyllotoxins (etoposide and teniposide), the anthracyclines (doxorubicin and daunorubicin), theVinca alkaloids (vincristine and vinblastine), and the taxanes (paclitaxel), out of cells (Pastan and Gottesman, 1991;Higgins, 1992). CSA is an effective MDR-reversing agent through its inhibition of the Pgp transport of anticancer agents (Nooteret al., 1990; Sonneveld and Nooter, 1990; Lehnert et al., 1993). Drawbacks to the use of CSA to reverse MDR in cancer therapy include its immunosuppressive action and the potential for renal side effects at the doses necessary to reverse MDR.
PSC is a new cyclosporine derivative that is superior to CSA as an MDR inhibitor. PSC is a very potent inhibitor of Pgp, being approximately 10-fold more potent than CSA, and has the safety advantage that it would not compromise patients because it lacks immunosuppressive activity and nephrotoxicity (Boesch et al., 1991; Twentyman and Bleehen, 1991; Te Boekhorst et al., 1992). In preclinical studies, the chemosensitizing effect of PSC was dose dependent. PSC significantly prolonged survival times of MDR-P388 tumor-bearing mice when combined with doxorubicin (Boesch et al., 1991) and increased the sensitivity toward etoposide of human carcinoma xenografts in nude mice (Keller et al., 1992). Furthermore, clinical treatment with PSC resulted in increased intracellular accumulation of doxorubicin and vincristine in Pgp-positive myeloma cells (Sonneveld et al., 1994).
The combination of PSC with a chemotherapeutic agent resulted in marked changes in the pharmacokinetics of the anticancer drugs. In clinical trials, administration of PSC resulted in increased neutropenia and significantly increased blood concentrations of doxorubicin, paclitaxel, and etoposide (Fisher and Sikic, 1995). Possible explanations for the reduced blood clearance of the anticancer agents in the presence of PSC include metabolic interactions at P450 and/or reduced elimination via the bile because of PSC inhibition of the biliary ATP-dependent carriers (Böhme et al., 1993).
PSC maintains a cyclic undecapeptide structure, like CSA. The clearance of CSA is dependent on biotransformation, and most drug-drug interactions are metabolic (Campana et al., 1996). PSC, like CSA, was slowly eliminated as metabolites in the bile in rats and dogs. PSC was also eliminated as metabolites after a single oral dose in human subjects, with <0.1% of the dose appearing as unchanged PSC in the urine and <14% (likely the result of unabsorbed PSC) appearing in the feces (Hauck C, personal communication).
Because the available data for PSC indicate a potential for drug-drug interactions similar to those of CSA and because PSC is intended to be used in combination therapy with anticancer agents, metabolic interactions were investigated using human liver tissue in vitro. Potential clinical consequences are predicted based on the enzyme(s) involved in metabolism and based on the reciprocal effects on biotransformation of PSC and potentially coadministered drugs.
Materials and Methods
Chemicals.
Unlabeled PSC, [3H]PSC (518 GBq/mmol), and [14C]PSC (1.96 GBq/mmol) [cyclo-[(N-methyl)-3-oxo-5-[1(E)-propenyl]-L-leucyl-L-[[2,3,4-3H] or [1-14C]]valyl-sarcosyl-(N-methyl)-L-leucyl-L-valyl-(N-methyl)-L-leucyl-L-alanyl-D-alanyl-(N-methyl)-L-leucyl-(N-methyl)-L-leucyl-(N-methyl)-L-valine]], the PSC metabolite M9[cyclo-[(N-methyl)-3-oxo-5-[1(E)-propenyl]-L-leucyl-L-valyl-sarcosyl-(N-methyl)-L-leucyl-L-valyl-(N-methyl)-L-leucyl-L-alanyl-D-alanyl-(N-methyl-4-hydroxy)-L-leucyl-(N-methyl)-L-leucyl-(N-methyl)-L-valine]], unlabeled and 14C-labeled tropisetron (1.98 GBq/mmol) [1H-indole-3-[14C]carboxylic acid (1"H,5"H)-8-methyl-8-azabicyclo[3.2.1]oct-3"-yl-ester], unlabeled and 14C-labeled ondansetron (1.60 GBq/mmol) [1,2,3,4-tetrahydro-9-methyl-3-[(2-methyl-1H-imidazol-1-yl)-[14C]methyl]carbazol-4-one], bromocriptine, CSA, etoposide, fluvastatin, diclofenac, and (S)-mephenytoin were all from Novartis Pharma Inc. (Basel, Switzerland). The purity of labeled PSC was >97% and that of labeled tropisetron and ondansetron was >98%, as assessed by HPLC.
[3H]CSA (374 GBq/mmol), [14C]doxorubicin (2.11 GBq/mmol), [14C]chlorzoxazone (2.18 GBq/mmol), [14C]tolbutamide (2.00 GBq/mmol), and (S)-[14C]mephenytoin (2.22 GBq/mmol) were obtained from Amersham International plc (Little Chalfont, UK). [3H]Paclitaxel (618 GBq/mmol) was obtained from Moravek Biochemicals (Brea, CA). Antipyrine, chlorpropamide, chlorzoxazone, cholchicine, cimetidine, dextromethorphan, doxorubicin, erythromycin, ethynylestradiol, glyburide, ketoconazole, nifedipine, [14C]phenacetin (0.46 GBq/mmol), quinidine, sparteine, paclitaxel, tolbutamide, vinblastine, and theophylline and its metabolites (1-methyluric acid, 1,3-dimethyluric acid, 3-methylxanthine, and 1-methylxanthine) were purchased from Sigma Chemical Co. (St. Louis, MO). [3H]Glyburide (1887 GBq/mmol) was from DuPont de Nemours (Brussels, Belgium). 4-Hydroxymephenytoin, bufuralol, and hydroxybufuralol were obtained from Ultrafine Chemicals (Manchester, UK). [14C]Theophylline (1.93 GBq/mmol) was obtained from Anawa Trading SA (Wangen, Switzerland), and unlabeled phenacetin was from Fluka (Buchs, Switzerland). Lovastatin was a gift from Merck Sharp and Dome (Rahway, NJ), and doxorubicinol was synthesized by Mercian (Tokyo, Japan). All other reagents were obtained from commercial sources and were of the highest grade available.
Human Liver Preparations.
Human liver tissue that could not be used for transplantation was obtained as either pieces or microsomes from the International Institute for the Advancement of Medicine (Exton, PA) (HHM-0011 and GGM-002) or from Vitron Inc. (Tucson, AZ) (HL-44). Microsomes from livers GGM-002 and M8 were prepared by differential centrifugation as previously described (Ball et al., 1992). Microsomal protein concentrations were determined by the method of Bradford (1976), and the P450 contents were determined from spectra obtained with carbon monoxide according to the method of Omura and Sato (1964). Total P450 contents were 0.89, 0.44, 0.29, and 0.23 nmol of P450/mg of protein for microsomal preparations HHM-0011, GGM-002, M8, and HL-44, respectively. Human liver S9 fractions EOH 368–04 and EJS 882–08 were obtained from Human Biologics Inc. (Phoenix, AZ) and exhibited P450 contents of 0.070 and 0.082 nmol/mg of protein, respectively.
Recombinant Human Proteins.
cDNA encoding either CYP3A4, CYP3A5, or CYP2D6 was cloned from human liver mRNA that was reverse-transcribed into first-strand cDNA and subjected to amplification by polymerase chain reaction. Specific oligonucleotides were used that spanned full-length P450, with CYP3A4-back and CYP3A4-forward primers (5′-agataagtaaggaaagtagtgatggc and 5′-tggatgaagcccatcttcatttcaga, respectively), CYP3A5-back and CYP3A5-forward primers (5′-agataagtaaggaaagtagtgatggc and 5′-tggatgaagcccatcttcatttcaga, respectively), and CYP2D6-back and CYP2D6-forward primers (gcaggtatggggctagaagcactggtg and agcaggctggggactaggtaccccattcta, respectively). The cDNA clones were subjected in their entirety to double-stranded sequencing. For CYP3A4 and CYP3A5, the sequences were in complete agreement with those previously reported (Gonzalez et al., 1988a; Aoyamaet al., 1989), whereas a difference of 1 base pair was found for CYP2D6, resulting in an arginine to cysteine exchange at position 140, compared with the published sequence (Gonzalez et al., 1988b). All three cDNAs were subcloned into an expression vector containing a cytomegalovirus promoter/enhancer unit from commercial vector pcDNA1 NEO (Invitrogen, Leek, The Netherlands). CHO-K1 cells were used as recipients for transfection. Stable geneticin-resistant cell clones with P450 activity were obtained and propagated in Dulbecco’s modified Eagle medium containing 25 mMN-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 1 mM sodium pyruvate, 4 mM glutamine, and 5.5 mM D-glucose. The medium also contained 100 μg/ml gentamicin, 0.4 mg/ml geneticin sulfate, and 0.4 mM L-proline and was supplemented with 10% fetal calf serum. Microsomes from cells expressing CYP3A4, CYP3A5, or CYP2D6 were prepared from CHO-K1 recombinant cells as for human liver microsomes. Isolated cells were lysed using a hand-held homogenizer, and cellular debris was removed by centrifugation at 10,000g for 20 min. The supernatant was centrifuged at 100,000g for 70 min, and the microsomal fraction was resuspended in 10 mM potassium phosphate, pH 7.4, 1 mM dithiothreitol, 20% glycerol, and stored at −80°C before use. The specific P450 contents, as measured in spectra obtained with carbon monoxide, were 70 and 134 pmol of P450/mg of microsomal protein for P4503A4 and CYP2D6, respectively. The specific CYP3A5 content was less than spectrally detectable. The corresponding catalytic activity for CYP2D6-mediated dextromethorphanO-demethylation was 80 nmol/hr/nmol of P450 (7.1 nmol/hr/mg of protein) with 5 μM dextromethorphan; the activities for CYP3A4- and CYP3A5-mediated CSA metabolism were 28 nmol/hr/nmol of P450 (2.1 nmol/hr/mg of protein) and 0.3 nmol/hr/mg of protein with 1 μM CSA, respectively.
Metabolism.
Human liver microsomal incubations were performed in 500 μl of 0.1 M phosphate buffer, pH 7.4, at 37°C, except for PSC and CSA (200 μl) and theophylline (100 μl). The substrate/inhibitor was added in dimethylsulfoxide or ethanol, except for the chlorzoxazone incubations, in which chlorzoxazone was dissolved in 60 mM KOH and PSC in polyethylene glycol 400. The final vehicle concentration did not exceed 1%. Metabolism was initiated by the addition of 0.2 mM β-NADPH and the NADPH-regenerating system, to yield final concentrations of 1 mM NADP, 5 mM isocitrate, 5 mM MgCl2, and 1 unit of isocitrate dehydrogenase. The reactions were stopped with an equal volume of cold methanol [CSA, (S)-mephenytoin, doxorubicin, and doxorubicinol], 70% perchloric acid (bufuralol), or 10% trichloroacetic acid, with cooling on dry ice. Control incubations were performed in the absence of β-NADPH and the regenerating system or in the absence of microsomal protein.
Additional typical incubation conditions, i.e. substrate concentration, incubation time, and microsomal protein content, respectively, were as follows: PSC, 0.1–25 μM, 15 min, 50 μg; bufuralol, 5 μM, 60 min, 100 μg; [14C]chlorzoxazone, 40 μM, 20 min, 50 μg; [3H]CSA, 1 μM, 15 min, 50 μg; dextromethorphan, 5 μM, 30 min, 50 μg; [14C]doxorubicin and doxorubicinol, 40 μM, 60 min, 2 mg of S9 protein; [3H]glyburide, 5 μM, 40 min, 100 μg; (S)-[14C]mephenytoin, 100 μM, 30 min, 500 μg; [14C]phenacetin, 20 μM, 30 min, 100 μg; [3H]paclitaxel, 1–100 μM, 20 min, 50 μg; [14C]theophylline, 50 μM, 60 min, 200 μg; [14C]tolbutamide, 150 μM, 40 min, 100 μg; [14C]tropisetron and [14C]ondansetron, 50 μM, 40 min, 1 mg. For identification of metabolites by LC/MS, higher protein concentrations and longer incubation times may have been used. For all kinetic studies, the incubations were performed under linear conditions with respect to time and protein concentration. IC50values for PSC (1 μM) metabolism were determined in the absence and presence of potential inhibitors (ranging in concentration from 0.1 to 1000 μM, depending on solubility). For the microsomes derived from the CYP3A4-, CYP3A5-, and CYP2D6-expressing cells, PSC and paclitaxel were incubated primarily as described above, except that 250 mM potassium phosphate buffer, pH 7.6, was used for CYP3A4 and CYP3A5.
HPLC Analysis.
Proteins were sedimented by centrifugation at 100,000g for 10 min at room temperature, and aliquots (200–350 μl) of the supernatant were injected into the HPLC system. HPLC separations were performed with a Kontron system (Kontron Instruments, Zürich, Switzerland), a LB 507A radioactivity monitor (Berthold AG, Wildbad, Germany), and a F1000 fluorescence detector (Merck, Darmstadt, Germany). On-line radioactivity monitoring was used unless stated otherwise. All compounds and their metabolites were characterized by their retention times, by comparison of the chromatograms with those of reference compounds, and/or by LC/MS analysis. The quantitation of metabolites of radiolabeled compounds was performed by calculating the relative peak areas of parent drug and metabolite peaks; for unlabeled compounds, concentrations were calculated from standard curves.
[3H or 14C]PSC and its metabolites were chromatographed at 75°C on LC 18 columns in series (5-μm particle size, one column of 20 × 4.6 mm and two of 250 × 4.6 mm; Supelco Inc., Bellefonte, PA). The mobile phases were 10 mM NH4HCO3/methanol (9:1, pH 8.1) as solvent A and acetonitrile/methanol (9:1) as solvent B. The proportion of solvent B was 0% for the first 1 min, was then increased linearly to reach 30% at 10 min, 40% at 20 min, 70% at 120 min, and 90% at 140 min, and remained constant until 160 min. The total flow rate was 0.8 ml/min. For LC/MS, mobile phase A was 50 mM ammonium acetate/methanol (9:1), and the proportion of solvent B was 0% for the first 10 min and was then increased linearly to reach 30% at 20 min, 70% at 157 min, and 90% at 165 min. The total flow rate was 1.2 ml/min.
Bufuralol and its metabolites were analyzed at room temperature on a LC 18-DB analytical column (5-μm particle size, 250 × 4.6 mm), with a total flow rate of 1 ml/min. The mobile phases were 65% 0.2 M NaClO4, pH 4/35% acetonitrile as solvent A and acetonitrile as solvent B. The proportion of solvent B was 0% for the first 5 min, was increased linearly to 60% at 15 min, remained constant until 20 min, and was then increased to 100% at 25 min. The fluorescence detector was set to an excitation wavelength of 252 nm and an emission wavelength of 302 nm.
[14C]Doxorubicin and its metabolites were chromatographed at 40°C on a LC 18 analytical column (5-μm particle size, 250 × 4.6 mm). The mobile phases were 0.28 M sodium formate (adjusted to pH 3.55 with formic acid) as solvent A and methanol as solvent B. The proportion of solvent B was 0% for the first 2 min, was increased linearly to reach 35% at 5 min, 60% at 65 min, and 100% at 67 min, and remained constant until 75 min. The total flow rate was 1 ml/min. Doxorubicinol metabolism was monitored at 254 and 490 nm.
[14C]Chlorzoxazone and its metabolite 6-hydroxychlorzoxazone were separated at room temperature on Spheri-5 phenyl columns (5-μm particle size; Brownlee, San Jose, CA),i.e. a guard column (30 × 4.6 mm) and an analytical column (100 × 4.6 mm). The mobile phase consisted of 90% 0.018 M ammonium acetate buffer, adjusted to pH 4 with acetic acid, and 10% acetonitrile. The flow rate was 1 ml/min.
[3H]Glyburide and its metabolites were separated at 40°C on Brownlee RP-18 columns (5-μm particle size; 30 × 2.1 mm and 100 × 2.1 mm). The mobile phases were 10 mM ammonium acetate, pH 4.3, as solvent A and acetonitrile as solvent B, with a total flow rate of 0.4 ml/min. The proportion of solvent B was 0% up to 2 min, was increased linearly to reach 45% at 55 min, 80% at 62 min, 100% at 72 min, and remained constant until 75 min.
(S)-[14C]Mephenytoin and its metabolites were separated at 45°C on Brownlee RP-18 columns (5-μm particle size, 30 × 4.6 mm and 100 × 4.6 mm). Solvent A was water and solvent B was methanol, which was increased from 30% at 0 min to 100% at 10 min. The total flow rate was 1 ml/min.
[14C]Phenacetin and its metabolites were analyzed at 35°C on LC 18-DB columns (5-μm particle size; Supelco),i.e. a precolumn (20 × 4.6 mm) and an analytical column (250 × 4.6 mm). The mobile phases were 5 mM tetrabutylammonium hydrogen sulfate, 10 mM Tris base, 5 mM ethylenedinitrilotetraacetic acid, pH 7.2, as solvent A and methanol as solvent B. The proportion of solvent B was 0% for the first 5 min, was increased linearly to reach 60% at 25 min, remained constant until 35 min, and then was increased to reach 100% at 45 min. The total flow rate was 1 ml/min.
[3H]Paclitaxel and its metabolites were chromatographed at room temperature on Spheri ODS columns (5-μm particle size, 30 × 4.6 mm and 100 × 4.6 mm. The mobile phases were water as solvent A and methanol as solvent B. The proportion of solvent B was 0% for the first minute, was then increased linearly to reach 50% at 5 min, 70% at 52 min, and 100% at 54 min, and remained constant until 65 min. The total flow rate was 1 ml/min.
[14C]Tolbutamide and its metabolite 4-hydroxytolbutamide were separated at 40°C on Spheri-5 RP-18 columns (5-μm particle size; Brownlee), i.e. a guard column (30 × 2.1 mm) and an analytical column (100 × 2.1 mm). The mobile phases consisted of 10 mM ammonium acetate buffer (adjusted to pH 4.3 with acetic acid) as solvent A and acetonitrile as solvent B. The proportion of solvent B was 5% for the first 1 min and was increased linearly to reach 40% at 40 min and 100% at 45 min. The flow rate was 0.4 ml/min.
[14C]Theophylline and its metabolites were monitored after HPLC separation at room temperature on Supelcosil LC 18-DB columns (5-μm particle size, 20 × 4.6 mm and 250 × 4.6 mm). The mobile phases were 10 mM sodium acetate, pH 4.5, as solvent A and acetonitrile as solvent B, with a total flow rate of 1 ml/min. The proportion of solvent B was 5% from 0 to 9 min, was increased linearly to reach 15% at 15 min, and remained constant until 19%. In a linear increase, solvent B reached 27% at 30 min and 100% at 32 min.
[14C]Tropisetron, [14C]ondansetron, dextromethorphan, and their metabolites were analyzed as described (Fischer et al., 1994), except that dextromethorphan was analyzed with a Supelcosil LC-DP column (5-μm particle size, 50 × 4.6 mm; Supelco), with a flow rate of 1 ml/min. [3H]CSA and its metabolites were analyzed as described by (Kronbach et al., 1988).
MS Analysis.
Positive-ion mass spectra were recorded with a TSQ-700 triple-stage quadrupole mass spectrometer (Finnigan MAT, San Jose, CA) equipped with an ESI interface. For PSC and its metabolites, 5 mM sodium acetate in water/methanol (8:2) was added to the column eluent at 0.3 ml/min. The total flow of 1.5 ml/min was then split ∼1:1, with 0.75 ml/min going to waste and 0.75 ml/min entering the ESI interface. For paclitaxel and its metabolites, 0.2% trifluoroacetic acid in methanol was added to the column eluent at 0.3 ml/min. The total flow of 1.8 ml/min was then split ∼2:1, with 1.2 ml/min going to waste and 0.6 ml/min entering the ESI interface. For doxorubicin and its metabolites, 0.2% trifluoroacetic acid in acetonitrile was added to the column eluent at 0.1 ml/min. The total flow of 1.0 ml/min was then split ∼3:1, with 0.75 ml/min being used for on-line radioactivity monitoring and 0.25 ml/min entering the ESI interface. Glyburide was detected in the negative-ion mode after addition of 0.1 ml/min acetonitrile, with 20-V additional skimmer octapol voltage to induce fragmentation.
Methanol (0.05–0.1 ml/min) was used as the sheath liquid and nitrogen served as the sheath and auxiliary gas. The ESI spray voltage was 3.5 kV for PSC, 3 kV for paclitaxel, and 4.5 kV for doxorubicin and glyburide, with a capillary temperature of 250°C. Selected-ion chromatograms for PSC and its metabolites were extracted from the raw data files. The mass traces of the respective isotopes were added to enhance the signal/noise ratio.
Pgp Interactions.
The paclitaxel-resistant MDA/T0.3 cells were selected from the sensitive human adenocarcinoma cell line MDA-435, which was obtained from the M. D. Anderson Cancer Center (Houston, TX) (Archinal-Mattheis et al., 1995). The drug concentration that inhibited growth by 50% was determined as described (Monkset al., 1991). Photoaffinity labeling with a [3H]cyclosporine derivative and inhibition studies of rhodamine-123 efflux from MDA/T0.3 cells were performed as described by Archinal-Mattheis et al. (1995).
Data Analysis.
For the calculation of metabolic rates, mean substrate concentrations over the incubation period were used. IC50 values were determined graphically by plotting the percentage of the control activity vs. the inhibitor concentration. The Michaelis-Menten parameters KM
andVmax and SEs were determined by nonlinear curve-fitting using Fig.P software (BIOSOFT, Cambridge, UK), with the following equation:
Using a model for mixed-type inhibition, theKi
values were calculated with the following equation (Segel, 1993):
Intrinsic clearance (CLint) was calculated by dividing the maximal rate of metabolism by the Michaelis constant, according to the equation
Results
Biotransformation Pathways.
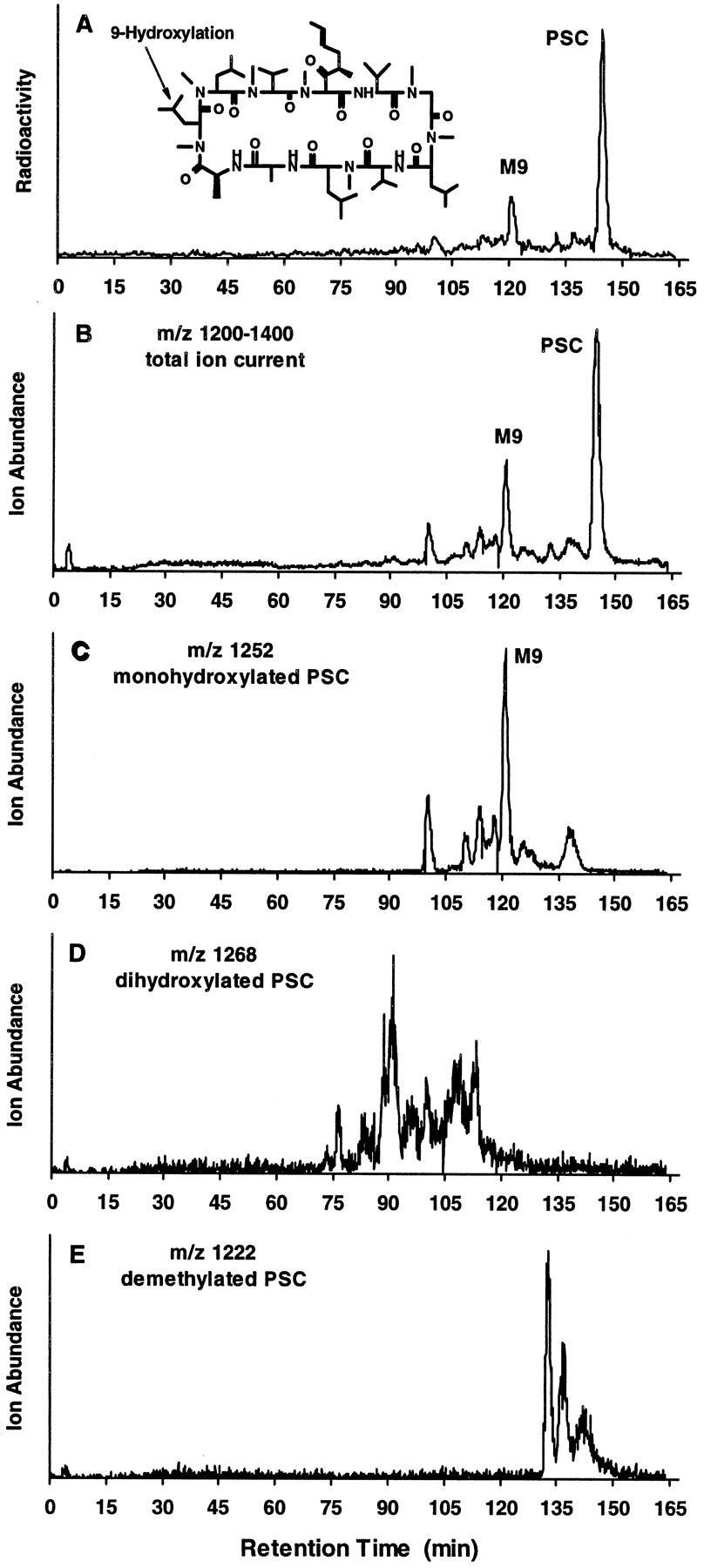
PSC was metabolized in human liver microsomal preparations to several metabolites (fig. 1). Structural information on these metabolites was obtained by LC/MS analysis with ESI (fig. 1, B–E). The chromatogram for the natriated ion of monohydroxylated metabolites (m/z1252) (fig. 1C) closely resembled the radioactivity profile (fig. 1A) and the total-ion current between m/z1200 and 1400 for PSC metabolites (fig. 1B), indicating that monohydroxylated products are predominantly formed under these conditions in human liver microsomal incubations. Additional structural information on the major monohydroxylated metabolite was suggested by its having a retention time (120 min) identical to that of syntheticM9, which is hydroxylated at amino acid 9 (data not shown). Additional natriated ions were detected at m/z 1268, indicating dihydroxylated products (fig. 1D), and atm/z 1222, suggesting N-demethylated products (fig. 1E).

PSC metabolite profile and identification in human liver microsomes.
A, typical HPLC separation, with radioactivity monitoring, of PSC and its metabolites formed in incubations of [3H]PSC (10 μM) with human liver microsomes (500 μg/ml) for 2 hr. B–E, LC/MS analysis of these metabolites using selected-ion monitoring, i.e.total ion current between m/z 1200 and 1400 (B), monohydroxylated PSC at m/z 1252 (C), dihydroxylated PSC at m/z 1268 (D), and demethylated PSC at m/z 1222 (E).
Metabolite Interaction with Pgp.
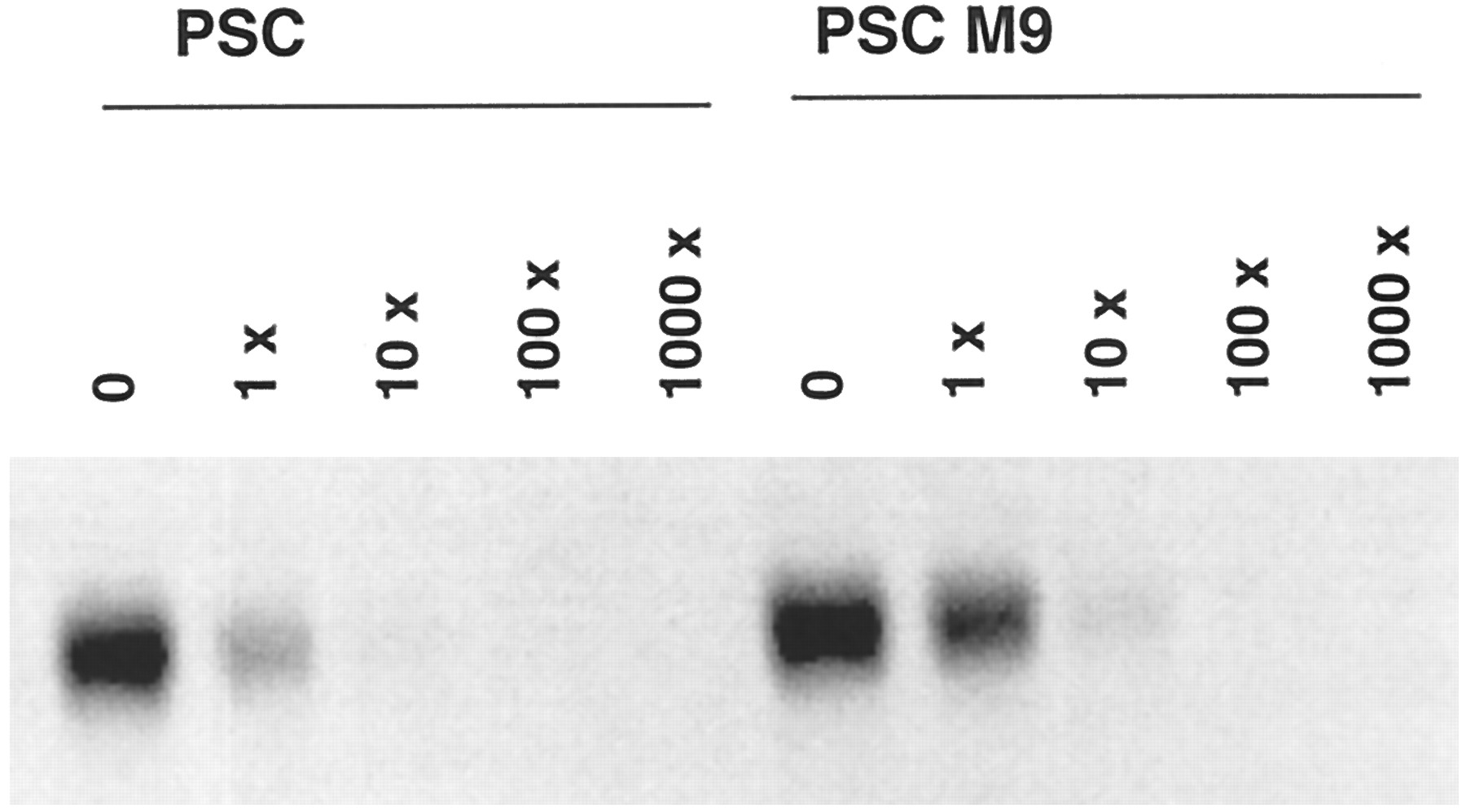
Because M9 was the major metabolite in human liver microsomes and in humans in vivo (Hauck C, personal communication), its ability to inhibit Pgp function was examined using the same human adenocarcinoma cell line (MDA/T0.3) as used previously for PSC (Archinal-Mattheis et al., 1995). These cells were selected for resistance to growth inhibition by paclitaxel (IC50 ∼ 1 μM). Exposure of the cells to paclitaxel in the presence of 1 μM M9 produced growth inhibition at an 8-fold lower concentration of paclitaxel (IC50 ∼ 0.12 μM) (data not shown). In comparison, PSC was much more potent and increased cellular sensitivity ∼300-fold (IC50 = 2.8 nM). M9 was also less potent than PSC in inhibiting rhodamine-123 efflux from MDA/T0.3 cells (fig. 2). Complete inhibition was obtained only with 5 μM M9, compared with 1 μM PSC. A similar difference in potency was observed when the binding of the photoaffinity label to Pgp was inhibited by PSC or M9(fig. 3). Densitometric evaluation of the labeled Pgp indicated 15-fold inhibition for PSC and 4-fold inhibition for M9, at equal (84 nM) concentrations, with respect to the [3H]cylosporine-derived label. With 10- and 100-fold excesses of PSC and M9, respectively, the binding of the photoaffinity label was completely inhibited.

Effect of PSC and M9 on rhodamine-123 retention in MDA cells.
Sensitive MDA-435 (black histograms) and paclitaxel-resistant MDA/T0.3 (white histograms) cells were pretreated with dimethylsulfoxide (A), 0.1, 1.0, 2.5, or 5.0 μM PSC (B), or 0.1, 1.0, 2.5, or 5.0 μMM9 (C) for 90 min. After incubation, the cells were washed and incubated with rhodamine-123 for an additional 90 min. The cellular fluorescence was analyzed by flow cytometry.

Photoaffinity labeling of membrane-enriched fractions from paclitaxel-resistant (MDA/T0.3) cells.
Cells were incubated with a [3H]cyclosporine derivative photoaffinity label (84 nM) in the presence of PSC or M9 at a 0-, 1-, 10-, 100-, or 1000-fold molar excess.
Intrinsic Clearance.
The rate of PSC metabolite formation was determined over the concentration range of 0.1–25 μM in two human liver microsomal preparations, i.e. GGM-002 and M8. Metabolism followed Michaelis-Menten kinetics, with similar KM values for the two livers (1.3 and 2.8 μM for GGM-002 and M8, respectively) (table 1). TheVmax values were 6.7 and 3.3 nmol/hr/mg of microsomal protein (15.2 and 11.4 nmol/hr/nmol of P450) for GGM-002 and M8, respectively. Consequently, the intrinsic clearance was ∼4-fold greater for GGM-002, compared with M8, when scaled to body weight.
Kinetic parameters for PSC metabolism
PSC was metabolized to the same metabolites as in human liver microsomes by microsomal preparations from CHO cells genetically engineered to express human CYP3A4 or CYP3A5 but not by those from cells expressing CYP2D6. The KM values for CYP3A4- and CYP3A5-containing microsomes were 1.1 and 12.6 μM, respectively, which were similar to those in liver microsomes. The maximal velocities of the reaction were 3.5 and 0.5 nmol/hr/mg of microsomal protein for CYP3A4 and CYP3A5, respectively. Consistent with these cells expressing only a single P450, the maximal velocity based on total P450 content was 4–5-fold greater for CYP3A4-expressing cells than for human liver. Because of low expression and a lack of P450 spectra, a similar comparison could not be made for cells expressing CYP3A5. However, the results suggested that both CYP3A4 and CYP3A5 are the enzymes responsible for PSC biotransformation.
Inhibition of PSC Metabolism.
PSC metabolism (1 μM) was investigated in the presence of characteristic P450 isoenzyme substrates/inhibitors and in the presence of compounds likely to be coadministered with PSC during therapy (table2). The most potent inhibitors of PSC metabolism were ketoconazole and CSA, with IC50values of 0.1 and 0.5 μM, respectively. Ketoconazole inhibited PSC competitively, with mean Ki values of 0.01–0.04 μM for both liver preparations. Ketoconazole at 0.1 μM produced a 3–7-fold increase in the apparentKM for PSC and at 0.5 μM produced a 23–67-fold increase in the apparent KM for PSC with M8 and GGM-002 preparations, respectively, with no change in the maximal velocity. CSA was also a competitive inhibitor; its presence increased the apparent KM for PSC 4–20-fold (Ki = 2.2 μM for GGM-002). However, CSA also exhibited a noncompetitive component, because the Vmax was decreased by factors of 2 and 3 at 3.5 and 10 μM CSA, respectively. Other compounds that inhibited PSC metabolism with an IC50 of ≤20 μM included the antineoplastic agents doxorubicin, paclitaxel, and vinblastine, the antiemetic ondansetron, the antihypertensive agent nifedipine, the antibiotic erythromycin, the antidiabetic glyburide, the antiparkinsonian agent bromocriptine, the steroid ethinyl estradiol, and the antihypercholesterolemic drug lovastatin (table 2). The IC50 value for M9 of 21 μM was also approximately 10-fold higher than theKM for PSC.
Inhibition of PSC metabolism
Effects of PSC on the Metabolism of Other Drugs.
The potential for PSC to inhibit the metabolism of other drugs was investigated with a series of compounds that are known to be characteristic for certain P450s. Additionally, drugs that are likely to be coadministered with PSC and that exhibited inhibition of PSC metabolism were studied (table 3). PSC had no effect (at concentrations up to 20 μM) on the metabolism of phenacetin or theophylline (CYP1A2), tolbutamide (CYP2C9/10), (S)-mephenytoin (CYP2C19), or chlorzoxazone (CYP2E1). Only a small effect was observed for the CYP2D6 substrates dextromethorphan and bufuralol, with maximal inhibition of 20 and 35%, respectively, at 20 μM PSC. However, the metabolism of the CYP3A substrate CSA was inhibited by PSC with an IC50 of 6.5 μM, consistent with CYP3A being involved in the metabolism of PSC.
Effects of PSC on the metabolism of characteristic CYP substrates and potentially coadministered compounds (IC50) in human liver microsomes or in S9 fractions
Of the anticancer drugs, paclitaxel has been reported to be metabolized by CYP2C8 and CYP3A4 (Cresteil et al., 1994; Rahman et al., 1994). In vitro and in humans, CYP2C8 catalyzes the formation of 6α-hydroxypaclitaxel, the major metabolite of paclitaxel (Harris et al., 1994a; Monsarrat et al., 1993). CYP3A4 has been suggested to be responsible for the formation of the second most important metabolite,p-hydroxyphenyl-C3′-paclitaxel (Harris et al., 1994b). p-Hydroxyphenyl-C3′-paclitaxel and 6α-hydroxypaclitaxel were confirmed to be M26 andM35, respectively, in human liver microsomes (fig.4). M35 and M26exhibited fragmentation patterns in LC/MS, as described previously (Bitsch et al., 1993). Additional metabolites monohydroxylated on the taxane ring were M29 andM32, and that monohydroxylated on the side chain wasM30. The metabolite eluting at 24 min was hydroxylated on both the taxane ring and the side chain. The involvement of CYP3A4 (Cresteil et al., 1994; Harris et al., 1994b) in the formation of p-hydroxyphenyl-C3′-paclitaxel but not that of 6α-hydroxypaclitaxel was confirmed with microsomes from CHO cells expressing human CYP3A4. p-Hydroxyphenyl-C3′-paclitaxel was formed at rates of ∼0.02 and ∼0.32 nmol/hr/mg of microsomal protein (∼0.29 and ∼4.57 nmol/hr/nmol of P450) with 1 and 100 μM paclitaxel, respectively. The HPLC peak corresponding top-hydroxyphenyl-C3′-paclitaxel did not increase in incubations with microsomes from CHO cells expressing either CYP3A5 or CYP2D6, compared with control incubations performed in the absence of NADPH.
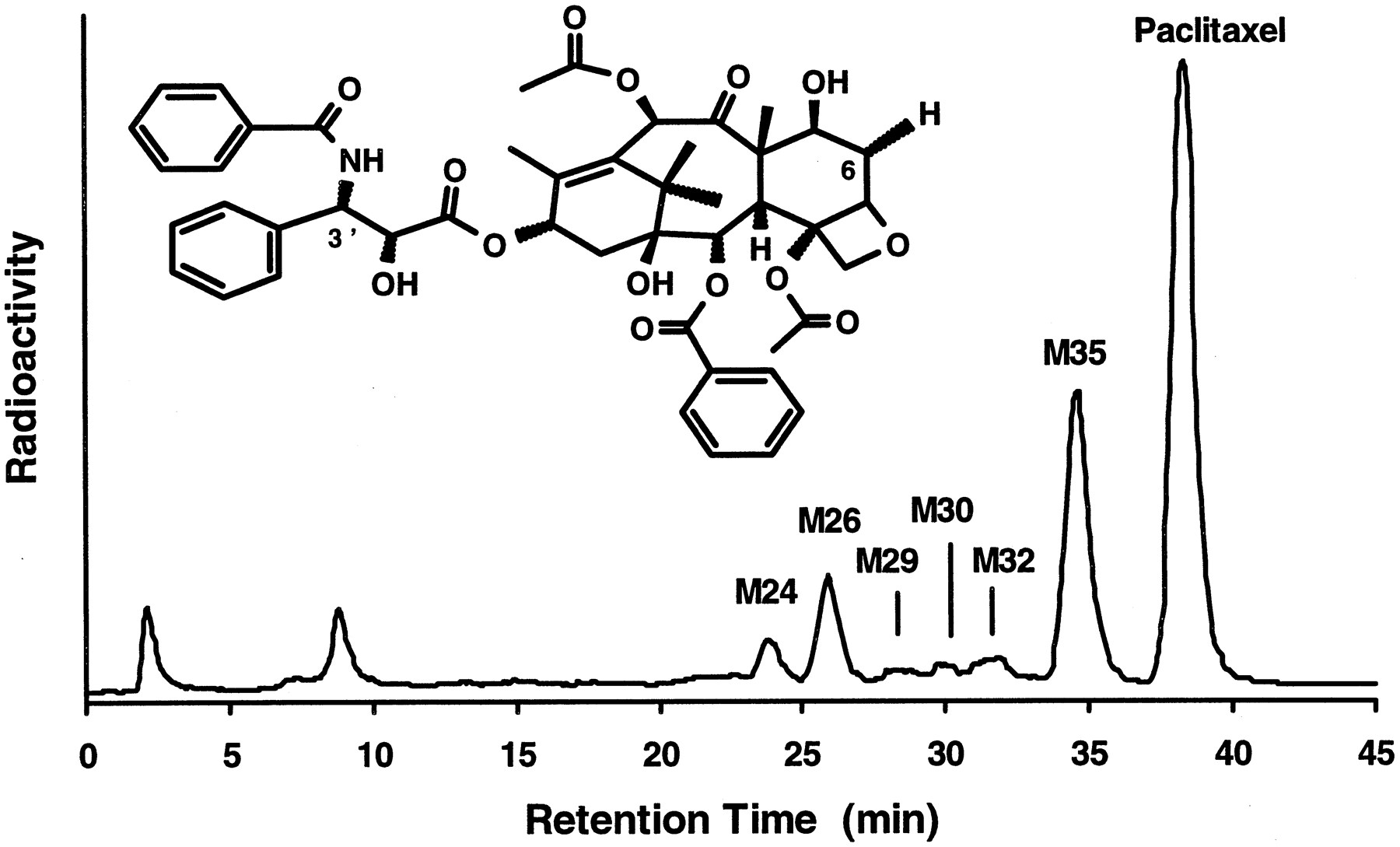
Paclitaxel metabolite formation.
A typical HPLC separation, with radioactivity monitoring, of paclitaxel and its metabolites found in incubations of [3H]paclitaxel (2.5 μM) with human liver microsomes is shown. M26 and M35 were assigned asp-hydroxyphenyl-C3′-paclitaxel and 6α-hydroxypaclitaxel, respectively.
With human liver microsomes (GGM-002, N = 4), paclitaxel (1–100 μM) exhibited Michaelis-Menten kinetics, as described (Cresteil et al., 1994; Sonnichsen et al., 1995), with apparent KM values of 57.9 ± 43.8 μM for overall metabolism, 16.0 ± 4.5 μM for p-hydroxyphenyl-C3′-paclitaxel formation, and 34.8 ± 28.5 μM for 6α-hydroxypaclitaxel formation.Vmax values were 277.3 ± 185.4, 15.8 ± 3.4, and 97.9 ± 58.3 nmol/hr/mg of microsomal protein for total metabolism, p-hydroxyphenyl-C3′-paclitaxel formation, and 6α-hydroxypaclitaxel formation, respectively. Intrinsic clearance values forp-hydroxyphenyl-C3′-paclitaxel and 6α-hydroxypaclitaxel formation (1.0 ± 0.1 and 3.1 ± 1.0 ml/hr/mg of microsomal protein, respectively) represented approximately 75% of total intrinsic clearance (5.1 ± 0.9 ml/hr/mg of microsomal protein). PSC selectively inhibited p-hydroxyphenyl-C3′-paclitaxel formation. The inhibition was of a mixed type (fig.5), with KM values approximately doubling while Vmaxvalues were decreased by ∼50% at 10 μM. The meanKi value was 1.15 μM, similar to theKM for PSC metabolism.
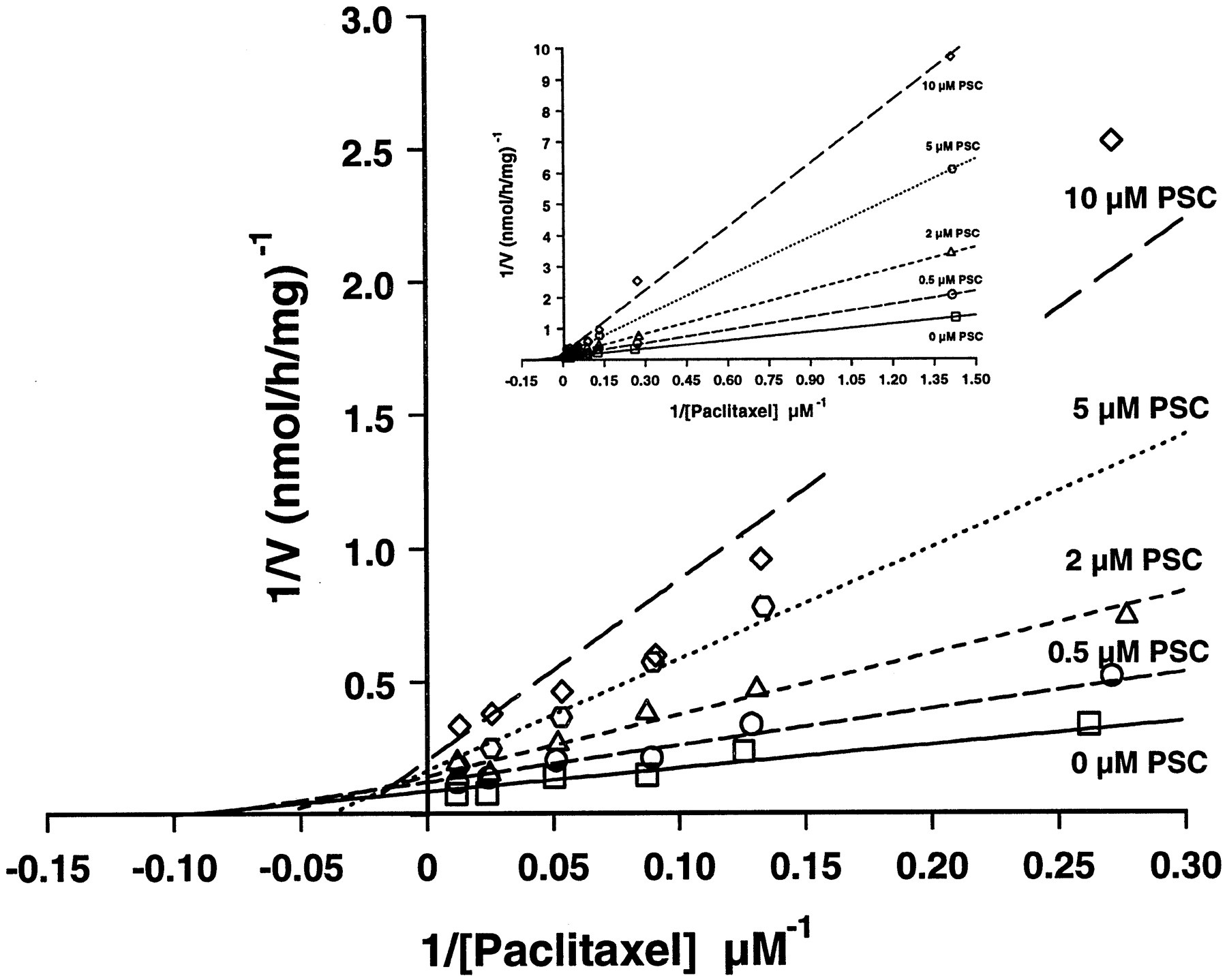
Lineweaver-Burk plots ofp-hydroxyphenyl-C3′-paclitaxel formation.
Human liver microsomal incubations with ∼0.7–100 μM paclitaxel (inset) were performed in the presence of 0 (■), 0.5 (○), 2 (▵), 5 (⬡), or 10 (⋄) μM PSC. Lines do not represent linear regression analyses but were drawn using the parameters obtained from nonlinear curve-fitting. The main panel is expanded for better visibility of the intercepts and does not show the low ∼0.7 μM concentration of paclitaxel.
In contrast, PSC (20 μM) had no effect on the metabolism of doxorubicin (another antineoplastic agent) or its metabolite doxorubicinol in two different human liver S9 preparations (table 3). Doxorubicinol is the major metabolite of doxorubicin formed by the ubiquitous cytoplasmic aldoketoreductase (Takanashi and Bachur, 1976). Both doxorubicin and doxorubicinol can also be converted by microsomal P450 reductase to the corresponding 7-deoxyaglycones (Bachur and Gee, 1976). The assignment of the HPLC peaks was in agreement with the molecular ions observed during LC/MS analysis (fig.6).
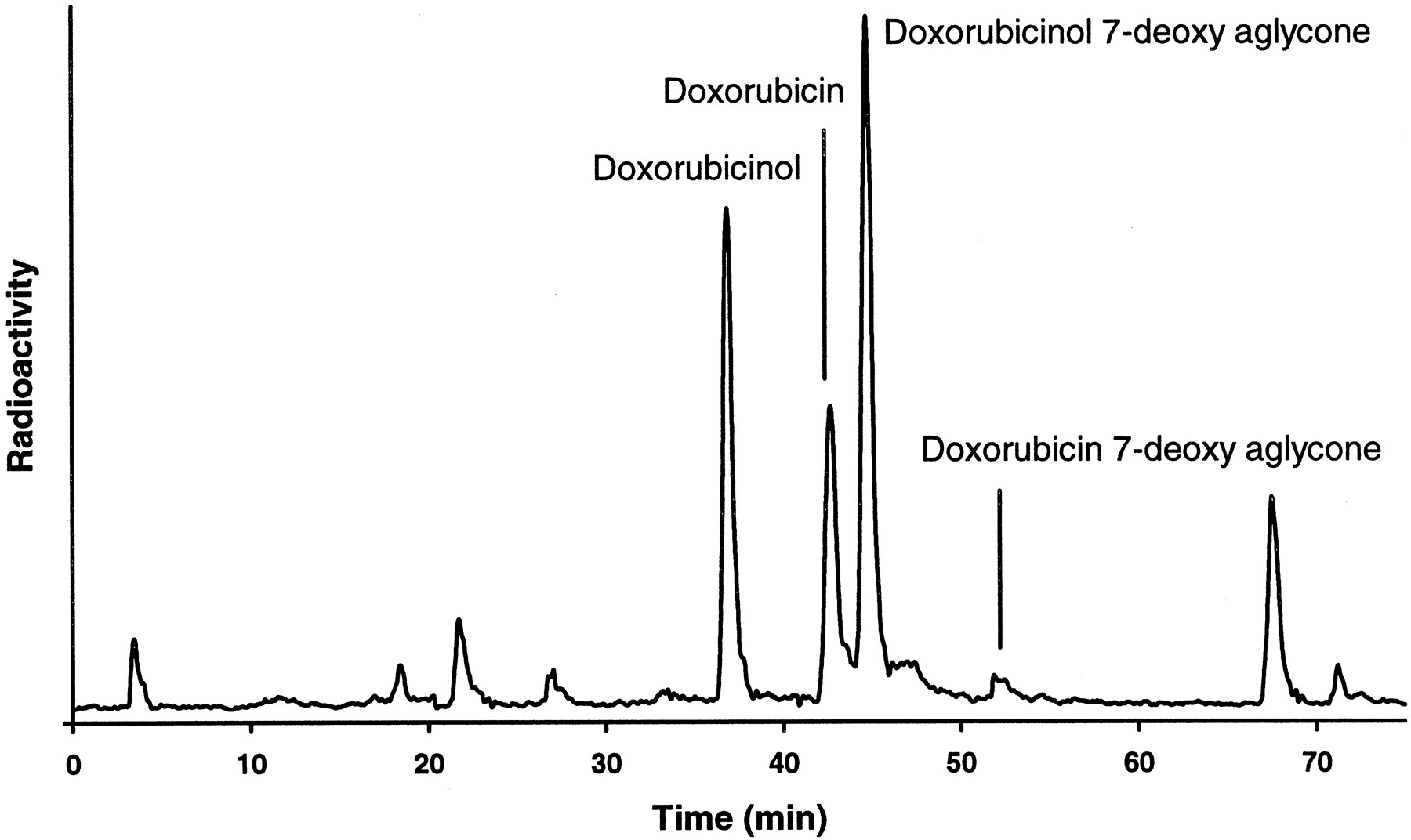
Doxorubicin metabolite formation.
A typical HPLC separation, with radioactivity monitoring, of doxorubicin and its metabolites found in incubations of [14C]doxorubicin (40 μM) with a human liver S9 preparation is shown.
Similarly, PSC would not be expected to have a clinically relevant effect on the metabolism of the antiemetics tropisetron and ondansetron. These compounds were metabolized primarily to 5/6/7-hydroxytropisetron (3.9 nmol/hr/mg) and 7/8-hydroxyondansetron (1.6 nmol/hr/mg), as previously described (Fischer et al., 1994). The inhibition was small but concentration dependent,i.e. 22% and 27% for tropisetron (50 μM) and ondansetron (50 μM), respectively (table 3).
The effect of PSC on the metabolism of glyburide was investigated because of the frequent use of glyburide as an antidiabetic agent and because of its potential to inhibit the metabolism of CSA (Pichardet al., 1990, 1996) and PSC. Previously, only 4-trans-hydroxyglyburide and 3-cis-hydroxyglyburide, which are hydroxylated on the cyclohexyl moiety, had been identified and isolated from human urine and feces (Rupp et al., 1969). However, in five different human liver microsomal preparations, glyburide was metabolized to several additional metabolites (fig. 7). LC/MS analysis did not result in detectable ions for M23(6.1 nmol/hr/mg of protein), but M44–M49 (13.6 nmol/hr/mg of protein) and M53 (6 nmol/hr/mg of protein) yielded ions of m/z 508, indicating monohydroxylation of these metabolites. The observed fragment of m/z 170 forM44–M53 and glyburide itself indicates that hydroxylation did not occur on the 5-chloro-2-methoxybenzaldehyde moiety. Cleavage of the urea amide bond resulted in fragments ofm/z 367 for glyburide and peaksM44–M49 or m/z 383 for peakM53. This indicates hydroxylation on the cyclohexyl ring for peaks M44–M49, which include the previously identified 4-trans-hydroxyglyburide and 3-cis-hydroxyglyburide, and hydroxylation on the phenylethyl moiety for peak M53. Therefore, the previously identified metabolites, which are considered to be the major metabolites in humans, represented only a fraction of the peaks hydroxylated on the cyclohexyl moiety, which are ∼50% of the total metabolites in human liver microsomes. In the presence of PSC (≤20 μM), cyclohexyl hydroxylation was not inhibited (table 3). In contrast, M23was already inhibited ∼40% with 5 μM PSC, and M53 was inhibited 50% with ∼3 μM PSC.

Glyburide metabolite formation.
A typical HPLC separation, with radioactivity monitoring, of glyburide and its metabolites formed in incubations of [3H]glyburide (5 μM) with human liver microsomes is shown.
Discussion
The metabolism of the cyclosporine derivative PSC in human liver is very similar, with respect to pathways and interactions with P450, to that of other cyclosporines such as CSA, IMM 125, and CSG. Therefore, for the prediction and interpretation of drug interactions with PSC (when it is used in combination therapy), the extensive clinical experience gained with CSA can be used as a guideline.
PSC is metabolized by human liver microsomes to monohydroxylated,N-demethylated, and dihydroxylated products, as determined by LC/MS. These pathways have also been found in vivo(metabolites detected in human plasma and urine) for PSC (Hauck C, personal communication) and are analogous for the other cyclosporines (Kronbach et al., 1988; Maurer, 1985; Vickers et al., 1995; Mangold et al., 1994). The apparentKM for PSC of the human liver microsomal enzymes is similar to those reported for CSA (1.7–10 μM) (Fischeret al., 1994; Pichard et al., 1990, 1996; Maurer, 1985), for IMM 125 (5.1 μM) (Vickers et al., 1995), and for CSG (5–9 μM) (Pichard et al., 1996). Furthermore, the intrinsic clearance of PSC determined with human liver microsomes (9–36 ml/min/kg of body weight) is within the range estimated for the other cyclosporines from literature values, as follows: CSA, 4–180 ml/min/kg of body weight (Fischer et al., 1994; Maurer, 1985); IMM 125, 4 ml/min/kg of body weight (Vickers et al., 1995); CSG, ∼9 ml/min/kg of body weight (Pichard et al., 1996).
The metabolism of PSC, like that of the other cyclosporines, was CYP3A dependent. PSC biotransformation was measurable only in cell lines expressing CYP3A4 and CYP3A5 and not CYP2D6. In addition, compounds that are known to inhibit CYP3A also inhibited PSC metabolism. Furthermore, the inhibition was mainly competitive for the CYP3A substrates CSA and ketoconazole. The potencies for inhibition of PSC metabolism by the compounds tested (table 2) compare well with the known potencies of these compounds to inhibit CYP3A. For example, reported Ki values for CYP3A substrate (CSA) inhibition are 0.7, 8, and 10 μM for ketoconazole, bromocryptine, and nifedipine, respectively (Pichard et al., 1990), and 31 μM for ondansetron (Fischer et al., 1994). In contrast, compounds such as phenacetin, quinidine, dextromethorphan, fluvastatin, and tolbutamide, which are known not to markedly inhibit CYP3A, had little effect on PSC metabolism. The selective inhibition of CYP3A-mediated reactions such as CSA metabolism andp-hydroxyphenyl-C3′-paclitaxel formation by PSC also is consistent with CYP3A being the major enzyme in PSC biotransformation. The observed trend toward CYP2D6 inhibition, although consistent for all investigated CYP2D6 substrates, i.e. dextromethorphan, bufuralol, tropisetron, and ondansetron (Fischer et al., 1994; Dayer et al., 1989; Yamazaki et al., 1994;Dixon et al., 1995), would be too small to be of any clinical relevance. Overall, PSC appears to be a specific inhibitor of CYP3A.
PSC inhibited the metabolism of glyburide, a potentially coadministered antidiabetic agent, to a limited extent. Only pathways that had not been previously reported, including hydroxylation at the phenylethyl moiety, were inhibited, indicating that these pathways must involve CYP3A. In contrast, the lack of inhibition by PSC of the formation of cyclohexyl-hydroxylated metabolites, such as 4-trans-hydroxyglyburide and 3-cis-hydroxyglyburide, indicates the involvement of enzymes other than CYP3A in these pathways. The limited involvement of CYP3A in glyburide metabolic clearance could explain why erythromycin, a CYP3A inhibitor, did not affect glyburide clearance in a clinical trial (Fleishaker and Phillips, 1991). Because PSC is also a selective CYP3A inhibitor, no clinically significant reduction in glyburide clearance can be expected when PSC is coadministered. Glyburide should also not influence PSC concentrations, because the IC50 toward PSC metabolism is >1 order of magnitude higher than its maximal plasma concentrations at a relatively high 20-mg dose (Coppack et al., 1990).
For the coadministration of PSC and paclitaxel, a result similar to that observed with CSA, which also selectively inhibitsp-hydroxyphenyl-C3′-paclitaxel formation in human liverin vitro (Harris et al., 1994b; Kumar et al., 1994), would be expected. Clinically, CSA increases paclitaxel exposure; indeed, a similar 50% dose reduction of paclitaxel was required when PSC was used instead of CSA (Fisheret al., 1994a,b). Inhibition of paclitaxel elimination by Pgp and/or inhibition of p-hydroxyphenyl-C3′-paclitaxel formation could contribute to the observed increase in plasma concentrations. However, it appears unlikely that either mechanism by itself can explain the extent of the observed clinical effect. Paclitaxel is eliminated predominantly by metabolic clearancevia the bile, and combined urinary and biliary excretion of unchanged paclitaxel was only ∼10% of the dose (Walle et al., 1995). Also, the major metabolite in 75% of human liver microsomal preparations appears to be 6α-hydroxypaclitaxel, whereasp-hydroxyphenyl-C3′-paclitaxel was the major metabolite in ∼25% of human liver microsomal preparations (Sonnichsen et al., 1995). Paclitaxel did not influence CSA or PSC exposure in humans, as expected based on the low plasma concentrations, compared with the paclitaxel Ki value for CYP3A inhibition (Fisher et al., 1994; Smith H, personal communication).
The clinically observed increases in doxorubicin exposure and reduced clearance in the presence of PSC (Erlichman et al., 1993;Bartlett et al., 1994) cannot be explained by a reduction in doxorubicin metabolism by PSC. Doxorubicin is not metabolized by CYP3A, and PSC had no effect on the formation of the major metabolite doxorubicinol by the ubiquitous aldoketoreductase or on the removal of the daunosamine sugar by the microsomal P450 reductase. Instead, a major portion (50%) of doxorubicin is excreted unchanged in the bile (Riggs et al., 1977), and changes in tissue distribution and reduced transport into the bile (Colombo et al., 1994; Speeg and Maldonado, 1994) could explain the increased exposure and reduced clearance observed in the clinical trials. Although doxorubicin was also found to inhibit PSC metabolism in vitro, with an IC50 value of 20 μM, the therapeutic range at steady state is at least 100-fold lower (Meyer, 1994), and the presence of doxorubicin should not lead to relevant changes in the metabolic clearance of PSC.
Etoposide is approximately equally excreted in urine and bile (Lumet al., 1992). Approximately 20–35% of the etoposide dose is excreted unchanged in the urine and ∼2% in bile. The remainder is metabolized, and CYP3A is the main enzyme catalyzing etoposideO-demethylation (Relling et al., 1994). Typical therapeutic concentrations of ∼17 μM (Lum et al., 1992) are approximately 5-fold below the KM for CYP3A (Relling et al., 1992) and 10-fold below the IC50 for PSC metabolism. Etoposide should therefore not be able to influence the metabolic clearance of PSC. Conversely, PSC at typical concentrations of 4 μM could inhibit theO-demethylation of etoposide. The observed increases in the plasma concentrations of etoposide when it is combined with PSC may be the result of a combination of inhibition of active secretion by Pgp and inhibition of metabolism (Boote et al., 1996). Because the pharmacokinetics of other epipodophyllotoxins, such as teniposide, are not significantly different from those of etoposide, similar interactions with PSC would be expected.
Vinca alkaloids are also metabolized primarily by CYP3A4, making their elimination susceptible to inhibition by PSC (Zhouet al., 1993; Zhou-Pan et al., 1993). However, little is known regarding whether Vinca alkaloids are excreted mainly unchanged or as metabolites. Therefore, it is difficult to determine the exact mechanism for the increases in plasma concentrations seen in clinical trials when PSC is coadministered withVinca alkaloids.
For the antiemetics tropisetron and ondansetron, metabolic drug interactions with PSC are not expected. Both are only partially metabolized by CYP3A, and their therapeutic plasma concentrations are 2–3 orders of magnitude below the concentrations at which they inhibit PSC metabolism in vitro (Fischer et al., 1994; Dixon et al., 1995).
In summary, the evidence indicates that PSC is metabolized in a manner analogous to that of other cyclosporines, by hydroxylation andN-demethylation to less active metabolites. As with the other cyclosporines, metabolism is mediated by CYP3A. The knowledge of the enzymes involved in the biotransformation of PSC and the proof of similarity to other cyclosporines will allow physicians to use the clinical experience gained with CSA for better prediction and interpretation of drug interactions with PSC.
Acknowledgments
We thank Drs. H. Andres, T. Moenius, and R. Voges for synthesis of the radiolabeled compounds, Dr. R. Wenger for synthesis of the main PSC metabolite, R. Nufer for LC/MS operation, and P. Kwon and N. Kohn for technical assistance.
Footnotes
-
Send reprint requests to: Dr. Volker Fischer, Novartis Pharmaceuticals Corp., 59 Route 10, East Hanover, NJ 07936.
- Abbreviations used are::
- MDR
- multidrug resistance
- Pgp
- P-glycoprotein
- CHO
- Chinese hamster ovary
- CSA
- cyclosporine A
- CSG
- cyclosporine G
- CYP or P450
- cytochrome P450
- ESI
- electrospray ionization
- PSC
- valspodar (PSC 833)
- Received November 21, 1997.
- Accepted March 31, 1998.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}