


Abstract
In an in vitro study, the cytochrome P-450 3A (CYP3A)-dependent metabolism and drug interactions of the 3-hydroxy-3-methylglutaryl-Co A reductase inhibitors lovastatin and pravastatin were compared. Lovastatin was metabolized by human liver microsomes to two major metabolites: 6′β-hydroxy [Michaelis-Menten constant (Km): 7.8 ± 2.7 μM] and 6′-exomethylene lovastatin (Km,10.3 ± 2.6 μM). 6′β-Hydroxylovastatin formation in the liver was inhibited by the specific CYP3A inhibitors cyclosporine (Ki, 7.6 ± 2.3 μM), ketoconazole (Ki, 0.25 ± 0.2 μM), and troleandomycin (Ki, 26.6 ± 18.5 μM). Incubation of pravastatin with human liver microsomes resulted in the generation of 3′α,5′β,6′β-trihydroxy pravastatin (Km, 4,887 ± 2,185 μM) and hydroxy pravastatin (Km, 20,987 ± 9,389 μM). The formation rates of 3′α,5′β,6′β-trihydroxy pravastatin by reconstituted CYP3A enzymes were (1,000 μM pravastatin) 1.9 ± 0.6 pmol·min−1·pmol CYP3A4 and 0.06 ± 0.04 pmol·min−1·pmol CYP3A5, and the formation rates of hydroxy pravastatin were 0.12 ± 0.02 pmol·min−1·pmol CYP3A4 and 0.02 ± 0.004 pmol·min−1·pmol CYP3A5. The specific CYP3A inhibitors cyclosporine, ketoconazole, and troleandomycin significantly inhibited hydroxy pravastatin formation by human liver microsomes, but only ketoconazole inhibited 3′α,5′β,6′β-trihydroxy pravastatin formation, suggesting that other CYP enzymes are involved in its formation. It is concluded that, compared with lovastatin [CLint formation 6′β-hydroxylovastatin (μl·min−1·mg−1): 199 ± 248, 6′-exomethylene lovastatin: 138 ± 104)], CYP3A-dependent metabolism of pravastatin [CLint formation 3′α,5′β,6′β-trihydroxy pravastatin (μl·min−1·mg−1): 0.03 ± 0.03 and hydroxy pravastatin: 0.02 ± 0.02] is a minor elimination pathway. In contrast to lovastatin, drug interactions with pravastatin CYP3A-catalyzed metabolism cannot be expected to have a clinically significant effect on its pharmacokinetics.
Hypercholesterolemia is a well established risk factor for arteriosclerosis, ultimately leading to coronary heart disease and stroke, both major causes of death in developed countries. Epidemiological studies have demonstrated the positive correlation between the development of arteriosclerosis and increased serum concentrations of low-density lipoprotein cholesterol and the concentration of total cholesterol as well as the inverse correlation between arteriosclerosis and concentrations of high-density lipoprotein cholesterol (Gordon et al., 1977, 1981;Castelli et al., 1986; Anderson et al., 1987). Lovastatin and pravastatin (Fig. 1) are inhibitors of HMG-CoA1reductase, the rate-limiting step in de novo cholesterol synthesis (Dietschy and Wilson, 1970), and are widely used to treat hypercholesterolemia (Hsu et al., 1995). Among their most important side effects are increases in creatinine kinase and myopathy (Hsu et al., 1995). In patients treated with lovastatin monotherapy, the incidence of skeletal muscle toxicity is 0.1 to 0.2% (Bradford et al., 1991). This risk dramatically increases when lovastatin is combined with drugs such as cyclosporine, itraconazole, or erythromycin (Hsu et al., 1995) and is as high as 30% within 1 year in patients treated with cyclosporine (Tobert, 1988).

Structures of (A) lovastatin and its major metabolites formed by CYP3A (Wang et al., 1991) and (B) pravastatin, its major metabolites, and degradation products (Everett et al., 1991).
Alternative names used for the pravastatin metabolites in the literature (Kitzawa et al., 1993; Quion and Jones, 1994) are shown.
Lovastatin is a prodrug and is converted by esterases from its lactone to the active, open acid form. Eighty-three percent of an oral lovastatin dose is eliminated in feces, and 10% is eliminated in urine (Henwood and Heel, 1988). In bile of lovastatin-treated patients, only metabolites were detected and there was no evidence for lovastatin or its ring-opened acid (Halpin et al., 1993). For the majority of metabolites recovered from human bile, cytochrome P-450 3A (CYP3A) enzymes were involved in their formation (Wang et al., 1991), demonstrating a key role of CYP3A-dependent drug metabolism in lovastatin elimination (Fig. 1A). All drugs reported to increase lovastatin plasma concentrations and, thus, potentially leading to skeletal muscle toxicity, such as cyclosporine, erythromycin, and itraconazole, are known CYP3A substrates and/or inhibitors (Rendic and DiCarlo, 1997). In healthy volunteers, itraconazole increased the lovastatin mean area-under-the-concentration time curve 20-fold compared with the period when the subjects received placebo in place of itraconazole (Neuvonen and Jalava, 1996).
In contrast to the prodrug lovastatin, pravastatin is administered as an acid, which is more hydrophilic and biochemically active (Quion and Jones, 1994). After i.v. administration of radiolabeled pravastatin, 47% was cleared by the kidneys and 53% was cleared by nonrenal routes. Seventy percent of the radioactivity in pooled human urine samples was attributed to the parent drug (Singhvi et al., 1990). 3′α-Iso-pravastatin (R416) and 6′-epi-pravastatin (R418) (Fig. 1B) had the highest concentrations of all metabolites in urine (Everett et al., 1991). Non-CYP-dependent pathways for formation of the major pravastatin metabolite 3′α-iso-pravastatin have been described, including acidic degradation in the stomach (Triscari et al., 1995) and sulfation in combination with subsequent degradation (Kitazawa et al., 1993). Muramatsu et al. (1992) demonstrated the involvement of CYP enzymes in the formation of 3′α,5′β,6′β-trihydroxy pravastatin but did not identify the CYP enzymes responsible. In an in vitro study, pravastatin inhibited CYP2C9-, 2D6-, and 3A4-dependent metabolism with inhibition constants Ki > 50 μM (Transon et al., 1996). Based on this study, in a recent review, Lennernäs and Fager (1997) listed pravastatin as a CYP3A substrate. However, the CYP enzymes involved in pravastatin have yet to be identified. Interaction of pravastatin with CYP3A-dependent metabolism (Transon et al., 1996) does not necessarily prove that pravastatin is also a CYP3A substrate.
It was our objective to identify the CYP enzymes involved in pravastatin metabolism and to compare pravastatin and lovastatin metabolism and CYP-dependent drug interactions, as drug interactions with HMG-CoA reductase inhibitors potentially cause serious skeletal muscle toxicity and are of major clinical concern.
Materials and Methods
Pravastatin, lovastatin, and their metabolites were quantified by using a 1090 M liquid chromatograph equipped with a diode-array detector and an autosampler (Hewlett-Packard, Waldbronn, Germany). Data were processed using the Hewlett-Packard ChemStation software (version C .02.02). Analytical high-performance liquid chromatography (HPLC) columns were packed with Hypersil C8 material of 3-μm particle size (Shandon; Chadwick, UK). Extraction columns were filled with C18 material of 25- to 40-μm particle size (LiChroprep; Merck/Recipe, Darmstadt, Germany). All solvents were of HPLC quality and purchased from Merck.
HPLC/electrospray-mass spectrometric (MS) analysis was carried out using a 1090 M HPLC apparatus (Hewlett-Packard) connected to a 5989B mass spectrometer equipped with an Iris Hexapole Ion Guide (Analytica of Branford, Branford, CT) by a 59987A electrospray interface. The mass spectrometer and interface were controlled and data were processed using ChemStation software revision A04.02 (all Hewlett-Packard).
Pravastatin and its metabolites 3′α-iso and 3′α,5′β,6′β-trihydroxy pravastatin were a kind gift from Bristol-Myers Squibb (Princeton, NJ), lovastatin was a gift from Merck, Sharp & Dohme (Rahway, NJ), and cyclosporine was a gift from Novartis Pharmaceuticals (Basel, Switzerland). Ketoconazole was purchased from RBI Research Biochemicals International (Natick, MA); nifedipine, mevastatin, and troleandomycin were obtained from Sigma Chemicals (Deisenhofen, Germany). Human liver samples were collected from the unused part of the liver after liver transplantation in children who received a part of an adult liver (Klinik für Abdominal- und Transplantationschirurgie, Medizinische Hochschule Hannover, Hannover, Germany). The ethics committee of the Medizinische Hochschule Hannover approved collection of tissue samples for in vitro metabolism studies. Only liver samples of donors who did not take drugs known to interact with CYP enzymes were included. Human liver (HL) samples were collected from two female (HLF1, HLF2) and two male patients (HLM1, HLM2). NADP, isocitric acid, and isocitrate dehydrogenase for an NADPH-generating system were from Boehringer (Mannheim, Germany). Recombinant CYP3A4 and CYP3A5 enzymes as well as CYP3A antibodies were purchased from Gentest (Woburn, MA).
Isolation of Microsomes.
Microsomes were isolated by differential centrifugation as described byGuengerich (1982) with the following modification: instead of Tris buffer, 0.1 M phosphate buffer (pH 7.4) was used. After ultracentrifugation, the supernatant was discarded and the residue was reconstituted in 4 times its volume of a buffer solution containing 0.1 mM Na+/K+ phosphate buffer, 0.1 mM pyrophosphate, 1 mM ethylenediaminetetraacetic acid, and 0.1 mM dithiotreitol and stored at −80°C. Protein concentration was determined using the bicinchoninic acid method described by Smith et al. (1985) with bovine serum albumin as standard. Protein concentration of the microsomal suspension was adjusted to 1.5 g/liter with 0.1 M phosphate buffer, pH 7.4. The CYP concentration was determined using the method described by Omura and Sato (1964) following the protocol ofEstabrook and Werringloer (1978).
Metabolism of Lovastatin and Pravastatin.
One milliliter of microsomal preparation (protein concentration adjusted using 0.1 M phosphate buffer, pH 7.4) and lovastatin (in acetonitrile/water, 2:1, v/v; final concentration: 5–100 μM) were preincubated for 5 min. The reaction was started by addition of 0.5 ml of an NADPH-producing system containing 2 mM ethylenediaminetetraacetic acid, 10 mM MgCl2, 0.84 mM NADP, 18 mM isocitric acid, and 700 U/liter isocitrate dehydrogenase in 0.1 M phosphate buffer (pH 7.4). The assays were incubated for 20 min, and the reaction was stopped by protein precipitation after addition of 0.5 ml of acetonitrile.
Compared with lovastatin, a higher microsomal protein concentration of 2.5 g/liter had to be used for pravastatin metabolism. The pravastatin concentrations incubated with the microsomes ranged from 100 to 2000 μM. Pravastatin was dissolved in water, pH 7.0. After the addition of the NADPH-producing system, assays were incubated for 60 min at 37°C. The reaction was stopped by the addition of 0.2 ml of 1 M zinc acetate.
Extraction of Lovastatin and Its Metabolites.
After the addition of the internal standard mevastatin (600 ng), the precipitated proteins were separated by centrifugation at 4°C (3400g, 5 min). The supernatant was extracted with 3 ml of ethyl acetate/acetone (2:1, v/v). The samples were vortexed for 15 s, and 2.5 ml of the organic phase was transferred into a glass centrifuge tube and evaporated under a stream of nitrogen at 40°C. The residues were reconstituted in 100 μl of acetonitrile. Fifty microliters of water was added, and the sample was transferred into a glass HPLC vial.
Extraction of Pravastatin and Its Metabolites.
The samples were centrifuged at 4°C (3400g, 5 min). The supernatant was separated from the precipitated proteins and used for liquid-solid extraction of pravastatin and its metabolites. Extraction columns filled with C18 material of 25- to 40-μm particle size (Merck/Recipe) were primed by drawing 2 ml of methanol and 2 ml of water through the columns. The flow rate was adjusted to 1 ml/min. The supernatants were loaded onto the extraction columns and the samples were washed with 2 ml of water. Pravastatin and its metabolites were eluted using 400 μl of acetonitrile/formic acid, pH 4, (2:1, v/v), and the eluates were transferred into HPLC vials.
Quantification of Lovastatin and Its Metabolites.
Lovastatin and its metabolites were quantified using an HPLC/UV assay. A 250 × 4-mm analytical C8 HPLC column was used, and the following acetonitrile/sulfuric acid (pH 3) gradient was run: 0 min, 15% acetonitrile; 25 min, 50% acetonitrile; and 45 min, 50% acetonitrile. From 46 to 53 min, the column was washed with 95% acetonitrile. Analysis was stopped after 53 min, and the column was re-equilibrated to the start conditions. The flow was 0.7 ml/min, and the column temperature was 40°C. The diode array detector was set to wavelengths of 205 nm, 239 nm, and 273 nm, which were detected in parallel. Lovastatin and its metabolites were quantified using an external lovastatin calibration curve after correction for losses during extraction using the internal standard mevastatin and for their different molar UV extinction coefficients (6′β-hydroxy lovastatin, 21,400 mol·liter−1·cm−1; 6′-exomethylene lovastatin, 32,200 mol·liter−1·cm−1; and lovastatin, 21,500 mol·liter−1·cm−1) as described by Vyas et al. (1990).
Quantification of Pravastatin and Its Metabolites.
Fifty microliters of extracted samples was injected into an HPLC/MS system using a 250 × 2-mm analytical column filled with Hypersil ODS2 C18 material of 5-μm particle size (Shandon). Formic acid (pH 4) and 2-propanol were used as eluents. The flow rate was set to 0.1 ml/min and the column temperature was set to 40°C. Pravastatin and its metabolites were eluted isocratically with 18% 2-isopropanol. The electrospray interface was adjusted to the following parameters (nomenclature according to the ChemStation software): nebulizer gas, nitrogen 5.0, 80 psi; drying gas, nitrogen 5.0; flow, 40 (arbitrary units), 350°C; Vcap, −4000 V; Vend, −3500 V; Vcyl, −6000 V; capillary exit voltage, 160 V. The following parameters were used for mass spectrometry analysis: quadrupole temperature, 150°C; multiplier voltage, 1,795 V; X-ray, 10,000 V. Positive ions [M+Na]+ of pravastatin and its metabolites were recorded in the single-ion mode:m/z = 447, pravastatin;m/z = 463, hydroxy pravastatin; andm/z = 481, 3′α,5′β,6′β-trihydroxy pravastatin. The dwell time for each ion was 100 ms.
Identification of Lovastatin and Pravastatin Metabolites.
HPLC/MS was used to identify the structures of lovastatin metabolites. The mass spectrometer was run in the scan mode (m/z = 50–600). Isolated metabolite fractions were introduced into the mass spectrometer by flow injection by using a manual injection valve connected between the analytical column and the electrospray interface. The electrospray interface and the mass spectrometer were adjusted as described above for quantification of pravastatin and its metabolites. In addition, lovastatin metabolites were identified by their characteristic UV absorption spectra (Vyas et al., 1990). For pravastatin metabolites, authentic standards were available.
Metabolism of Cyclosporine and Quantification of Cyclosporine and Its Metabolites.
The effect of lovastatin and pravastatin on cyclosporine metabolism was evaluated. Cyclosporine (12.5 μM) was incubated with 1 mg of microsomal protein and NADPH-producing system. Cyclosporine and its metabolites, AM1 and AM9, were extracted and quantified by HPLC as described previously (Christians et al., 1988).
Determination of Km andVmax.
To determine Km andVmax of metabolite formation, microsomes were incubated with lovastatin concentrations (n = 4 for each concentration) of 5, 10, 15, 20, 30, 40, and 50 μM or pravastatin concentrations of 250, 500, 750, 1000, 1250, 1500, and 2000 μM. Heat-denatured microsomes were used as negative controls. Km andVmax were determined after data fitting using Lineweaver-Burk plots and SigmaPlot software (version 4.0; Jandel Scientific, San Rafael, CA).
Identification of the CYP Enzymes Involved in Pravastatin Metabolism.
The CYP enzymes involved were identified by inhibition of pravastatin metabolite formation by using specific CYP inhibitors and specific CYP antibodies, and by metabolism of pravastatin with isolated, recombinant CYP enzymes. The effects of the following specific CYP inhibitors on pravastatin metabolism were assessed: phenacetin (CYP1A1/2), coumarin (CYP2A6), tolbutamide (CYP2C8/9), S-(+)-mephenytoin (CYP2C19), debrisoquine (CYP2D6), chlorzoxazone (CYP2E1), and nifedipine (CYP3A) (Guengerich, 1995). All inhibitors, except debrisoquine, which was dissolved in water, were dissolved in methanol. Ten microliters resulting in final concentrations of 1, 10, 100, and 500 μM (n = 4) was added to the microsomal suspension containing 1000 μM pravastatin. Microsomes to which 10 μl of the vehicles, methanol, or water were added were used as controls. The mixtures were incubated and analyzed as described above. The effect of the inhibitors on pravastatin metabolite formation was evaluated statistically by using analysis of variance (GLM procedure, SAS, version 6.10, SAS Institute, Cary, NC).
Inhibition of pravastatin metabolism by specific CYP antibodies.
One hundred micrograms of microsomal protein isolated from human liver was incubated with 0, 2, 4, 6, 8, or 10 μl of CYP3A antibody solution (1 μl ≅ 10 μg of protein; Gentest) on ice for 15 min. Then, 1000 μM pravastatin and the NADPH-producing system were added, and the samples were incubated and extracted as described above.
Metabolism of lovastatin and pravastatin by recombinant CYP3A enzymes.
Thirty picomoles of recombinant CYP3A4 or CYP3A5 was incubated with 100 μM lovastatin or 1000 μM pravastatin and an NADPH-producing system as described for HLMs. Incubations of pravastatin with microsomes isolated from wild-type baculovirus-infected insect cells (Gentest) were used as negative controls.
Inhibition of Lovastatin and Pravastatin Metabolism by CYP3A Inhibitors.
Drug interactions with the CYP3A-dependent metabolism of lovastatin and pravastatin metabolism were evaluated using the specific CYP3A inhibitors cyclosporine, troleandomycin, and ketoconazole (Guengerich, 1995). All inhibitors were dissolved in methanol, and 10 μl was added to the microsomal assays, resulting in the following final concentrations: 0.1, 1, 10, and 100 μM for ketoconazole and, additionally, 500 μM for troleandomycin. To the controls, 10 μl of methanol was added. Cyclosporine and ketoconazole were preincubated with 1 ml of the microsomal preparation and 0.5 ml of the NADPH-producing system at 37°C for 15 min before lovastatin or pravastatin was added. Troleandomycin was preincubated with the microsomes and the NADPH-producing system for 20 min. The results of these assays were used to determine whether or not cyclosporine, ketoconazole, or troleandomycin was an effective inhibitor of lovastatin or pravastatin metabolism and, if so, to determine the concentration range of the inhibitors to be used for determination of the inhibition constant Ki.
When statistical analysis (analysis of variance, GLM procedure, SAS) showed a significant inhibitory effect, the inhibition constant (Ki) was determined. Lovastatin and pravastatin concentrations, as described for determination ofKm, were used. The inhibitors were added at the following concentrations: cyclosporine, 2.5, 5, and 10 μM; ketoconazole, 0.1, 0.25, 0.5, and 1 μM; and troleandomycin, 25, 50, and 100 μM. The Ki values were determined using secondary plots after Lineweaver-Burk analysis of metabolite concentrations. In secondary plots, the slopes of the regression lines were plotted versus the inhibitor concentrations. TheKi value was determined by the intersection of the fitted line of the secondary plot and the x-axis (SigmaPlot, version 4.0).
Results
Quantification of Lovastatin and Pravastatin.
The lower limit of quantitation of the HPLC assay for lovastatin was 100 μg/liter. Linearity was established up to a concentration of 2400 μg/liter. The regression equation was y = 0.0019 × +0.16 with r = 0.9997 (Spearman regression, REG procedure; SAS). After extraction from liver microsomes, the day-to-day variability was 2.7% (n = 24; 3 days). The recovery of the ring-open form (range, 50–60%) was significantly lower than that of lovastatin, its metabolites, and the internal standard (range, 95–100%) (analysis of variance, GLM procedure; SAS). The recoveries of lovastatin, its metabolites, and the internal standard did not differ from each other. Stability for the extracted samples at room temperature (autosampler stability) was established for 48 h and, for stock solutions and extracted samples at −20°C, for 56 days.
The lower limit of quantitation of the pravastatin HPLC/MS assay was 1 μg/liter, and linearity of the calibration curve was established up to 1250 μg/liter. The regression equation was y = 5,691 × +287 (r = 0.995) (Spearman regression, REG procedure; SAS). Interassay variability was 8.3%. The recoveries of pravastatin and its metabolites were >90%. Stability of pravastatin for 48 h was established in acetonitrile/water (2:1, v/v), which was used during the extraction procedure and in 2-isopropanol/formic acid, pH 4 (3:7, v/v), which was used as mobile phase for HPLC/MS. Extracted samples were stable at −20°C for at least 3 weeks.
Identification of Lovastatin Metabolites Generated by Human Liver Microsomes.
After incubation with human liver microsomes, two major lovastatin metabolites were formed. Based on 1) the UV spectra recorded using the diode array detector, 2) the known elution sequence from the reversed-phase analytical column as described previously (Vyas et al., 1990), and 3) the mass spectra of the isolated metabolites, these metabolites were identified as 6′β-hydroxy and 6′-exomethylene lovastatin.
Identification of Pravastatin Metabolites Generated by Human Liver Microsomes.
After incubation of pravastatin with liver microsomes, two metabolites were detected using HPLC/MS in the single-ion mode. HPLC/UV was not sensitive enough to detect pravastatin metabolites. A representative ion chromatogram is shown in Fig. 2. Because of the low concentrations of pravastatin metabolites generated, full-scan MS analysis of their structures was not possible. Full-scan analysis of pravastatin showed that, under the HPLC/MS conditions used, more than 90% of the ions formed were sodium adducts and no significant fragmentation occurred. The two metabolite peaks were formed neither by controls containing heat-inactivated microsomes nor during incubations with microsomes without NADPH (Fig. 2B). The metabolite with a retention time of 8.7 min (Fig. 2A) had a molecular ion [M + Na]+ of m/z = 481, which was compatible with 3′α,5′β,6′β-trihydroxy pravastatin (metabolite 12) described by Everett et al. (1991). The structure of this metabolite was confirmed by comparison of the retention time and mass spectrum with those of authentic standard material injected into the HPLC/MS system under the same conditions. The structure of the metabolite with a retention time of 9.5 min and an [M + Na]+ at m/z = 463 was hydroxy pravastatin. Because no authentic standard material was available and, due to the small concentration, full-scan HPLC/MS analysis was not possible, the exact structure was not further identified.

Ion chromatograms after incubation of pravastatin with human liver microsomes.
A, incubation of pravastatin with human liver microsomes and an NADPH-producing system. B, incubation of pravastatin with human liver microsomes without NADPH (negative control). Single ions were recorded (m/z = 447, pravastatin; m/z = 463, hydroxy pravastatin; m/z = 481, 3′α,5′β,6′β-trihydroxy pravastatin), and ion chromatograms were overlaid. Typical ion chromatograms (total n = 4) are shown. The run time of the ion chromatograms (x-axis) is given in minutes.
Enzyme Kinetics of Lovastatin and Pravastatin Metabolite Formation by Human Liver Microsomes.
The apparent binding constant as determined by UV differential spectroscopy of lovastatin to CYP enzymes (KS) was 18 μM. Formation of lovastatin metabolites by human liver microsomes was linear over an incubation period of 20 min. The apparent Km andVmax values of 6′β-hydroxy and 6′-exomethylene lovastatin formation are shown in Table1. Lovastatin concentrations > 100 μM resulted in substrate inhibition.
Apparent Km and Vmax values for the formation of pravastatin and lovastatin metabolites by human liver microsomes
The apparent binding constant (KS) of pravastatin to CYP enzymes in human liver microsomes was 35,000 μM. Formation of 3′α,5′β,6′β-trihydroxy pravastatin and hydroxy pravastatin was linear over a 60-min incubation period. The apparentKm and Vmax as estimated using Lineweaver-Burk analysis are shown in Table 1.
Identification of the CYP Enzymes Involved in Pravastatin Metabolism.
Of the specific inhibitors used, only nifedipine (CYP3A) had a significant effect on 3′α,5′β,6′β-trihydroxy pravastatin and -hydroxy pravastatin formation (P < .05). The half-maximal inhibition constant for 3′α,5′β,6′β-trihydroxy pravastatin was 51 ± 9 μM (mean ± S.D.) and that for hydroxy pravastatin was 80 ± 18.4 μM (n = 4). Addition of 500 μM nifedipine reduced formation of 3′α,5′β,6′β-trihydroxy and hydroxy pravastatin by 70 and 86%, respectively. Inhibitors of CYP1A1/2 (phenacetin), CYP2A6 (coumarin), CYP2C8/9 (tolbutamide), CYP2C19 (S-(+)-mephenytoin), CYP2D6 (debrisoquine), and CYP2E1 (chlorzoxazone) had no significant effect, and formation of pravastatin metabolites was not statistically different from the uninhibited controls.
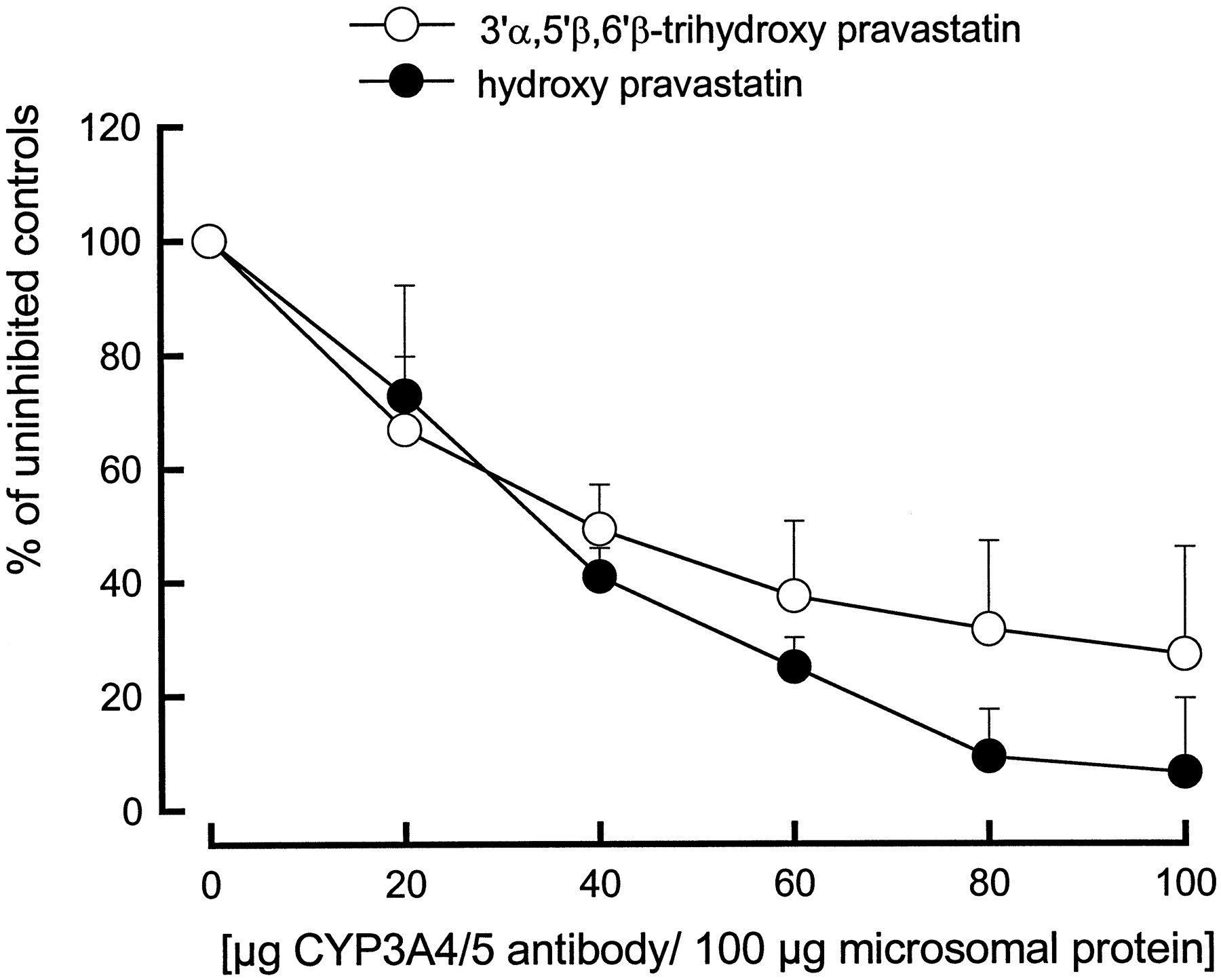
Antibodies against human CYP3A enzymes inhibited pravastatin metabolism by human liver microsomes with an IC50 of 20 μg of antibody per 100 μg of human liver microsomal protein. Compared with uninhibited controls, 100 μg of antibody per 100 μg of human liver microsomal protein reduced 3′α,5′β,6′β-trihydroxy and hydroxy pravastatin formation by 73 and 94%, respectively (Fig.3).

CYP3A4/5 antibody concentration-dependent immunoinhibition of pravastatin metabolite formation by human liver microsomes.
One hundred micrograms of microsomal protein and different volumes of antibody solution (Gentest) were preincubated for 15 min. The reaction was started by addition of 1000 μM pravastatin and NADPH-generating system. The assays were incubated at 37°C for 30 min. Concentrations of pravastatin metabolites were determined by HPLC/MS as described inMaterials and Methods. Each data point represents the mean ± S.D. (n = 4).
Isolated recombinant CYP3A4 incubated with 1000 μM pravastatin generated 3′α,5′β,6′β-trihydroxy pravastatin at a rate of 1.9 ± 0.6 pmol·min−1·pmol−1CYP3A4 and hydroxy pravastatin at a rate of 0.12 ± 0.02 pmol·min−1·pmol−1CYP3A4. Isolated recombinant CYP3A5 generated 3′α,5′β,6′β-trihydroxy pravastatin at a rate of 0.062 ± 0.042 pmol·min−1·pmol−1CYP3A5 and hydroxy pravastatin at a rate of 0.016 ± 0.004 pmol·min−1·pmol−1CYP3A5 (all mean ± S.D., n = 4). In comparison, isolated recombinant CYP3A4 incubated with 100 μM lovastatin produced 6′β-hydroxy lovastatin at a rate of 98.0 ± 2.9 pmol·min−1·pmol−1CYP3A4 and 6′-exomethylene lovastatin at a rate of 44.3 ± 1.2 pmol·min−1·pmol−1CYP3A4. CYP3A5 was 10-fold less active: 6′β-hydroxy lovastatin, 8.2 ± 0.2 pmol·min−1·pmol−1CYP3A5 and 6′-exomeyhylene lovastatin, 2.8 ± 0.05 pmol·min−1·pmol−1CYP3A5.
Drug Interactions with the Metabolism of Lovastatin and Pravastatin by Human Liver Microsomes.
Cyclosporine, ketoconazole, and troleandomycin were effective inhibitors of lovastatin metabolism. All inhibitors significantly reduced formation of both lovastatin metabolites with comparableKi values. Cyclosporine inhibited 6′-exomethylene lovastatin formation with aKi of 7.6 μM and hydroxy lovastatin formation with a Ki of 10.3 μM. TheKi values for ketoconazole were 0.3 μM 6′-exomethylene lovastatin and 0.4 μM 6′β-hydroxy lovastatin, and those for troleandomycin were 26.6 μM 6′-exomethylene lovastatin and 31.0 μM 6′β-hydroxy lovastatin. Ketoconazole (10 μM) and 100 μM troleandomycin completely inhibited lovastatin metabolism by human liver microsomes. Cyclosporine (100 μM) reduced 6′-exomethylene formation to 43.4 ± 4.3% (mean ± S.D.) and 6′β-hydroxy lovastatin formation to 54.1 ± 16.3% compared with uninhibited controls (100%).
Cyclosporine significantly inhibited formation of hydroxy pravastatin with a Ki of 5.5 μM but failed to inhibit formation of 3′α,5′β,6′β-trihydroxy pravastatin as tested by analysis of variance. Compared with uninhibited controls, 100 μM cyclosporine reduced hydroxy pravastatin formation to 27.2 ± 7.9%, whereas formation of 3′α,5′β,6′β-trihydroxy pravastatin was 99.1 ± 39.7%. Like cyclosporine, troleandomycin inhibited hydroxy pravastatin formation (Ki, 0.6 μM) but failed to affect formation of 3′α,5′β,6′β-trihydroxy pravastatin to a statistically significant extent. Troleandomycin (μM) abolished hydroxy pravastatin formation, whereas formation of 3′α,5′β,6′β-trihydroxy pravastatin was still 67.2 ± 36.2% of the uninhibited controls. Like nifedipine (vide supra) and in contrast to cyclosporine and troleandomycin, ketoconazole inhibited metabolism of pravastatin to both its hydroxy metabolite (Ki 0.27 μM) and its 3′α,5′β,6′β-trihydroxy metabolite (Ki0.5 μM). However, although in the presence of 10 μM ketoconazole no hydroxy pravastatin was formed, a ketoconazole concentration as high as 500 μM did not completely inhibit 3′α,5′β,6′β-trihydroxy pravastatin generation (26.9 ± 7.8% of uninhibited controls).
Inhibition of Cyclosporine Metabolism by Pravastatin and Lovastatin.
Lovastatin inhibited formation of AM1 as well as formation of AM9 with a Ki of 8.5 μM. In the presence of 1000 μM lovastatin, cyclosporine metabolites were not detected. In contrast, pravastatin at a concentration of as high as 5000 μM failed to affect formation of the major cyclosporine metabolites AM1 and AM9.
Discussion
We demonstrated that in the human liver, CYP3A4 and CYP3A5 are involved in the biotransformation of pravastatin to its hydroxy and 3′α,5′β,6′β-trihydroxy metabolites. However, compared with lovastatin, the binding constant of pravastatin to CYP enzymes and the Michaelis-Menten constants of its metabolite formation are 1000-fold higher. Thus, higher microsomal protein and substrate concentrations were required in the pravastatin than in the lovastatin in vitro metabolism assays. In addition, HPLC/UV was not sensitive enough to detect and quantify pravastatin metabolites after incubation with liver microsomes, and development of a 100-fold more sensitive HPLC/MS assay was required.
It must be noted that only apparent enzyme kinetic parameters were determined for the following reasons. Microsomes do not represent an isolated enzyme but a mixture of different enzymes, and more than one CYP enzyme is involved in the metabolism of lovastatin and pravastatin. Additionally, some of the inhibitors tested, such as cyclosporine, are also CYP3A substrates and are metabolized during incubation. In addition, the Km values of pravastatin metabolite formation, as determined after linearization according to Lineweaver-Burk analysis, were higher than the highest pravastatin concentration used in the kinetic analysis. Therefore, Michaelis-Menten kinetic analysis was rerun with pravastatin concentrations close to or higher than theKM (range, 250–10,000 μM). The resultingKm values confirmed those presented in Table 1. Because microsomal preparations different from those for lovastatin had to be used, these data are not shown in detail.
Although the structure of the hydroxy pravastatin metabolite ultimately could not be identified, human liver microsomes most likely generated 3"-hydroxy pravastatin. A total of three pravastatin metabolites with an [M + Na]+m/z = 463 have been described: 3"-hydroxy pravastatin, 5,6-epoxy-3′α-iso pravastatin, and 7-OH-3′α-iso pravastatin (Everett et al., 1991). In contrast to 3"-hydroxy pravastatin, which is directly derived from pravastatin, 5,6-epoxy-3′α-iso pravastatin and 7-OH-3′α-iso pravastatin require generation of 3′α-iso pravastatin as a precursor. 3′α-Iso pravastatin is generated by either phase II metabolism or acidic degeneration (Kitzawa et al., 1993; Triscari et al., 1995), and, therefore, formation of these two metabolites under the incubation conditions used was unlikely (Everett et al., 1991).
After administration of [14C]pravastatin to healthy volunteers, pravastatin constituted most of the radioactivity (Everett et al., 1991) recovered in urine and feces. In addition to the isomers 3′α-iso pravastatin and 6′epi pravastatin, which accounted for 13% of the radioactivity after p.o. and 4% of the radioactivity after i.v. administration, at least 15 metabolite fractions were isolated from urine. None of these accounted for more than 6% of the total urinary radioactivity. In contrast to lovastatin, whose major metabolic pathways are catalyzed by CYP3A, pravastatin undergoes several different reactions, including acidic degeneration, β-oxidation of the side chains, glucuronidation, conjugation with glutathione (Muramatsu et al., 1992), and sulfation with subsequent epimerization (Kitazawa et al., 1993). Muramatsu et al. (1992) demonstrated glutathione conjugation and formation of a dihydrodiol metabolite (3′α,5′β,6′β-trihydroxy pravastatin) in isolated rat hepatocytes and proposed 4′αβ,5′β epoxide pravastatin as a precursor for both metabolites. 4′αβ,5′β-Epoxide pravastatin, from which 3′α,5′β,6′β-trihydroxy pravastatin is formed by a nonenzymatic reaction (Muramatsu et al., 1992), was not detected in our study.
Because in our study the specific CYP3A inhibitors cyclosporine, troleandomycin, and ketoconazole were effective inhibitors of hydroxy pravastatin formation in the human liver and, at high concentrations, abolished its formation, it is concluded that CYP3A is mainly responsible for its formation. Although nifedipine, ketoconazole, and CYP3A4/5 antibodies were effective inhibitors, none of these inhibitors, even at the highest concentrations, reduced 3′α,5′β,6′β-trihydroxy pravastatin formation by more than 75%, indicating the involvement of CYP enzymes other than members of the CYP3A subfamily. This is supported by the results of Muramatsu et al. (1992). In addition to phenobarbital, an inducer of CYP enzymes 3A1 and 3A2 in the rat (Soucek and Gut, 1992), Muramatsu et al. (1992)successfully induced formation of 3′α,5′β,6′β-trihydroxy pravastatin by 3-methylcholanthrene, an inducer of CYP enzymes 1A1, 1A2, 2A1, 2A2, and 2D1 in the rat (Soucek and Gut, 1992). In addition to CYP3A inhibitors, we studied the effects of specific inhibitors of CYP1A1/2, CYP2A6, CYP2C8/9, CYP2C19, CYP2D6, and CYP2E1 on 3′α,5′β,6′β-trihydroxy pravastatin formation. Because none of these inhibitors affected pravastatin metabolism, a significant involvement of one or more of these CYP enzymes in 3′α,5′β,6′β-trihydroxy pravastatin formation seems unlikely.
The quantitative differences between CYP-dependent metabolism of lovastatin and pravastatin in the liver became especially clear when the intrinsic clearances (Vmax/Km) for the two major in vitro metabolites of either drug were calculated. The intrinsic clearance is a parameter commonly used for quantitative in vitro-in vivo allometric scaling (Ashforth et al., 1995;Iwatsubo et al., 1996; Houston and Carlile, 1997). The intrinsic clearances for the two pravastatin metabolites (hydroxy pravastatin, 0.024 ± 0.022 μl·min−1·mg−1; 3′α,5′β,6′β-trihydroxy pravastatin, 0.03 ± 0.03 μl·min−1·mg−1; mean ± S.D.) were 10,000-fold lower than those for the lovastatin metabolites (6′β-hydroxy lovastatin, 199 ± 248 μl·min−1·mg−1; 6′-exomethylene lovastatin, 138 ± 104 μl·min−1·mg−1), indicating that CYP-dependent metabolism of pravastatin is quantitatively negligible compared with that of lovastatin.
The different intrinsic clearances of the structurally closely related HMG-CoA reductase inhibitors pravastatin and lovastatin may be explained by the fact that the 6′ position is the major lovastatin hydroxylation site and pravastatin already has a hydroxy group in this position (see Fig. 1).
According to Everett et al. (1991), 3"-hydroxy pravastatin is only a minor metabolite accounting for less than 1% of the radioactivity in urine and plasma after a [14C]pravastatin dose. In addition, as demonstrated by inhibition of pravastatin metabolism by specific CYP3A substrates and CYP3A antibodies, there is strong evidence that CYP enzymes other than CYP3A are involved in the formation of 3′α,5′β,6′β-trihydroxy pravastatin. Taking into account the low intrinsic clearance of 3′α,5′β,6′β-trihydroxy pravastatin, which predicts a hepatic clearance of 0.024 ± 0.025 ml·min−1·kg−1, CYP-dependent metabolism of pravastatin to its 3′α,5′β,6′β-trihydroxy metabolites does not seem sufficient to explain the 3′α,5′β,6′β-trihydroxy pravastatin concentrations in urine (Everett et al., 1991), suggesting the existence of alternative formation pathways.
Our in vitro study supports the results of a recently reported clinical itraconazole-pravastatin drug interaction study in healthy volunteers (Neuvonen et al., 1998). Although the CYP3A inhibitor itraconazole increased lovastatin (Neuvonen and Jalava, 1996) and simvastatin (Neuvonen et al., 1998) area-under-the-concentration values 20-fold, as compared with results of the period when the volunteers received placebo instead of itraconazole, itraconazole did not significantly affect pravastatin pharmacokinetics.
Furthermore, it is interesting to note that although several cases of skeletal muscle toxicity have been described when CYP3A inhibitors such as cyclosporine were coadministered with lovastatin (Hsu et al., 1995), no such case of toxicity has been reported when pravastatin and CYP3A inhibitors were coadministered.
The clinical study by Neuvonen et al. (1998) supports the conclusion from our in vitro study that CYP3A-dependent metabolism of pravastatin is unlikely to be a clinically relevant elimination pathway and that, in contrast to lovastatin, inhibition of its CYP3A-dependent metabolism should not significantly impact pravastatin pharmacokinetics.
Footnotes
-
Send reprint requests to: Uwe Christians, Department of Biopharmaceutical Sciences, School of Pharmacy, University of California at San Francisco, 513 Parnassus Avenue, Room S-834, San Francisco, CA 94143-0446. E-mail: uwec{at}itsa.ucsf.edu
-
This study was supported by Deutsche Forschungsgemeinschaft Grants SFB265/A7, SFB280/A8, and Ch95/6–1 and National Institutes of Health Grant GM26691.
- Abbreviations used are::
- CYP
- cytochrome P-450
- HLF
- human liver (female)
- HLM
- human liver (male)
- HMG
- 3-hydroxy-3-methylglutaryl
- HPLC
- high-performance liquid chromatography
- MS
- mass spectrometry
- NADPH
- reduced nicotinamide adenine dinucleotide phosphate
- Received April 8, 1998.
- Accepted July 30, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}