


Abstract
Testosterone, terfenadine, midazolam, and nifedipine, four commonly used substrates for human cytochrome P-450 3A4 (CYP3A4), were studied in pairs in human liver microsomes and in microsomes from cells containing recombinant human CYP3A4 and P-450 reductase, to investigate in vitro substrate-substrate interaction with CYP3A4. The interaction patterns between compounds with CYP3A4 were found to be substrate-dependent. Mutual inhibition, partial inhibition, and activation were observed in the testosterone-terfenadine, testosterone-midazolam, or terfenadine-midazolam interactions. However, the most unusual result was the interaction between testosterone and nifedipine. Although nifedipine inhibited testosterone 6β-hydroxylation in a concentration-dependent manner, testosterone did not inhibit nifedipine oxidation. Furthermore, the effect of testosterone and 7,8-benzoflavone on midazolam 1′-hydroxylation and 4-hydroxylation demonstrated different regiospecificities. These results may be explained by a model in which multiple substrates or ligands can bind concurrently to the active site of a single CYP3A4 molecule. However, the contribution of separate allosteric sites and conformational heterogeneity to the atypical kinetics of CYP3A4 can not be ruled out in this model.
Human cytochrome P-450 3A4 (CYP3A4)3, one of the major cytochrome P-450s in human liver, is responsible for the metabolism of a large number of therapeutic agents and endogenous substrates (Wrighton and Stevens, 1992; Guengerich, 1995). This enzyme has been shown to be allosteric, difficult to reconstitute from the purified components, and subject to modulation by buffers, detergent, cytochrome b5, and other factors (Guengerich et al., 1986; Kitada et al., 1987; Shimada and Guengerich, 1989; Imaoka et al., 1992; Gillam et al., 1993; Shet et al., 1993, 1995; Lee et al., 1995; Yamazaki et al., 1996; Maenpaa et al., 1998). Molecular modeling and metabolism studies suggest that the active site of CYP3A4 has the capacity to accommodate large molecules and more than one substrate (Shou et al., 1994; Lewis et al., 1996).
The ability of CYP3A4 to metabolize numerous substrates accounts for the large number of documented drug interactions associated with CYP3A4 inhibition (Venkatesan, 1992; Wilkinson, 1996; Ameer and Weintraub, 1997; Bertz and Granneman, 1997; Lin and Lu, 1997a,b). Whenever two or more drugs are administered concurrently, the possibility of metabolism-based drug interaction exists if drugs are metabolized by the same cytochrome P-450. As a result, many in vitro studies were conducted in recent years using human liver microsomes or recombinant human cytochrome P-450 systems as screening tools to evaluate the potential drug-drug interaction in vivo, based on the assumption that these drugs will compete for the same enzyme catalytic site. The clinical significance of a metabolic drug interaction will depend on the relative Km andKi values of the drugs, the magnitude of the change in parent drug and/or metabolite concentrations at the site of pharmacological action, and the therapeutic index of the drugs.
Because of the unique properties of CYP3A4, substrate interactions involving this enzyme do not always follow typical competitive inhibition kinetics (Korzekwa et al., 1998). The in vitro metabolism of one substrate can be either inhibited or stimulated by another substrate. In a recent study (Wang et al., 1997), we reported that CYP3A4-catalyzed testosterone 6β-hydroxylation and erythromycinN-demethylation involves competitive inhibition of both substrates. At high substrate concentration, either one of the substrates can bind to the substrate-enzyme complex to form the S1·E·S2 complex, which is catalytically competent. Thus, higher concentrations of erythromycin do not necessarily result in greater inhibition of testosterone 6β-hydroxylation. Instead, only partial inhibition was observed.
To determine whether partial inhibition represents a typical mechanism involving other CYP3A4-catalyzed reactions, we selected several CYP3A4 substrates and studied their metabolism in pairs. The results indicate that CYP3A4 is indeed a very complex enzyme and that interaction patterns are substrate-dependent.
Experimental Procedures
Materials.
Testosterone, 6β-hydroxy testosterone, nifedipine, terfenadine, glucose 6-phosphate, NADP, and glucose 6-phosphate dehydrogenase were purchased from Sigma (St. Louis, MO). Oxidized nifedipine was obtained from Gentest Corp. (Woburn, MA). Midazolam, 1′-hydroxy midazolam, and 4-hydroxy midazolam were gifts from Hoffmann-La Roche, Inc. (Nutley, NJ). [N-butyl-4-3H]terfenadine and hydroxy terfenadine were synthesized by the Labeled Compound Synthesis Group, Merck Research Laboratories (Rahway, NJ). All other reagents and solvents were of high analytical grade and supplied by Fisher Scientific Co., (Fairlawn, NJ). Human liver microsomal preparations were provided by Dr. Judy Raucy (Agouron Institute, La Jolla, CA). Protein concentrations and P-450 contents were determined using the bicinchoninic acid procedure (Smith et al., 1985) and according to Omura and Sato (1964), respectively. Microsomes from human B-lymphoblastoid cells expressing human CYP3A4 and NADPH-cytochrome P-450 reductase (CYP3A4/OR) were obtained from Gentest Corp.
Microsomal Incubation.
Human liver microsomal samples (0.2 mg) or microsomes from cells containing recombinant human CYP3A4/OR (0.4 mg) were incubated with various concentrations of a pair of CYP3A4 substrates in 100 mM potassium phosphate buffer (pH 7.4) with 1 mM EDTA, 6 mM MgCl2, and an NADPH-generating system consisting of 10 mM glucose 6-phosphate, 1 mM NADP, and 0.14 U glucose 6-phosphate dehydrogenase in a total volume of 0.2 ml. Incubations were carried out in a 37°C shaking water bath for 5 min, except for testosterone-nifedipine interaction experiment, which was 10 min. Reactions were stopped by adding 0.2 ml of methanol. Samples were then centrifuged at 14,000g for 10 min, and the supernatants were directly injected for HPLC analysis. Each set of incubation was carried out with six to eight concentrations of substrate and inhibitor. The control samples that contain only one substrate were performed in duplicate. Four additional human liver microsomes were used in the testosterone-nifedipine interaction experiments.
HPLC Analysis.
An aliquot of the supernatant (50 μl) from each incubation was injected onto a Zorbax SB C8 column (4.6 × 75 mm; 3.5 μM; Mac-Mod Analytic, Inc., Chadds Ford, PA) and eluted at a flow rate of 2 ml/min by a linear gradient with the mobile phase, which consisted a mixture of buffer A (10 mM ammonium acetate) and buffer B (10 mM ammonium acetate in 90% acetonitrile and 10% methanol). Chromatographic peaks of testosterone, midazolam, nifedipine, and their metabolites were monitored at 254 nm. Terfenadine and its alcohol metabolite (t-butyl-hydroxy terfenadine) were monitored with an on-line β-RAM radioactivity detector (IN/US, Tampa, FL). The linear gradient condition for each assay, and the retention times of the substrate and its metabolite, are as follows: testosterone 6β-hydroxylation (buffer B from 25–60% in 7 min, 6β-hydroxy testosterone 3.1 min, and testosterone 6.6 min); midazolam 1′-and 4-hydroxylation (buffer B from 25–65% in 7 min, 4-hydroxy midazolam 4.5 min, 1′-hydoxy midazolam 5 min, and midazolam 6.5 min); Nifedipine oxidation (buffer B from 25–40% in 10 min, oxidized nifedipine 8 min, and nifedipine 9.2 min); and terfenadinet-butyl-hydroxylation (buffer B from 25–65% in 35 min,t-butyl-hydroxy terfenadine 10.5 min, and terfenadine 22 min).
Results
General Considerations.
Testosterone, terfenadine, midazolam, and nifedipine, four commonly used probes for CYP3A4, were selected as substrates in this study. Erythromycin, the substrate used in the previous study (Wang et al., 1997), is known to be converted to a metabolic intermediate that can form a metabolite-CYP3A4 complex (Franklin, 1977). Formation of this complex could further complicate the interaction study, so erythromycin was not included as a test substrate.
The Km and Vmaxvalues for all four substrates were determined (Table1). Both human liver microsomes and a recombinant CYP3A4 and P-450 reductase system were used as the enzyme source in separate inhibition experiments. Because competition of both substrates for a single enzyme is concentration-dependent, a wide range of concentrations of both substrates was used. Thus, any lack of inhibitory effect can not be attributed simply to the differences inKm values for different substrates.
Kinetic parameters for various CYP3A4 substrates
To obtain more consistent data, initial reaction rates were obtained simultaneously for both substrates in a single incubation mixture. Although six to eight concentrations of each substrate were routinely used in the interaction study, only part of the data is displayed in the figures for clear presentation. Because the results from human liver microsomes and the recombinant system are similar, most of the data obtained from the recombinant CYP3A4 system is not presented.
Testosterone-Terfenadine Interaction.
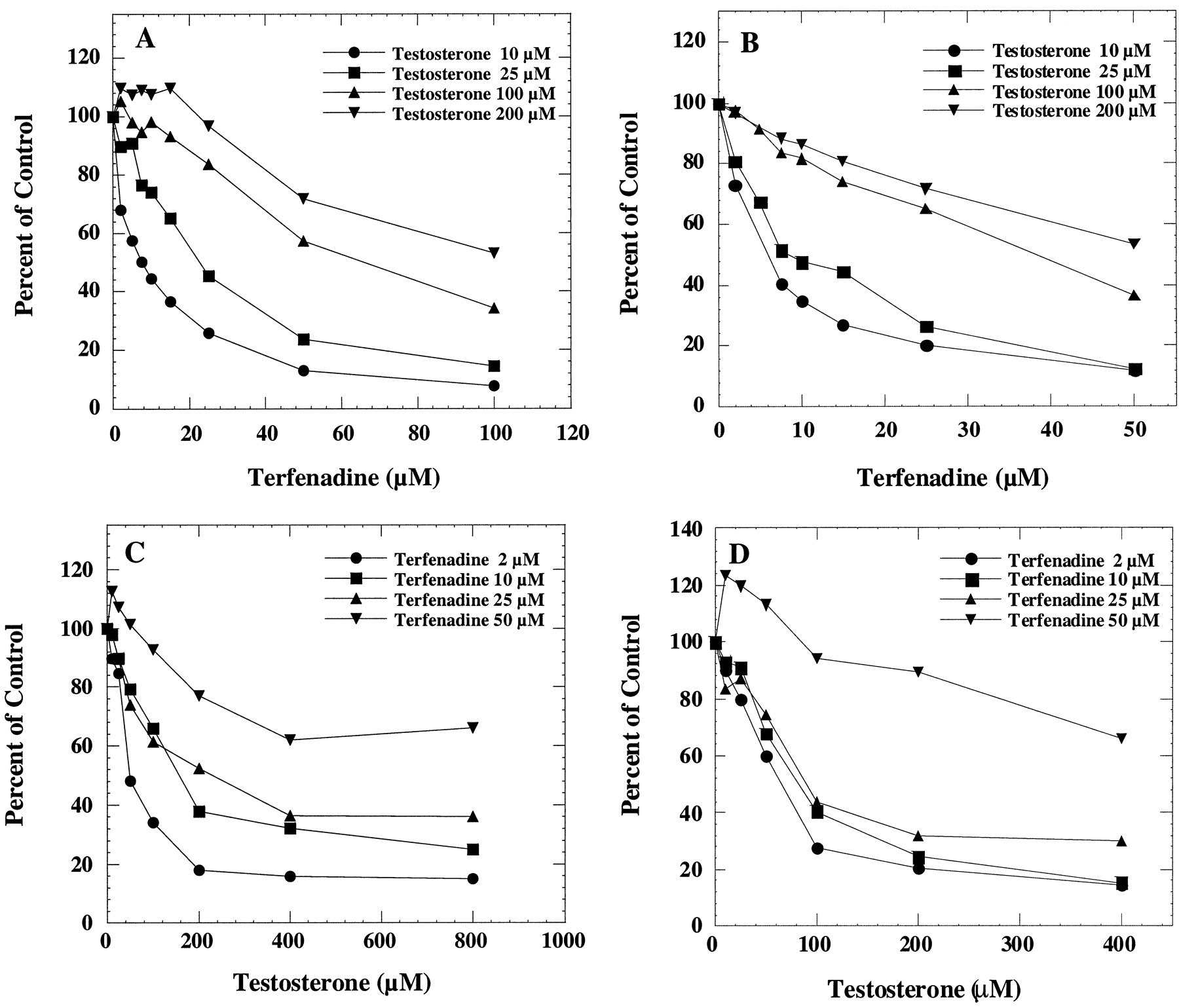
Terfenadine has been reported to be metabolized to at least three major metabolites in human liver microsomes and in the CYP3A4/OR fusion protein (Rodrigues et al., 1995). In this study, the formation oft-butyl-hydroxy terfenadine, one of the major metabolites, was determined. At low substrate concentrations, testosterone 6β-hydroxylation was inhibited in a concentration-dependent manner by terfenadine in human liver microsomes (Fig.1A) and in the recombinant CYP3A4/OR system (Fig. 1B). In the same incubation mixtures, the rates of formation of t-butyl-hydroxy terfenadine were also determined in the presence of testosterone in human liver microsomes (Fig. 1C) and in the recombinant CYP3A4 system (Fig. 1D). The formation of t-butyl-hydroxy terfenadine at 50 μM terfenadine concentration was slightly stimulated by low concentrations of testosterone. Partial inhibition, indicated by the lack of additional inhibition at higher inhibitor concentrations, was more evident for testosterone inhibition of terfenadine t-butyl-hydroxylation but less so for terfenadine inhibition of testosterone 6β-hydroxylation. These results suggest that the testosterone-CYP3A4-terfenadine complex, formed at higher substrate concentrations, favors the testosterone 6β-hydroxylation pathway rather than the terfenadine hydroxylation pathway.
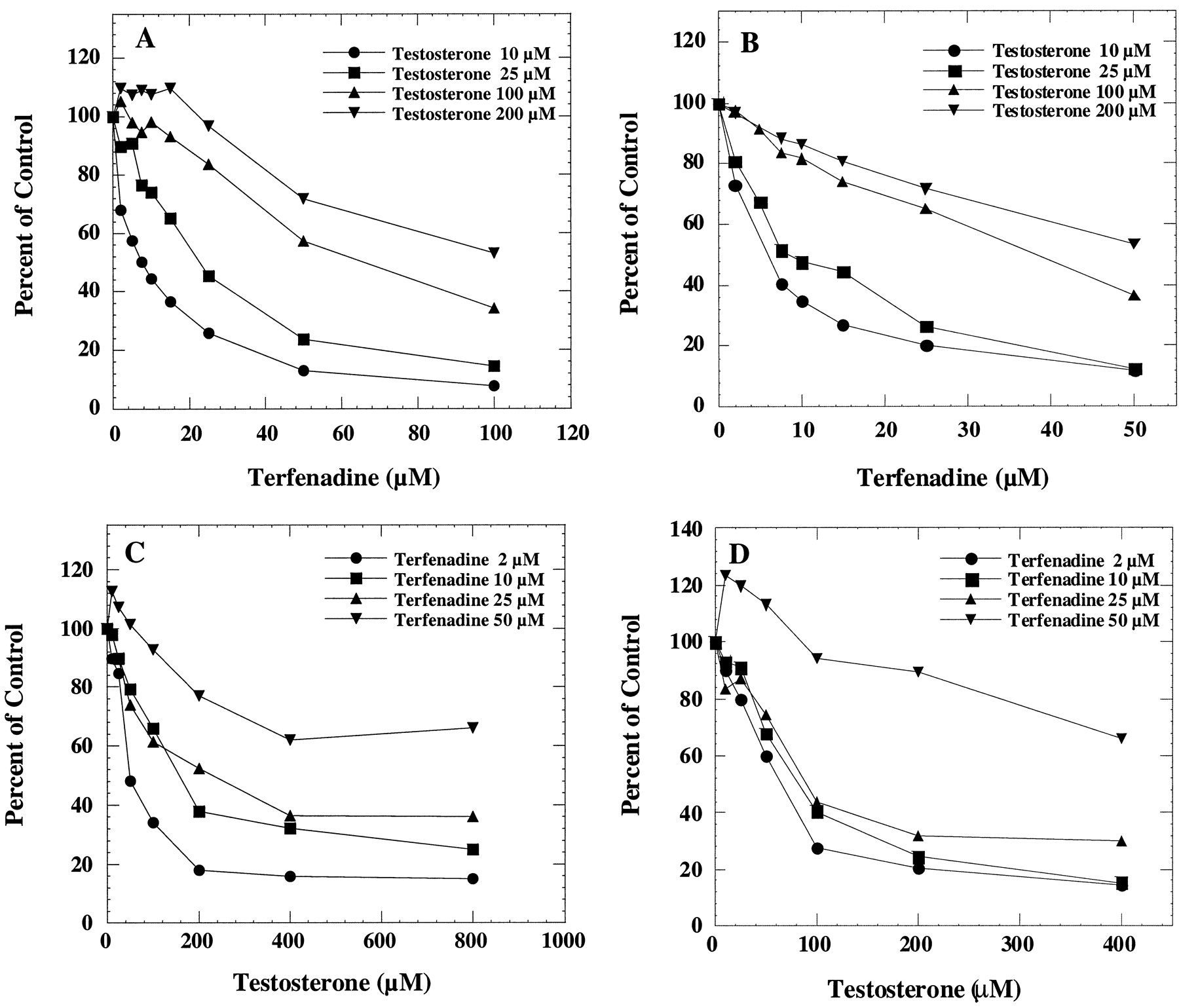
Interaction of testosterone and terfenadine in human liver microsomes and microsomes prepared from cells expressing CYP3A4/OR.
The effect of terfenadine on testosterone 6β-hydroxylation was determined by incubating human liver microsomes (A) or microsomes containing CYP3A4/OR (B) with 10, 25, 100, and 200 μM testosterone and various concentrations of terfenadine. The corresponding control activities were 1.1, 2.2, 6.5, and 8.6 nmol/min/mg, respectively, in human liver microsomes. The corresponding control activities were 0.4, 1.1, 2.5, and 3.1 nmol/min/mg in CYP3A4/OR. The effect of testosterone on terfenadine t-butyl-hydroxylation was determined by incubating human liver microsomes (C) or microsomes containing CYP3A4/OR (D) with 2, 10, 25, and 50 μM terfenadine and various concentrations of testosterone. The corresponding control activities were 0.07, 0.17, 0.31, and 0.28 nmol/min/mg, respectively, in human liver microsomes. The corresponding control activities were 0.02, 0.06, 0.09, and 0.07 nmol/min/mg in CYP3A4/OR.
Testosterone-Midazolam Interaction.
Midazolam is metabolized by CYP3A4 to 1′-hydroxy (major metabolite) and 4-hydroxy (minor metabolite) products (Kronbach et al., 1989; Gorski et al., 1994; Ghosal et al., 1996). We confirmed that midazolam metabolism was inhibited by anti-CYP3A4 peptide antibodies (Wang et al., 1999). In human liver microsomes, midazolam inhibited testosterone 6β-hydroxylation (Fig. 2A) and testosterone inhibited midazolam 1′-hydroxylation (Fig. 2B). Partial inhibition of midazolam 1′-hydroxylation by testosterone was more distinct than testosterone 6β-hydroxylation by midazolam. Similar results were obtained with the recombinant CYP3A4 system (data not shown).
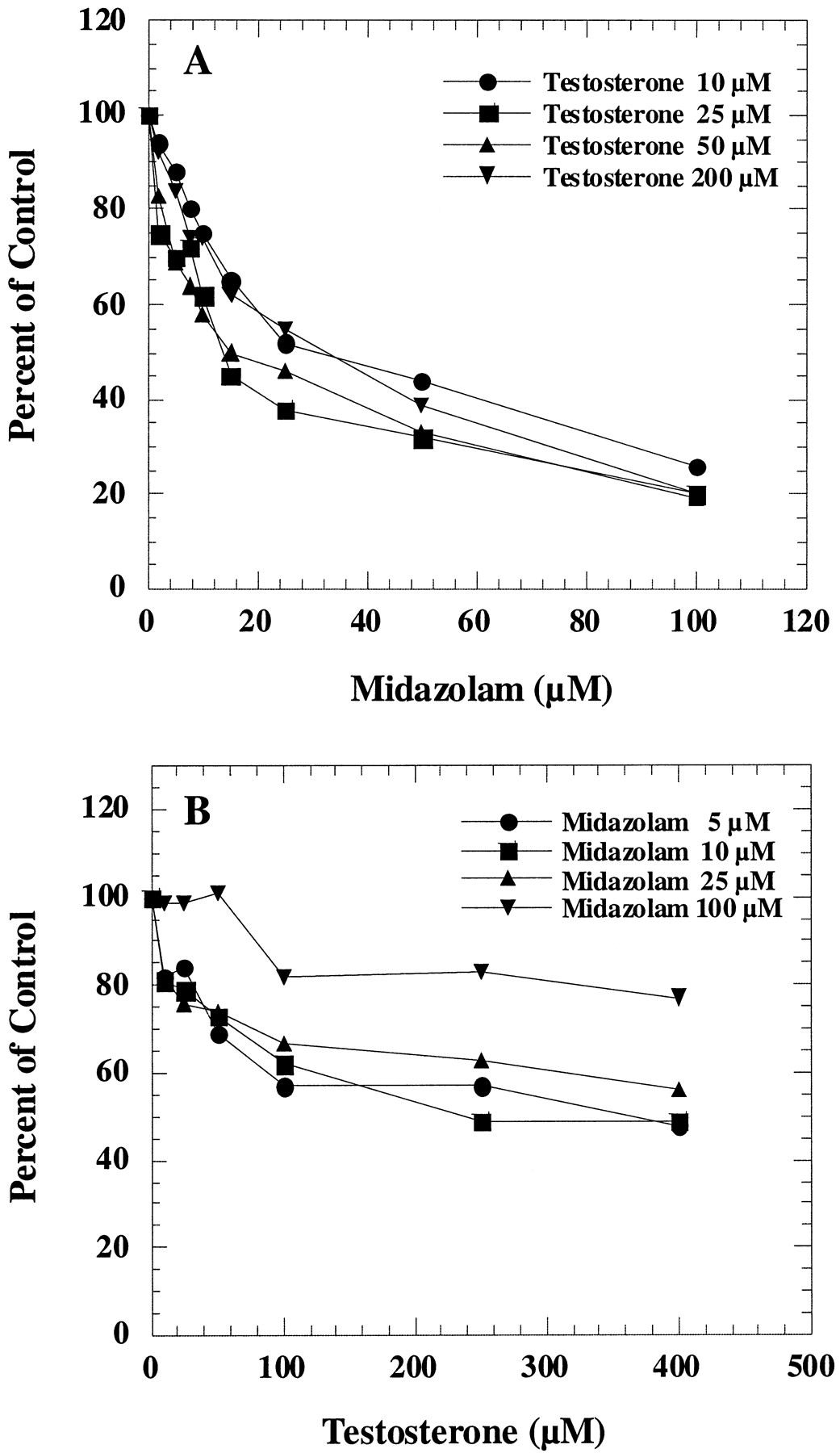
Interaction of testosterone and midazolam in human liver microsomes.
A, effect of midazolam on testosterone 6β-hydroxylation was determined by incubating microsomes with 10, 25, 50, and 200 μM testosterone and various concentrations of midazolam. The corresponding control activities were 0.9, 2.8, 4.8, and 9.8 nmol/min/mg, respectively. B, effect of testosterone on midazolam 1′-hydroxylation was determined by incubating microsomes with 5, 10, 25, and 100 μM midazolam and various concentrations of testosterone. The corresponding control activities were 0.97, 1.77, 2.31, and 2.94 nmol/min/mg, respectively.
Midazolam is metabolized by CYP3A4 to two different products, so we investigated the effect of testosterone on these two different pathways in human liver microsomes. As shown in Fig.3A, testosterone inhibited the major midazolam metabolic pathway (1′-hydroxylation) but stimulated formation of the minor product (4-hydroxylation). Maximum activation and inhibition were observed at approximately 50 to 100 μM testosterone. Partial inhibition kinetics for 1′-hydroxy midazolam formation was observed. Complete inhibition was not observed even at high testosterone concentrations. Although only the result from incubation with 25 μM midazolam was presented in the figure, similar inhibition and activation patterns were also observed at various concentrations of midazolam.
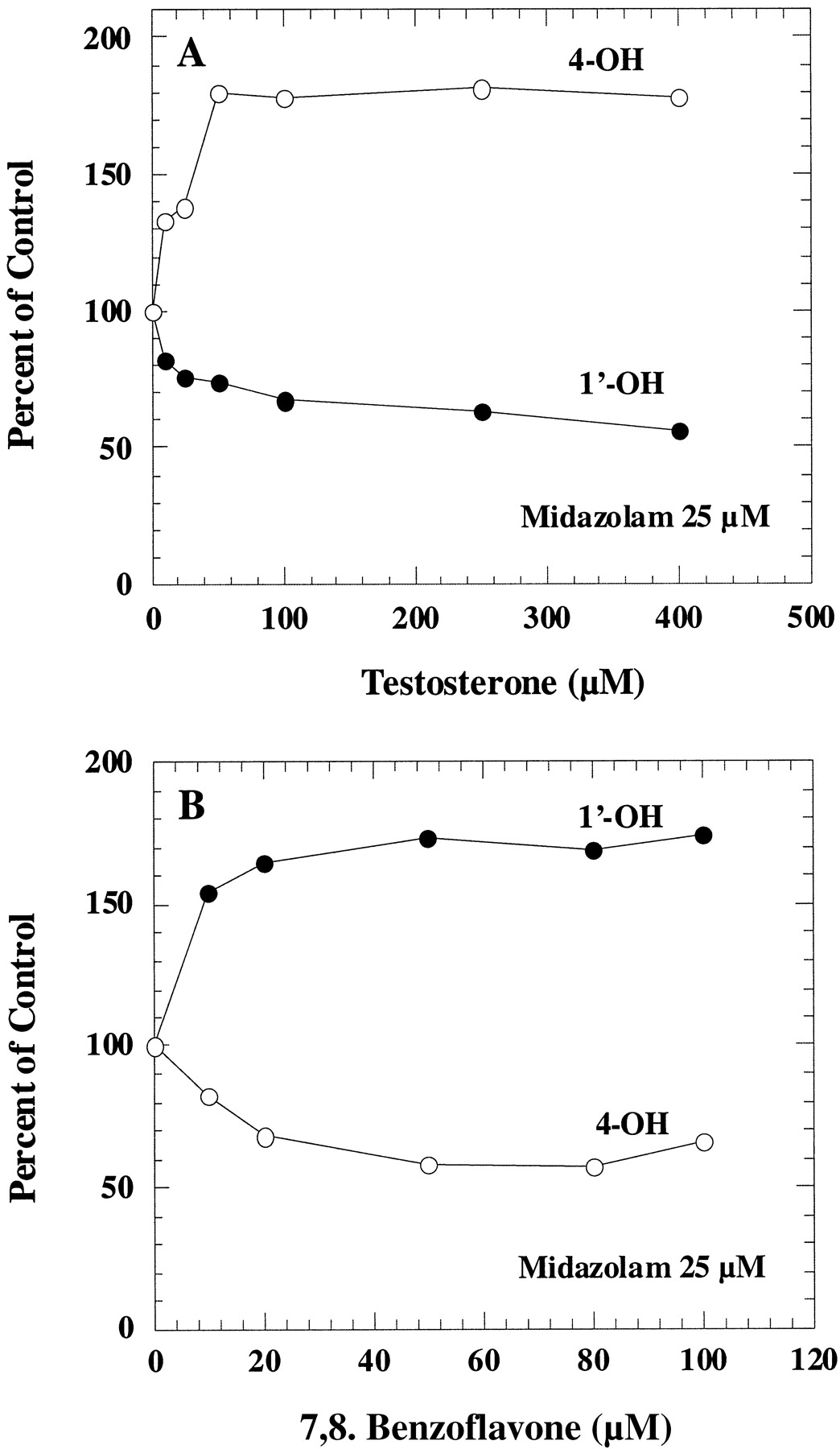
The effect of testosterone and 7,8-benzoflavone on midazolam 1′- and 4-hydroxylation in human liver microsomes.
Human liver microsomes were incubated with 25 μM midazolam in the presence of testosterone (A) and 7,8-benzoflavone (B). The corresponding control activities were 2.31 and 0.42 nmol/min/mg for 1′-hydroxylation and 4-hydroxylation, respectively.
7,8-Benzoflavone, a compound known to activate the metabolism of several CYP3A4 substrates (Shou et al., 1994; Ueng et al., 1997;Korzekwa et al., 1998), activated 1′-hydroxylation, but partially inhibited 4-hydroxylation of midazolam (Fig. 3B). Maximum effects were observed at about 20 to 50 μM 7,8-benzoflavone when midazolam concentration was at 25 μM.
Terfenadine-Midazolam Interaction.
As shown in Fig. 4A, low concentrations of midazolam slightly activated terfenadinet-butyl-hydroxylation in human liver microsomes, but inhibited the reaction at higher concentrations. Midazolam 1′-hydroxylation was partially inhibited by terfenadine at all the concentrations tested (Fig. 4B).

Interaction of terfenadine and midazolam in human liver microsomes.
A, effect of midazolam on terfenadinet-butyl-hydroxylation was determined by incubating microsomes with 5, 10, 25, and 100 μM terfenadine and various concentrations of midazolam. The corresponding control activities were 0.12, 0.18, 0.26, and 0.22 nmol/min/mg, respectively. B, effect of terfenadine on midazolam 1′-hydroxylation was determined by incubating microsomes with 5, 10, 25, and 100 μM midazolam and various concentrations of terfenadine. The corresponding control activities were 1.6, 3.1, 4.8, and 5.3 nmol/min/mg, respectively.
Testosterone-Nifedipine Interaction.
The most surprising results were obtained when testosterone and nifedipine oxidations were studied. Nifedipine inhibited testosterone 6β-hydroxylation in human liver microsomes (Fig.5A), but nifedipine oxidation was unaffected by testosterone (Fig. 5B) regardless of the concentrations of either substrate. In fact, nifedipine oxidation was slightly stimulated by testosterone. These unexpected results prompted us to repeat the experiment with the recombinant CYP3A4 system, because the possibility exists that testosterone and nifedipine may be metabolized in human liver microsomes by different CYP3A isoforms. The results obtained with the recombinant CYP3A4 system confirmed the results obtained from human liver microsomes (data not shown). Thus, CYP3A4 is very unique in its handling of testosterone and nifedipine as substrates, unlike the other pairs examined in this study.

Interaction of testosterone and nifedipine in human liver microsomes.
A, effect of nifedipine on testosterone 6β-hydroxylation was determined by incubating microsomes with 50, 100, 200, and 400 μM testosterone and various concentrations of nifedipine. The corresponding control activities were 1.1, 1.9, 2.7, and 3 nmol/min/mg, respectively. B, effect of testosterone on nifedipine oxidation was determined by incubating microsomes with 40, 100, 200, and 400 μM nifedipine and various concentrations of testosterone. The corresponding control activities were 1.9, 2.8, 3, and 3.1 nmol/min/mg, respectively.
If the observed results with testosterone and nifedipine involve some kind of slow equilibrium or slow conformational change due to interaction between the substrate and CYP3A4, then the order of substrate addition should make a difference in results. To address this question, two different experimental conditions were used. In one set of incubations, various concentrations of nifedipine were preincubated with human liver microsomes at 37°C for 10 min. Testosterone was then added along with the NADPH-generating system. The rate of metabolism of both testosterone and nifedipine was determined from the same incubation. In a parallel experiment, testosterone was preincubated with microsomes first and then nifedipine was added. In both cases, the results confirmed the earlier findings (Fig. 5, A and B) that testosterone 6β-hydroxylation was strongly inhibited by nifedipine, but testosterone did not inhibit nifedipine oxidation regardless of the order of substrate addition.
To determine whether testosterone was an unique case, other CYP3A4 substrates, terfenadine, midazolam, and erythromycin, were also examined for their effects on nifedipine oxidation. Figure6 shows that these three substrates all inhibited nifedipine oxidation to different degrees.

The effect of other CYP3A4 substrates on nifedipine oxidation in human liver microsomes.
Human liver microsomes were incubated with 200 μM nifedipine in the presence of testosterone, erythromycin, midazolam, and terfenadine. The corresponding control activities was 3.8 nmol/min/mg.
Discussion
In a recent study (Wang et al., 1997), we reported the partial inhibition kinetics with CYP3A4 for the interaction between testosterone and erythromycin. Partial inhibition kinetics can be best displayed by plotting the rates of metabolism at several fixed concentrations of the first substrate in the presence of increasing concentrations of the second substrate. These plots clearly demonstrate the effect of saturating concentrations of the second substrate on the metabolism of the first substrate. In fact, the extent of inhibition is governed by α, a constant of proportionality for theKm values of a given substrate in the presence and absence of the second substrate. If the concentration of the first substrate is fixed at 10 times itsKm value, then the maximum inhibition by the second substrate is 50% when α = 10, 25% when α = 3.33, and negligible when α = 1 (Segal, 1975).
To determine whether partial inhibition represents a typical mechanism involving other CYP3A4-catalyzed reactions, four CYP3A4 substrates were selected and studied in pairs for more detailed investigation. The results of our study indicate that partial inhibition kinetics could be demonstrated for the interaction between testosterone and terfenadine, testosterone and midazolam, and terfenadine and midazolam. Consistent with published data by other investigators (Shou et al., 1994; Irshaid et al., 1996; Ueng et al., 1997, Korzekwa et al., 1998; Maenpaa et al., 1998), we observed unusual kinetic characteristics of CYP3A4 involving two substrates that were complex and substrate-dependent. These observations included activation of the metabolism of the first substrate by the second substrate (either only at low concentrations in some cases or at all concentrations in other cases), mutual inhibition, partial inhibition, and the alteration of regiospecificity. This type of interaction was not observed, however, for testosterone and nifedipine. Although nifedipine inhibited testosterone 6β-hydroxylation, testosterone did not inhibit nifedipine oxidation, even though other CYP3A4 substrates, terfenadine, midazolam, and erythromycin all inhibited nifedipine oxidation. In the literature, it is rare to find an example in which two substrates of the same enzyme do not show mutual inhibition kinetics. One similar, but not identical, example was the recent report by Ueng et al., (1997), which stated that 7,8-benzoflavone activates the metabolism of aflatoxin B1 but aflatoxin B1 does not inhibit 7,8-benzoflavone metabolism.
Several models have been proposed to explain the unusual kinetic characteristics with CYP3A4 involving two substrates. Based on the results from kinetic studies, it has been proposed that two substrates can simultaneously bind to the CYP3A4 active site (Shou et al., 1994,Wang et al., 1997). Korzekwa et al. (1998) proposed a two-site (or multiple site) model in which the enzyme can bind two molecules of one substrate or one molecule each of the two substrates, or one molecule each of the substrate and effector. In this model, the relative orientation of the substrates in the active site is not defined, but both substrates must have access to the heme-bound reactive oxygen for metabolism to occur. This two-site model can describe many of the atypical CYP3A4 kinetics, including activation, autoactivation, partial inhibition, substrate inhibition, and biphasic saturation curves. A recent report on the analysis of CYP3A4 cooperativity using L211F/D214E double mutant of CYP3A4 (Harlow and Halpert, 1998) suggested that the effector site is part of the active site, but substrate oxidation occurs only in the substrate binding site and not in the effector binding site.
The activation or inhibition by 7,8-benzoflavone of the metabolism of several CYP3A4 substrates, including aflatoxin B1, were studied by Ueng et al. (1997). They proposed an allosteric model with a substrate binding site and a distinct allosteric site for CYP3A4. Both substrate and effector can bind to the allosteric site, resulting in a change of the affinity of the substrate-binding site for the substrate. The proximity of the putative allosteric site to the catalytic site is unknown. To explain the differential effects of 7,8-benzoflavone on the two oxidative pathways of aflatoxin B1, they also proposed that CYP3A4 can bind aflatoxin B1 in different configurations and that the presence of 7,8-benzoflavone can shift the equilibrium of aflatoxin B1 binding to favor one particular metabolic pathway. This allosteric model can explain most of the experimental data with CYP3A4, but the authors stated that this model can not account for the lack of effect of aflatoxin B1 on 7,8-benzoflavone metabolism.
Using flash photolysis techniques, Koley et al. (1995) observed that the rate of CO binding to the total mixture of CYP3A4 conformers is increased in the presence of nifedipine and erythromycin, decreased by quinidine, testosterone, and warfarin, and unaffected by 17α-ethynylestradiol, suggesting that different conformers have distinct substrate specificities. Another study showed that nifedipine and quinidine bind to different CYP3A4 species with distinct conformations (Koley et al., 1997c). When both drugs are present simultaneously, nifedipine interacts with only one CYP3A4 conformer. Furthermore, when the kinetics of CO binding to various cytochrome P-450s were studied, the data strongly suggest that the heme environment of CYP3A4 exists in dynamic equilibrium between conformational states in the absence of substrates, and substrates (or ligands) may perturb this equilibrium to favor one conformational state over another (Koley et al., 1997a,b). These experiments do not unambiguously prove that substrates (or ligands) perturb the equilibrium between different CYP3A4 conformers, but they provide a physical link to the kinetic data with a reasonable suggestion that different substrates (or ligands) bind more tightly to different conformations.
The atypical CYP3A4 kinetics, including activation, mutual inhibition, partial inhibition, and alteration of regiospecificity, observed in substrate oxidation and substrate-substrate interaction studies has been explained by the two-substrate model (Korzekwa et al., 1998), the cooperativity model (Ueng et al., 1997), and the multiple conformer model (Koley et al., 1995). As for the unusual testosterone-nifedipine interaction, one can explain the results with the two-substrate model by postulating that nifedipine has freedom of movement and can bind to multiple sites, including the testosterone binding site in the CYP3A4 active site, whereas testosterone is fixed at a certain part of the active site. Consequently, nifedipine can inhibit testosterone 6β-hydroxylation, but inhibition of nifedipine oxidation by testosterone can not be demonstrated kinetically. With the multiple conformer model, the testosterone-nifedipine interaction can be explained by assuming that nifedipine has high affinity (or access) to multiple CYP3A4 conformers (including the ones interacting with testosterone), but testosterone has only limited affinity (or access) to certain conformers. Although the two-substrate scheme of Korzekwa et al. (1998) represents an attractive and simpler model to explain the atypical CYP3A4 kinetics, we can not rule out the significant contributions of an allosteric site or multiple conformers in the proposed model at the present time. Perhaps one should consider a modified model taking multiple binding sites, allosteric site, and multiple conformations into account. More studies are needed to distinguish these possibilities. Regardless of the mechanisms by which CYP3A4-dependent drug interactions occur, the results provide a systematic comparison of the interactions between several substrates. The results indicate that the effect of a single substrate on the metabolism of a second is dependent on the identity of the latter. Apparently, there is no hierarchical relationship where in the presence of a specific drug has a consistent effect on the metabolism of all others.
Acknowledgments
We thank Dr. Frank Tang for the synthesis of 3H-labeled terfenadine, Dr. Dennis Dean and Michael Wallace for synthesis of terfenadine metabolites, Dr. Judy Raucy for providing human liver microsomes, Dr. Ralph Stearn for editing the manuscript, Dr. Su Huskey for valuable discussion, and Terry Rafferty for her assistance in preparing the manuscript.
Footnotes
-
Send reprint requests to: Regina W. Wang, Department of Drug Metabolism, RY80-D100, Merck Research Laboratories, P.O. Box 2000, Rahway, NJ 07065. E-mail: regina_wang{at}merck.com
-
↵1 Parts of this work were presented at 11th International Conference on Cytochrome P450 in Sendai, Japan.
-
↵2 Current address: Laboratory for Cancer Research, College of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, New Jersey 08854.
- Abbreviations used are::
- CYP3A4
- human cytochrome P-450 3A4
- CYP3A4/OR
- cytochrome P-450 3A4 and NADPH-cytochrome P-450 reductase
- Received July 27, 1999.
- Accepted November 29, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}