


Abstract
This is a report of a symposium held at the March 1997 meeting of the American Society for Pharmacology and Therapeutics in San Diego. Our understanding of the events that control first-pass drug elimination in humans has increased tremendously by two sequential discoveries. First, cytochrome P-450s 3A4 and 5 are expressed at high concentrations in both hepatocytes and upper intestinal enterocytes, and therefore limit the systemic availability of many drugs. Second, P-glycoprotein is expressed at the lumenal surface of the intestinal epithelium and therefore also acts to oppose the absorption of unchanged drug. The following discussion brings together our current understandings of these interrelated phenomena to aid a more complete picture of how they may contribute both qualitatively and quantitatively to first-pass elimination.
CYP3A4 is the most abundant cytochrome P-450 (CYP)2 present both in the liver (Guengerich et al., 1986; Shimada et al., 1994) and in the epithelial cells (enterocytes) that line the lumen of the small bowel (Watkins et al., 1987; Kolars et al., 1992). Although the importance of hepatic drug metabolism is well established, it has recently become clear that some substrates for CYP3A4 can also undergo substantial metabolism in enterocytes during absorption from the lumen of the bowel (Regardh et al., 1989; Wang et al., 1989; Kolars et al., 1991; Wu et al., 1995; Paine et al., 1996; Lown et al., 1997). Although hepatic drug metabolism can make a major contribution to systemic drug elimination, the combination of hepatic and intestinal drug metabolism appears to have a large influence on presystemic or first-pass drug biotransformation.
It has recently become apparent that another factor that must also be considered in the absorption of many CYP3A substrates is intestinal P-glycoprotein (P-gp). P-gp is a versatile transporter with broad substrate specificity that allows it to pump a wide variety of xenobiotics including cyclosporin A, FK506, Taxol, diltiazem, dexamethasone, lidocaine, erythromycin, and protease inhibitors (Bech-Hanson et al., 1976; Hofsli and Nissen-Meyer, 1989;Mechetner and Roninson, 1992; Roninson, 1992; Ueda et al., 1992; Saeki et al., 1993a, b; Kim et al., 1998). P-gp has been shown to be one of the major factors responsible for the resistance of many cancer cells to chemotherapy agents. In the intestine, P-gp is located almost exclusively within the brush border on the apical (luminal) surface of mature enterocytes, where it functions to pump xenobiotics from the cytoplasm to the exterior of the cell (i.e., from the enterocyte back into the intestinal lumen) (Hsing et al., 1992; Saitoh and Aungst, 1995; Thiebaut et al., 1997). The close cellular location of P-gp and CYP3A4 expression in mature enterocytes and their similar substrate specificity suggest that the function of these two proteins may be complimentary and form a coordinated intestinal barrier. Hence, a more complete model of first-pass elimination of CYP3A substrates is likely to include the drug getting past the outward directed transport function of P-gp in the enterocyte plasma membrane, avoiding metabolism by CYP3A4 in the enterocyte cytoplasm, and then finally escaping metabolism by the large mass of CYP3A4 in the liver. As with CYP3A4, there is significant interindividual variation in the intestinal expression of P-gp, up to 4-fold variation in healthy volunteers and up to 10-fold in medical patients (Lown et al., 1995).
Role of Enterocyte CYP3A in the First-Pass Extraction of Midazolam (K.E.T., M.F.P.)
The systemic availability of an orally administered drug is dependent on a number of physical and biological factors. These include disintegration and dissolution properties of the drug formulation, solubility of the drug molecule in the lumenal environment of the gastrointestinal tract, plasma membrane permeability, and the susceptibility of the molecule to various plasma membrane transporters and intra- or extracellular biotransformation enzymes. The various factors that influence the absolute systemic availability (F) of a drug can be described by eq. 1:
We have recently studied first-pass intestinal metabolism in humans using the CYP3A probe midazolam. CYP3A-catalyzed 1′ and 4-hydroxylation eliminate midazolam from the body almost exclusively (Kronbach et al., 1989). The drug is well absorbed after oral administration (Heizmann and Ziegler, 1981) and can be recovered in urine as glucuronide conjugates of its primary oxidative metabolites (Dundee et al., 1984). To assess the extent of intestinal metabolism after i.v. and oral administration, a single dose of midazolam was administered to liver transplant patients during the anhepatic phase of their operation (Paine et al., 1996). During an anhepatic period that lasted approximately 60 min, a 2-mg dose administered directly into the duodenal lumen was rapidly absorbed into the body. There was extensive intestinal first-pass metabolism, such that approximately 43% of the midazolam dose (14–59%) reached the hepatic portal blood as the primary 1-hydroxy metabolite. In contrast, after i.v. administration, roughly 8% (0–26%) of midazolam delivered to the intestinal mucosa by arterial blood was extracted during each passage through the organ. The extensive 1′-hydroxylation that occurred after intraduodenal midazolam administration suggests a high metabolic capacity for the proximal small intestine. Indeed, results from a pharmacokinetic study in healthy volunteers suggest that the first-pass extraction ratio for the gut and liver are comparable (43 and 44%, respectively), yielding an overall oral bioavailability of 30% (Thummel et al., 1996).
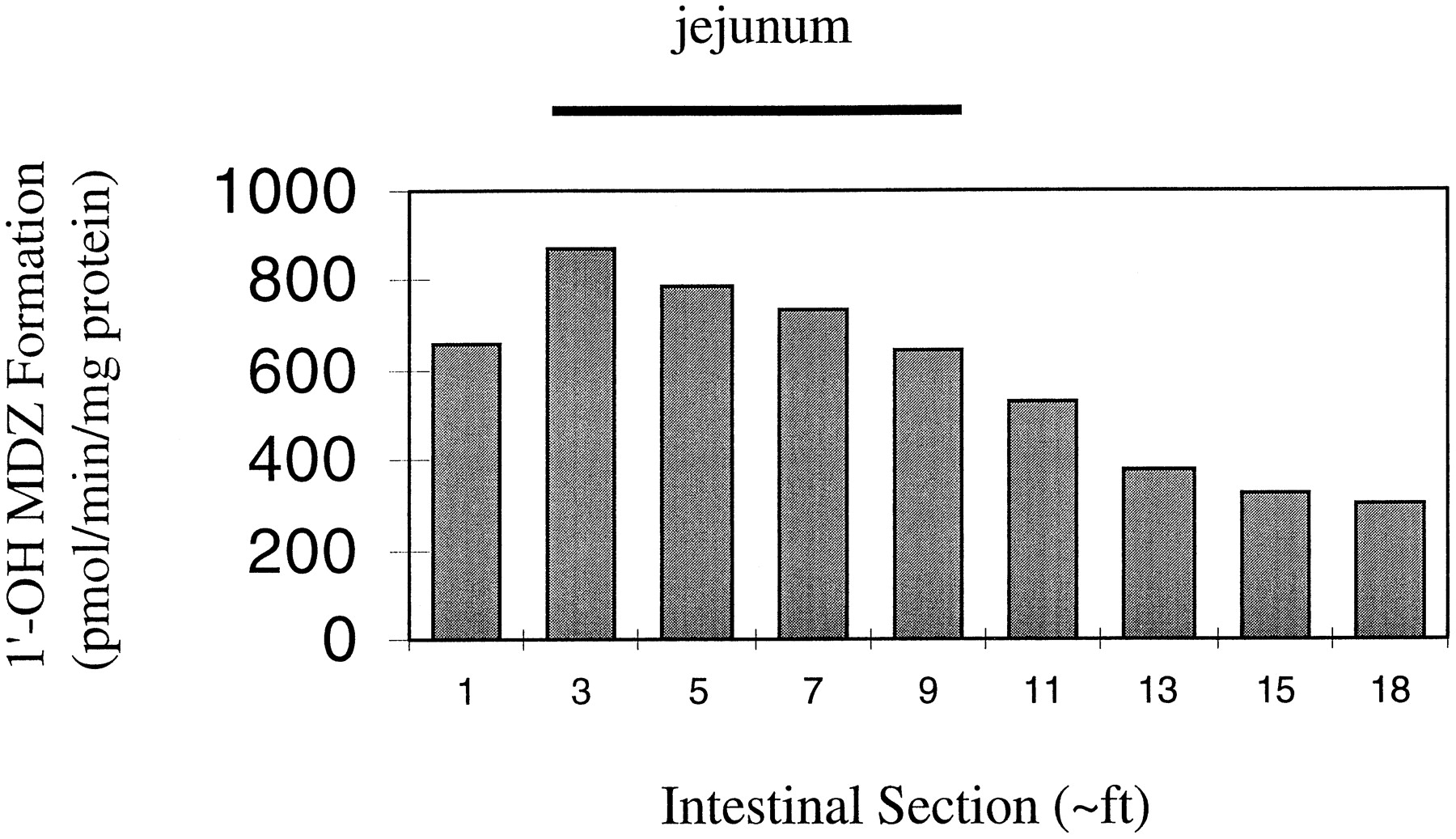
To more fully understand the enzymatic basis for an extensive yet variable first-pass intestinal metabolism, we characterized CYP3A content and midazolam intrinsic metabolic clearance in mucosal tissue from 15 full-length human small intestines. Total mucosal homogenate and microsomes were prepared from three regions, namely, the duodenum, jejunum, and ileum. The amount of CYP3A detected in total mucosal homogenate was strongly correlated with microsomal CYP3A levels (rs = 0.93, 0.93, and 0.83 for each intestinal region, respectively). This finding provided justification for using the more stable microsomal preparation for subsequent catalytic studies. The degree of interdonor variability in homogenate and microsomal CYP3A was large, and exceeded 18-fold for each region of small intestine. Within small intestine of a given individual, the microsomal CYP3A content and the associated midazolam hydroxylation activity were generally highest in the proximal region of the small intestine and declined distally (Fig. 1). In contrast, the Km for microsomal 1′-hydroxylation was similar for all regions of the small intestine; 3.8, 3.7, and 4.5 μM for duodenum, jejunum, and ileum, respectively. Thus, the intrinsic metabolic clearance (Vmax/Km) for the proximal intestine was higher than that for the distal intestine. The median total amount of CYP3A protein in the human small intestine was estimated to be 70 nmol, the majority of this residing in the jejunum (Table 1). The associated 1′-hydroxymidazolam formation clearance was also most concentrated in the jejunum. Surprisingly, the total unbound intrinsic clearance for the entire small intestine was low at approximately 1.4% of an average human liver (0.21 liters/min versus 15.8 liters/min). To rationalize the apparent discrepancy between an in vitro measure of hepatic and intestinal metabolic efficiency and in vivo measures of first-pass and systemic organ extraction ratios, we postulate that the high degree of plasma protein binding (∼98%) modulates the clearance of midazolam within the hepatocyte and enterocyte when drug is delivered to the site of metabolism via the systemic blood circulation. However, it does not influence enterocyte metabolic extraction when drug is presented to the enzyme via direct absorption from the intestinal lumen. That is, the rate of first-pass, CYP3A-dependent midazolam metabolism by enterocytes is a function of total intracellular midazolam concentration rather than a concentration reduced by the plasma-free fraction. In conclusion, the aggregate of results from our studies with midazolam suggest that the average first-pass intestinal extraction of other CYP3A substrates might be readily predicted from simple in vitro measurements of intestinal metabolic intrinsic clearance.

Distribution of mucosal midazolam 1′-hydroxylation activity for the human small intestine.
Mucosal microsomes were prepared from lengths (∼1 foot) of intestine obtained from a single organ donor (HI-35). Midazolam 1′-hydroxylation rates were measured after incubation of 50–100 μg of protein with 8 μM midazolam and NADPH for 4 min. Product was measured by gas chromatography-mass spectrometry as described (Thummel et al., 1996).
Cumulative CYP3A content and unbound 1′-hydroxymidazolam intrinsic formation clearance for the human small intestine and liver
Assessing the Role of Intestinal CYP3A4 in Limiting Oral Delivery of Drugs: New Approaches (P.B.W., D.G.B., K.S.L., R.J.F.)
Determination of the extent to which a given CYP3A4 substrate undergoes metabolism in intestine is not straightforward at present. In part, this may reflect the observation that there does not appear to be coordinate regulation of CYP3A4 expression in liver and intestine (Lown et al., 1994). A person can therefore have relatively high CYP3A4 activity in the liver while having relatively low CYP3A4 activity in the intestine and vice versa. For a given drug, it is theoretically possible that the liver might be the major site of metabolism in one patient, whereas the intestine could be the major site for the metabolism of the same drug in a different patient. It may therefore be necessary to study many subjects to be sure that the results obtained are representative of the population as a whole.
A recent approach to assessing the degree of first-pass intestinal metabolism requires measurement of the pharmacokinetics of the CYP3A4 substrate after it is administered both orally and i.v. (Hebert et al., 1992; Gomez et al., 1995; Thummel et al., 1996). To calculate intestinal metabolism, the extent of absorption of the drug must be known or assumed to be complete, and the rate of liver blood flow and hematocrit must be estimated. It must be assumed that systemic elimination does not involve intestinal metabolism and that CYP3A4 is the only enzyme capable of metabolizing the drug in vivo. For these reasons, this method generally does not provide unequivocal answers.
A second approach utilizes the [14C-N-methyl]erythromycin breath test (ERMBT) to measure CYP3A4 activity in the liver (reviewed in Watkins, 1994). Oral clearance values calculated for a CYP3A4 substrate can then be correlated with liver CYP3A4 activities among the study subjects. Because intestinal CYP3A4 activity does not appear to correlate with liver CYP3A4 activity (Lown et al., 1994), a good correlation between the ERMBT values and oral clearance of the substrate would suggest that the major site of metabolism of the substrate is the liver and not the intestine. The feasibility of this approach has been recently demonstrated with the CYP3A4 substrate cyclosporin A (Lown et al., 1997). Interpatient variation in liver CYP3A4 (as measured by the ERMBT) accounted for 56% of the variation in oral clearance of cyclosporin A, supporting the idea that the liver is generally the major site of metabolism of this substrate. However, this observation does not exclude possible substantial and variable intestinal metabolism.
An alternate, or complimentary, means of assessing the relative contribution of liver and intestinal CYP3A4 to first-pass metabolism would result if it were possible to selectively knock out intestinal CYP3A4 without influencing liver CYP3A4 activity. Oral kinetics of a drug would be determined before and after this selective manipulation, and the contribution of intestinal CYP3A4 could be directly assessed. Our recent study (Lown et al., 1997) of the dihydropyridine calcium channel antagonist felodipine, a well established substrate for CYP3A4 (Guengerich et al., 1991), suggests that grapefruit juice may be useful for this purpose.
In this study, 10 healthy volunteers received 8 oz of grapefruit juice three times a day for 6 days. Before and after receiving grapefruit juice, each subject underwent small bowel mucosal biopsies to directly measure intestinal CYP3A4, and the ERMBT was conducted to measure liver CYP3A4 activity. The pharmacokinetics of oral felodipine were examined on three occasions: with water before grapefruit juice treatment, with the first glass of grapefruit juice, and on the 6th day after the 16th glass of grapefruit juice. When taken with water felodipine is completely absorbed, as measured by the excretion of radioactivity after oral and i.v. administration, but has only 15% systemic oral bioavailability (Edgar et al., 1985). Grapefruit juice inhibited first-pass felodipine metabolism. Single-dose felodipine area under the plasma concentration time curve (AUC) and maximum plasma concentration (Cmax) after the last glass of grapefruit juice were 370% (range, 156–539%) and 538% (range, 182–1080%), respectively, compared to the values with water. Although the magnitude of the interaction was highly variable among individuals, the overall effect was to significantly decrease the coefficient of variation of absolute values for felodipine AUC from 61% to 36% and Cmax from 61% to 32% (Lown et al., 1997). In other words, grapefruit juice not only increased the oral availability of felodipine, but also improved the reliability of its delivery to the systemic circulation.
A surprising finding in this study was that grapefruit juice treatment resulted in a marked fall in the actual amount of CYP3A4 present in small bowel epithelial cells or enterocytes (Lown et al., 1997). Although it was not completely eliminated or knocked out, mean CYP3A4 protein concentration measured 4 h after the last glass of grapefruit juice was reduced to 38% of pretreatment levels. More recent studies (Schmeidlin-Ren et al., 1997) suggest that the components in grapefruit juice responsible for this loss of CYP3A4 protein are furanocoumarins. These compounds appear to be both competitive and mechanism-based inactivators of the enzyme (Schmeidlin-Ren et al., 1997). The loss of CYP3A4 enzymatic activity in the intestine after grapefruit juice ingestion therefore probably exceeds that estimated from the loss of enzyme protein. Although further studies are needed, a knock out equivalent may in fact be achieved by the juice.
The effect of grapefruit juice appeared to be selective for intestinal CYP3A4 in that liver CYP3A4 activity, as measured by the ERMBT, was not altered by the juice (Lown et al., 1997). In addition, concentrations of intestinal P-gp, villin, CYP1A1, and CYP2D6 did not change. However, measurements of the catalytic activities of these enzymes was not performed.
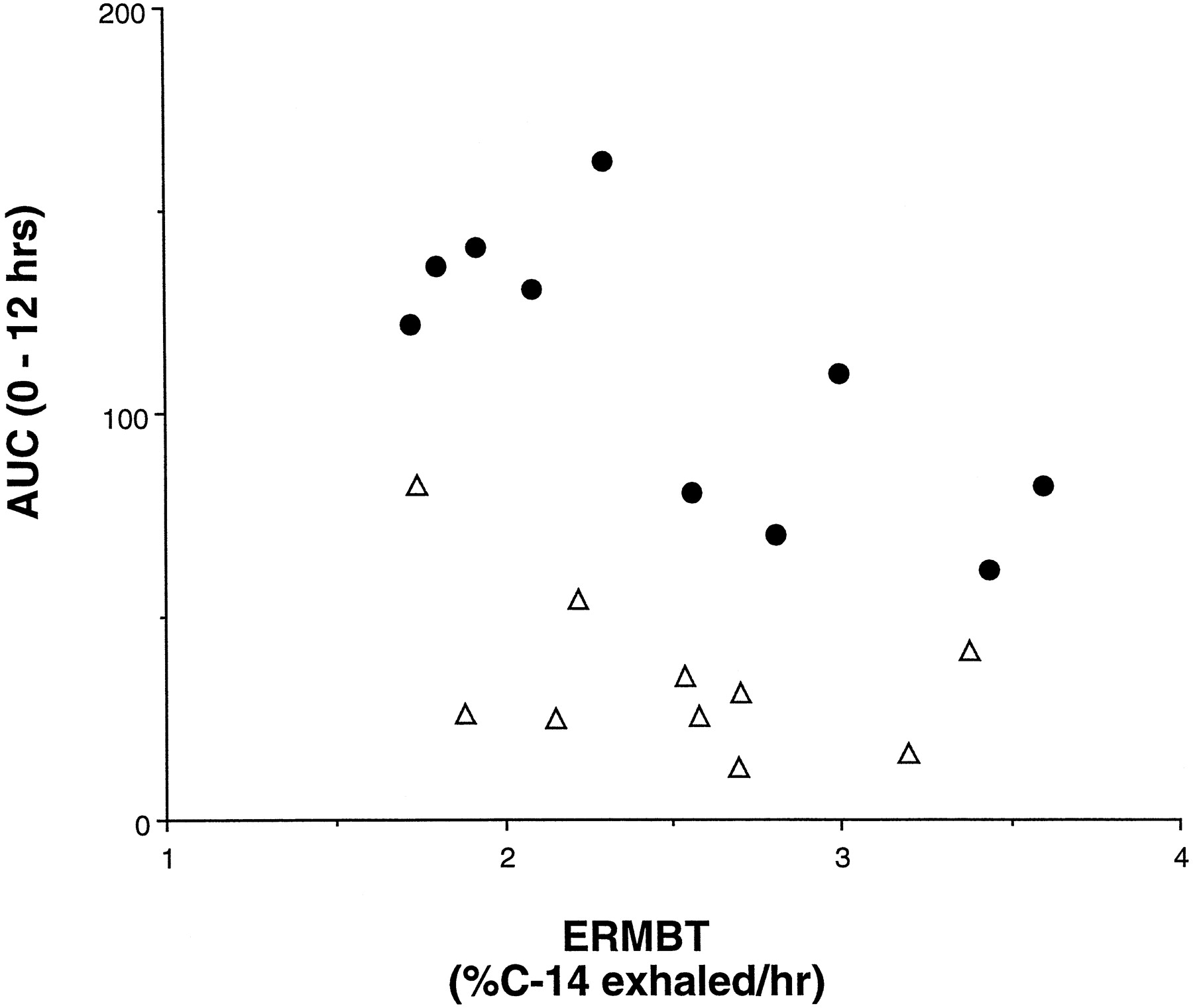
If the effect of grapefruit juice is to knock out intestinal CYP3A4 while having a negligible effect on liver CYP3A4, three observations might be expected from our data. First, because subjects with high enterocyte concentrations of CYP3A4 should have marked first-pass metabolism of felodipine at the level of the intestine (when taken with water), they would tend to have the low felodipine peak concentrations (Cmax) with water. We did in fact observe a weak but significant inverse correlation between enterocyte CYP3A4 concentration and Cmax (r = 0.66, p = .05, Spearman rank nonparametric correlation, not shown). Second, it might be expected that individuals with the highest intestinal levels of CYP3A4 would experience the greatest increase in felodipine Cmax with grapefruit juice. This was also observed (Fig.2). Finally, the bulk of felodipine metabolism might be anticipated to shift from the intestine to the liver when the drug is taken with grapefruit juice. Hence, correlation between liver CYP3A4 activity, as measured by the ERMBT, and the felodipine AUC should improve when the drug is ingested with grapefruit juice. This was observed (Fig. 3). Although these observations make intuitive sense, they should be confirmed in a larger study.

Correlation between enterocyte CYP3A4 protein concentration and magnitude of the effect of grapefruit juice treatment on felodipine Cmax in 10 healthy volunteers.
Enterocyte concentrations of CYP3A4 were measured in small bowel biopsies obtained before the subjects received grapefruit juice. Shown are the fold increases in Cmax demonstrated with the first 8-ounce glass of grapefruit juice relative to that observed when the felodipine was taken with water (•, r = 0.67,p = .043, 02, Spearman rank correlation) and the fold increases observed after 6 days (16 glasses) of grapefruit juice relative to water (▵, r = 0.71, p = .02, Spearman rank correlation). Subjects with the highest enterocyte CYP3A4 concentrations had the greatest response to grapefruit juice.

Correlation between liver CYP3A4 activity and AUC (0–12 h) for felodipine when taken with water and grapefruit juice.
The [14C-N-methyl] ERMBT was administered to each subject at the time felodipine kinetics were performed. Shown is the correlation observed when felodipine was taken with water (A) (▵,r = 0.34, p = .30, Spearman rank correlations) and with the 16th glass of grapefruit juice (B) (•,r = 0.64, p = .05, Spearman rank correlation). The improvement in correlation caused by grapefruit juice supports the idea that its effect is to shift the major site of metabolism of felodipine from the intestine to the liver.
Based on all available data, two hypotheses now seem reasonable and should be tested. First, the absence of an effect of grapefruit juice on oral clearance of a CYP3A4 substrate should mean that intestinal metabolism by the enzyme is not significant. Also, if variation in oral clearance of this substrate correlates well with liver CYP3A4 activity, liver CYP3A4 is likely to be the major catalyst of metabolism and locus for drug interactions. Second, a marked enhancement of oral availability by grapefruit juice would signify that substantial intestinal metabolism by CYP3A4 is occurring. This would be further supported if the juice caused improvement in the correlation between liver CYP3A4 activity and oral clearance. However, this second hypothesis is less well founded than the first because it assumes that grapefruit juice does not contain inhibitors of other enzymes or intestinal transport proteins (such as P-gp), which has yet to be tested.
In summary, grapefruit juice (or the active component(s) therein), and probe based tests such as the ERMBT, appear to be promising tools to complement traditional pharmacokinetic approaches when estimating the relative contributions of intestine and liver to the oral kinetics of CYP3A4 substrates. As pharmaceutical companies are increasingly performing grapefruit juice interaction studies with CYP3A4 substrates, data relevant to the two hypotheses proposed should be forthcoming.
The Emerging Role of Intestinal P-Glycoprotein (MDR1) in Oral Drug Availability (K.S.L., R.R.M., D.K.T., P.B.W.)
To directly evaluate the relative roles of intestinal and hepatic CYP3A4 and intestinal P-gp in determining oral cyclosporin A bioavailability, we recently performed a study in 25 stable renal transplant patients (Lown et al., 1997). Each patient had liver CYP3A4 activity measured using the ERMBT, underwent endoscopy to obtain small bowel biopsies for determination of the enterocyte content of CYP3A4 and P-gp, and had oral cyclosporin A pharmacokinetic parameters determined over 24 h.
Of the cyclosporin A pharmacokinetic parameters we measured, Cmax is the one most relevant to this discussion because it largely reflects the first-pass effects of the liver and intestine and is the parameter most likely to be influenced by intestinal CYP3A4 or P-gp. Consistent with our previous studies, liver CYP3A4 was significantly inversely correlated with the cyclosporin A Cmax (p = .012). That is, patients with the highest liver CYP3A4 activity as measured by the ERMBT tended to have the lowest cyclosporin A Cmax values. Liver CYP3A4 explained 32% of the variation in cyclosporin A Cmax.
In a stepwise forward multiple regression analysis, liver CYP3A4 was the most significant independent predictor of peak cyclosporin A levels. The next step of the regression incorporated the log of the enterocyte content of P-gp into the model. The correlation between intestinal P-gp and Cmax was the inverse; higher P-gp expression was associated with lower Cmax levels. No other independent variable, including enterocyte CYP3A4 level, was selected as significant in the regression analysis. P-gp accounted for an additional 30% of the variability in the model for Cmaxand was highly significant (p = .0024). The relationship between the observed log Cmax/dose values versus those predicted by the final model [log (Cmax/dose) = −0.236(ERMBT) − 0.533(log P-gp) + 3.686] is shown in Fig.4.
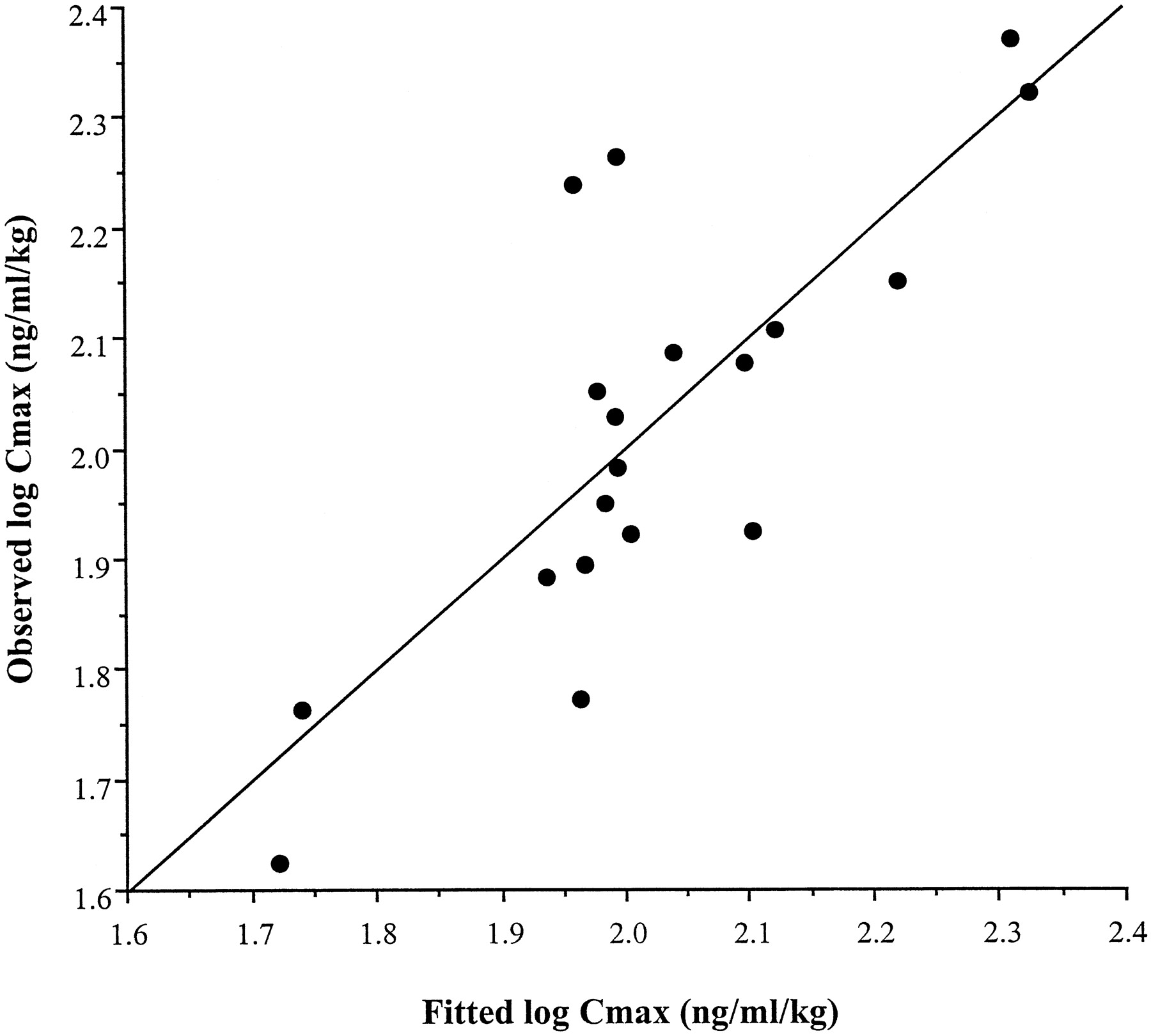
Comparisons of the observed versus predicted values from the multiple regression equations for cyclosporin A Cmax.
The regression model was derived from the ERMBT results and intestinal P-gp measurements.
We found similar results when we examined the apparent oral clearance of cyclosporin A. There was a highly significant correlation between liver CYP3A4 and the apparent oral clearance of cyclosporin A (p = .0003). Patients with the highest liver CYP3A4 activity as measured by the ERMBT tended to have the highest apparent oral clearance values. In a stepwise forward regression analysis, liver CYP3A4 was selected as most predictive of variation in cyclosporin A oral clearance and accounted for 56% of the variability in oral clearance. In the next step of the regression, enterocyte content of P-gp was again selected as next most predictive and accounted for an additional 17% of the variation in oral clearance (p = .0059). As expected, P-gp was positively correlated with oral clearance, i.e., the higher the intestinal P-gp level, the higher the oral clearance. No other variable, including enterocyte CYP3A4 levels, was significantly predictive.
In the past, the poor and highly variable oral absorption of cyclosporin A was felt to be due to complex interactions between many factors including bile salt solubilization, fat absorption, gastrointestinal motility, and lipid partitioning. One surprising result of this study is that none of these appear to be major factors in determining the availability of cyclosporin A. Using only the values for liver CYP3A4 and intestinal P-gp, we are able to explain up to three-fourths of the variation in cyclosporin A oral kinetics. As summarized in Table 2, two-thirds of the variation in Cmax was explained by just the ERMBT and intestinal P-gp, with P-gp explaining an amount essentially equal to hepatic CYP3A4. Almost three-fourths of oral cyclosporin A clearance was accounted for by these two variables. Not surprisingly, oral clearance was less dependent on intestinal P-gp than was Cmax. Clearance is more dependent than Cmax on the elimination of cyclosporin A after it has entered the systemic circulation, a process less likely to reflect the activity of P-gp on the lumenal surface of the intestine.
Significance of ERMBT and intestinal P-gp in cyclosporin A Cmaxand oral clearance
Another surprising result of this study was that intestinal CYP3A4 activity was not significantly associated with any of the oral cyclosporin A kinetic parameters. There are several possible explanations for this finding. One is that the proximal intestinal CYP3A4 levels that we measured may not be representative of the level of CYP3A4 expression in the distal intestine where significant cyclosporin A absorption occurs. This seems unlikely, however, because the intestinal levels of CYP3A4 appear to drop slowly and in a relatively linear pattern along the proximal to distal axis (Fig. 1). Another possibility is that the intestinal CYP3A4 activity was incorporated into the ERMBT result and therefore would not have appeared as an independent variable. However, we have never seen a correlation between the ERMBT result and intestinal CYP3A4 levels in any of our studies, and we have also shown that the enterocyte concentration of CYP3A4 did not significantly correlate with the variation in Cmax of another orally administered CYP3A4 substrate, felodipine (Lown et al., 1997).
This is not to suggest, however, that intestinal CYP3A4 is not important. There is considerable data to support the occurrence of substantial first-pass metabolism of cyclosporin A in human intestine (Hebert et al., 1992; Kolars et al., 1992; Gomez et al., 1995; Wu et al., 1995). However, our data does suggest that the extent of intestinal metabolism of cyclosporin A does not depend on the level of intestinal CYP3A4. A likely explanation for this finding is that intestinal P-gp may be the rate-limiting step in intestinal cyclosporin A metabolism/absorption. It is presumed that P-gp functions to keep cyclosporin A out of the body by keeping it within the lumen of the small bowel. Thus, whatever cyclosporin A that does get past P-gp and enter the enterocyte is then metabolized by enterocyte CYP3A4, i.e., the limiting step is entry of the cyclosporin A into the enterocyte.
The importance of intestinal P-gp in determining oral cyclosporin A kinetics may also have accounted for the results of another recent study and may have led to an overestimation of the amount of intestinal metabolism of cyclosporin A. It was noted that treatment with ketoconazole, a potent inhibitor of CYP3A4, increased the oral bioavailability of cyclosporin A, which was estimated to exceed 65% in healthy volunteers (Gomez et al., 1995) and 75% in kidney transplant recipients (Wu et al., 1995). These investigators assumed that the only effect of ketoconazole was to inhibit CYP3A4 and concluded that there was substantial metabolism of cyclosporin A in the intestinal wall. An alternate hypothesis for the effect of ketoconazole on the bioavailability of cyclosporin A would be that it was inhibiting P-gp function because ketoconazole has been shown to be a potent inhibitor of P-gp in a highly drug-resistant cancer cell line (Siegsmund et al., 1994).
The finding that intestinal P-gp rather than CYP3A4 is a primary determinant of oral cyclosporin A availability has several important clinical implications. Because most CYP3A4 inhibitors are also inhibitors of P-gp activity, it is entirely possible that drug interactions with cyclosporin A previously ascribed to CYP3A4 inhibition (i.e., the elevation of cyclosporin A blood levels when patients are concomitantly treated with nicardipine, ketoconazole, or erythromycin) could result in part from inhibition of P-gp activity.
Conversely, we have shown that intestinal expression of P-gp is markedly increased in humans with oral administration of rifampin (Wille et al., 1997). It is, therefore, quite possible that drug interactions with cyclosporin A previously attributed to induction of CYP3A4 (i.e., decreased cyclosporin A blood levels when given concomitantly with rifampin) may in part be due to induction of intestinal P-gp expression.
Finally, as noted above, inhibition of P-gp would be expected to improve the oral availability of cyclosporin A and potentially other P-gp substrates. This may be an important clinical means for improving the absorption of drugs with poor oral availability. Moreover, this strategy holds the potential to reduce intersubject variability in cyclosporin A clearance by removing the second most important variable (after liver CYP3A4 activity) from our predictive models.
In summary, the currently available data supports a substantial role for intestinal P-gp in determining the oral availability of cyclosporin A. As a result, it is likely that some of the drug interactions with cyclosporin A are mediated by inhibition or induction of P-gp. Whether these findings are applicable to the oral pharmacokinetics of other P-gp substrates in humans is an increasingly important question for future studies.
Overlapping Substrate Specificities of CYP3A4 and P-Glycoprotein: Implications in Drug Delivery (L.Z.B.)
In 1995, our laboratory noted a marked overlap between substrates and inhibitors for CYP3A and the multiple drug resistance protein P-gp (Wacher et al., 1995). The importance of CYP3A and P-gp to oral drug delivery was further suggested by their shared location in small intestinal enterocytes. These similarities do not appear to translate into coordinated regulation, however, as no correlation has been found between enterocyte P-gp levels and enterocyte CYP3A concentrations in healthy volunteers or kidney transplant recipients. The spatial relationship of P-gp traversing the plasma membrane and CYP3A inside the cell on the endoplasmic reticulum suggests that P-gp may act to control exposure of substrates to metabolism by the CYP3A enzymes. That is, the transporter, by serving as an efflux pump into the intestinal lumen, would allow CYP3A substrates to repeatedly encounter the enzyme by passive absorption processes, followed by further extrusion back into the gut lumen.
A series of studies in our laboratory relating to the availability of cyclosporine from the Sandimmune (Novartis, Summit, NJ) formulation seem to support this coordinated protective activity of CYP3A and P-gp in the intestine in limiting drug bioavailability. Concomitant administration of rifampin, an inducer of CYP3A and P-gp in humans, with cyclosporine resulted in a 2.7-fold decrease in cyclosporine bioavailability. In contrast, concomitant administration of ketoconazole (an inhibitor of CYP3A and P-gp in humans) resulted in a 2.5-fold increase in cyclosporine bioavailability. Our analysis of this data (Wu et al., 1995) suggests that the low bioavailability from the Sandimmune formulation of cyclosporine is not due to poor absorption, but rather due to marked intestinal metabolism of the drug. On an average, we calculate that 86% of cyclosporine from the Sandimmune formulation is absorbed, but that 51% of the cyclosporine dose is metabolized by intestinal CYP3A, on first-pass through the gut. Further studies of concomitant dosing of Sandimmune with Liqui-E (Tocopherol Polyethylene Glycol 1000 Succinate), a compound which has no inhibiting effects on CYP3A enzyme but does increase the rate of cyclosporine absorption, yielded an average 60% increase in cyclosporine bioavailability (Chang et al., 1996). All of these studies led us to conclude that on a qualitative basis, changes in the extraction ratio in the gut can be predicted depending upon the ratio of the rate constant for metabolism in the gut (kmg) to the sum of the rate constant of metabolism in the gut plus the absorption rate constant (kmg + ka). Thus, oral bioavailability can be increased for CYP3A substrates by only increasing ka, as is the case for Liqui-E and cyclosporine. In contrast, for rifampin, which induceskmg and slows ka (by inducing P-gp), a pronounced effect on decreasing cyclosporine bioavailability is observed. Since ketoconazole has opposite effects on CYP3A and P-gp from that of rifampin, a greater increase in bioavailability would be expected, as we observed.
Our laboratory hypothesizes that this coordinated activity of an enzyme, which metabolizes a drug, and a transporter, which controls the access to the enzyme, is a very logical protective mechanism which we expect to find at other sites in the body and with other enzymes and transporters. However, at the present time, we believe that understanding the importance of the interactive nature of CYP3A and P-gp will be of importance in defining, controlling, and improving oral bioavailability of CYP3A substrates. Further activities related to modeling of these processes are ongoing in our laboratory.
Footnotes
-
Send reprint requests to: Stephen D. Hall, Ph.D., Clinical Pharmacology, 320 OPW Wishard Hospital, 1001 West Tenth Street, Indianapolis, IN 46202. E-mail: sdhall{at}iupui.edu
-
↵1 Conflict of Interest Statement: Dr. Watkins owns equity in Metabolic Solutions, Inc., (Merrimack, NH) which manufactures and sells the ERMBT kit and he is also Chairman of the Scientific Advisory Board of Avmax.
-
Supported by National Institutes of Health Grants GM38149-11 (P.B.W.), GM53095-01 (K.S.L.), and MO1 RR00042 (University of Michigan General Clinical Research Center), and Grant MA-11584 from the Medical Research Council of Canada.
- Abbreviations used are::
- CYP
- cytochrome P-450
- ERMBT
- erythromycin breath test
- AUC
- area under the plasma concentration time curve
- Cmax
- maximum plasma concentration
- P-gp
- P-glycoprotein
- Vmax
- maximal rate of metabolism
- ka
- absorption rate constant
- kgm
- rate constant for gut wall metabolism
- Received August 19, 1998.
- Accepted September 1, 1998.
- The American Society for Pharmacology and Experimental Therapeutics
References




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Role of Enterocyte CYP3A in the First-Pass Extraction of Midazolam (K.E.T., M.F.P.)
- Assessing the Role of Intestinal CYP3A4 in Limiting Oral Delivery of Drugs: New Approaches (P.B.W., D.G.B., K.S.L., R.J.F.)
- The Emerging Role of Intestinal P-Glycoprotein (MDR1) in Oral Drug Availability (K.S.L., R.R.M., D.K.T., P.B.W.)
- Overlapping Substrate Specificities of CYP3A4 and P-Glycoprotein: Implications in Drug Delivery (L.Z.B.)
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters