


Abstract
In an attempt to understand the species-selective toxicity of felbamate (2-phenyl-1,3-propanediol dicarbamate, FBM), which is thought to result from bioactivation to 2-phenylpropenal, FBM metabolism was evaluated in rats and humans. The formation of 2-phenylpropenal was monitored by the amount of its mercapturates excreted in urine. The data show a relative 5-fold increase in mercapturate excretion in patient urine as a result of differences in metabolism through P450-, esterase-, and aldehyde dehydrogenase-mediated pathways. To compensate for the significant species differences in FBM metabolism, and to produce toxic levels of 2-phenylpropenal in rat comparable to humans levels, monocarbamate felbamate (2-phenyl-1,3-propanediol monocarbamate, MCF), was administered to rats in the hopes of eliciting a toxic response. The desired result, an increase in mercapturate excretion, was not observed in MCF-treated rats due to the identification of a new FBM metabolite, 2-phenyl-1,3-propanediol monocarbamate-α-d-glucuronic acid (MCF-glucuronide). Formation of MCF-glucuronide is significant and represents about 80% of MCF metabolites in MCF-dosed rats, 3% of FBM metabolites in FBM-dosed rats, and about 11% of FBM metabolites in FBM patients. To overcome the protective effect of glucuronidation, uridine diphosphoglucuronosyltransferase (UGT)-deficient Gunn rats were treated with FBM and MCF, which surprisingly had no effect on the amount of MCF-glucuronide formed. Given the known UGT polymorphisms and the fact that MCF glucuronidation contributes to the elimination of a 2-phenylpropenal precursor, the correlation between poor UGT activity and an increase in mercapturates excretion was evaluated in patients. The result of the first 34 patients screened suggests that a patient with poor UGT activity is not necessarily at risk for FBM toxicity.
Insights into the mechanistic understanding of idiosyncratic reactions continue to augment classic toxicology and the toxicological screening of new therapeutic agents. Adverse drug reactions account for 140,000 deaths in the United States annually (Classen et al., 1997). Often, idiosyncratic adverse reactions are not realized until after widespread drug distribution resulting in the limited usefulness of some new therapies. Examples of idiosyncratic adverse reaction inducing agents include acetaminophen, troglitazone (Gitlin et al., 1998), tolcapone (Assal et al., 1998), diclofenac (Schapira et al., 1986), tienilic acid (Homberg et al., 1984), halothane (Davidson et al., 1966), and felbamate (FBM2; Pennell et al., 1995;O'Neil et al., 1996). Evidence suggests two critical characteristics of idiosyncratic reactions: 1) the bioactivation of the drug to a reactive intermediate capable of covalently modifying proteins–the “hapten hypothesis” and 2) an immune-mediated response to the protein-drug conjugate (Uetrecht, 1999). To date there is no good predictive model for assessing the potential idiosyncratic toxicities for new therapeutics. Idiosyncratic toxicities are not observed preclinically; they are most likely a result of differences in species metabolism/bioactivation and immune responsiveness. We have taken a mechanistic approach to determining species-specific differences in the bioactivation of the antiepileptic drug FBM, 2-phenyl-1,3-propanediol dicarbamate, which may be applied to new compounds with structural and metabolic similarities.
Previous research has identified atropaldehyde (2-phenylpropenal) as a reactive metabolite of FBM (Thompson et al., 1997). The aim of this study is to explore differences in the bioactivation of FBM to atropaldehyde and challenge the question of why FBM does not cause toxicity in animal models. Even after exhaustive efforts of high-dose long-term treatment of FBM or its esterase-mediated metabolite monocarbamate felbamate (MCF, 2-phenyl-1,3-propanediol monocarbamate) in GSH-depleted rats, we were not able to demonstrate toxicity. This lack of toxicity is, in part, the result of the formation of a newly identified metabolite, 2-phenyl-1,3-propanediol monocarbamate-α-d-glucuronic acid (MCF-glucuronide), which we propose to be protective. Our results suggest that species differences in the bioactivation of FBM have important implications for the observed idiosyncratic reactions and speak to the limited usefulness of small animals as preclinical indicators of idiosyncratic drug reactions.
Materials and Methods
Chemicals and Instruments.
The chemicals were purchased from either Sigma (St. Louis, MO) or Aldrich (Milwaukee, WI) and were of the highest grade available unless noted otherwise. The urinalysis was performed on a Waters 2690 Separations Module HPLC using a Waters Symmetry C8 (2.1 × 150 mm) column. The flow was directed into a Waters 486 Tunable absorbance detector set at λ = 214 nm and then into a Finnigan Mat LCQ ion trap electrospray ionization mass spectrometer. NMR spectra were obtained on a 300 MHz General Electric QE300 spectrometer; chemical shifts are reported in ppm.
Synthesis.
2-Phenyl-(1,1,3,3-tetradeuterio)-1,3-propanediol, 2-phenyl-(1,1,3,3-tetra-deuterio)-1,3-propanediolmonocarbamate, 3-carbamoyl-(3,3-di-deuterio)-2-phenylpropionic acid, 2-phenyl-(1,1,3,3-tetra-deuterio)-1,3-propanediol dicarbamate, N-d3-acetyl-l-cysteine, N-d3-acetyl-S-(2-phenylpropan-3-ol)-l-cysteine, and N-d3-acetyl-S-(2-phenyl-propanoic acid)-l-cysteine.2-Phenyl-(1,1,3,3-tetradeuterio)-1,3-propanediol, 2-phenyl-(1,1,3,3-tetra-deuterio)-1,3-propanediol monocarbamate, 3-carbamoyl-(3,3-di-deuterio)-2-phenyl-propionic acid, 2-phenyl-(1,1,3,3-tetra-deuterio)-1,3-propanediol dicarbamate,N-d3-acetyl-l-cysteine,N-d3-acetyl-S-(2-phenylpropan-3-ol)-l-cysteine, andN-d3-acetyl-S-(2-phenylpropanoic acid)-l-cysteine were synthesized and in spectroscopic agreement with previously published results (Thompson et al., 1999). The compound purity and isotopic purity of each compound was determined by 1H NMR and liquid chromatography/electrospray ionization-mass spectrometry (LC/ESI-MS) and found to be ≥95% pure.
2-Phenyl-1,3-propanediol monocarbamate.
2-Phenyl-1,3-propanediol monocarbamate was synthesized as previously published (Thompson et al., 1999). The purity of this compound was determined to be ≥98% pure by 1H NMR and LC/ESI-MS.
6-[3-(Aminocarbonyloxy)-2-phenylpropoxy]-3,4,5-trihydroxyperhydro-2H-pyran-2-carboxylic acid (MCF-glucuronide).
The synthesis of MCF-glucuronide was adapted from published procedures (Lacy and Sainsbury, 1995). Briefly, 4,5-diacetyloxy-2-bromo-6-(methoxycarbonyl)-perhydro-2H-pyran-3-yl acetate (Sigma) was combined with 2-phenyl-1,3-propanediol monocarbamate (250 mg) and AgCO3 on Celite (50%, 2.0 g) in toluene under nitrogen. The reaction was brought to reflux and monitored by thin layer chromatography. After 30 min, the crude reaction mixture was loaded directly onto silica gel (300 g) and eluted with ethyl ether. The 2-phenyl-1,3-propanediol monocarbamate-aceto-α-d-glucuronic acid methyl ester was saponified in methanol with 1 ml of 0.1 M NaOH for 1 h. The reaction mixture was acidified to pH = 3, concentrated to 1 ml, loaded onto silica gel (40 g), and eluted with methanolic chloroform (30%). The compound was additionally purified on a Waters Oasis SEP cartridge and eluted with 0.1% HOAc in water. The purity was determined to be ≥95% as determined by 1H NMR and LC/ESI-MS. 1H NMR (d4-methanol) 7.20 to 7.05 (m, 5H), 5.35 (d, 1H, J = 7 Hz), 4.70 (d, 1H, J = 8 Hz), 4.35 (t, 1H, J = 8 Hz), 4.04 (d, 1H, J = 10 Hz), 3.84 (t, 1H, J = 10 Hz), 3.75 to 3.25 (m, 4H), 3.20 (m, 1H). LC/ESI-MS: parent [M + 1H]+ m/z = 372. Liquid chromatography/electrospray ionization-tandem mass spectrometry (LC/ESI-MS/MS): daughters [M + 1H]+m/z = 354 (loss of water) and [M + 1H]+ m/z = 196 (MCF).
6-[3-(Aminocarbonyloxy)-2-phenyl-1,1,3,3-tetradeuteriopropoxy]-3,4,5-trihydroxyperhydro-2H-pyran-2-carboxylic acid (d4-MCF-glucuronide).
The 2-phenyl-(1,1,3,3-tetra-deuterio)-1,3-propanediol monocarbamate-α-d-glucuronic acid was synthesized as above using 2-phenyl-(1,1,3,3-tetra-deuterio)-1,3-propanediol monocarbamate. The purity was determined to be ≥95% as determined by1H NMR and LC/ESI-MS. 1H NMR (d4-methanol) 7.20 to 7.05 (m, 5H), 5.35 (d, 1H, J = 7 Hz), 3.75 to 3.25 (m, 4H), 3.20 (m, 1H). LC/ESI-MS: parent [M + 1H]+m/z = 376. LC/ESI-MS/MS: daughters [M + 1H]+ m/z = 358 (loss of water) and [M + 1H]+ m/z= 200 (d4-MCF).
Animals.
Animal studies were carried out under a protocol approved by the University of Virginia Animal Research Committee. The rats used in these experiments were either 250-g male Sprague-Dawley rats or 250-g Gunn rats obtained from Harland. The rats were acclimated for 24 h before dosing. Throughout the experiments, the animals were maintained on a 12-h light/dark cycle and were able to access food and water at liberty. The urine was collected for 18-h post dose in metabolic cages and stored frozen at −20°C until prepared for analysis.
FBM Sprague-Dawley Rat Study.
Nine rats were divided into three groups (n = 3). The first group was dosed daily via gavage with 350 mg/kg FBM formulated as an oral suspension containing 600 mg of FBM/5 ml of suspension (Wallace Laboratories, Cranbury, NJ) throughout the experiment. The second group (study group of rats) was dosed with FBM in the same manner as the first group with the exception that on day 5, 30 mMl-buthionine-[S,R]-sulfoximine (BSO, Sigma) in water was exchanged for the drinking water to deplete GSH (Manning et al., 1991) for the remainder of the experiment. The third group was a control group whose drinking water was also exchanged for 30 mM BSO on day 5. Urine samples were collected for 18-h post dose biweekly and stored frozen at −20°C. The rats were dosed in this manner for 4 weeks at which time the animals were sacrificed. Liver and bone (sternum) samples were harvested and fixed in formalin for histological examination.
MCF Sprague-Dawley Rat Study.
Fifteen rats were divided into three groups (n = 5). The first group was dosed daily via gavage from a 25 mg/ml MCF aqueous solution. The rats were dosed daily: days 1 to 24, 50 mg/kg; days 25 to 42, 70 mg/kg; days 43 to 50, 100 mg/kg; days 51 to 58, 200 mg/kg; days 59 to 63, 300 mg/kg. The dosage was incrementally increased in an attempt to demonstrate a toxic effect. The maximal dose, 300 mg/kg/day, corresponds to about 14 times an average dose of FBM (3000 mg/70 kg patient/day). An increase in dosage corresponded to an increase in the amount of excreted metabolites and is not thought to have saturated the metabolic pathways. The second group of rats was dosed similarly to those in the first group with the exception that on day 8 their drinking water was exchanged with a 30 mM aqueous BSO solution to deplete their GSH. The third group of rats was a control group given 30 mM BSO on day 8 to replace their drinking water. Urine samples were collected for 18-h post dose biweekly and stored frozen at −20°C. At the end of the 9-week experimental period, the animals were sacrificed. Liver and sternum samples were harvested and fixed in formalin for histological preparation and examination.
FBM and MCF Gunn Rat Study.
Fifteen rats were divided into five groups (n = 3). The drugs (FBM or MCF) were formulated as aqueous suspensions in a 30% (w/v) PEG-3350 (Sigma). The first group was dosed daily via gavage with FBM: days 1 to 15, 600 mg/kg; days 16 to 36, 1200 mg/kg. The third group was dosed daily via gavage with MCF: days 1 to 15, 300 mg/kg; days 16 to 21, 600 mg/kg; days 22 to 36, 900 mg/kg. The second group of rats was dosed similarly to those in the first group and the fourth group was dosed similarly to those in the third group with the exception that on day 8 their drinking water was exchanged with a 30 mM aqueous BSO solution to deplete their GSH. The fifth group of rats was given 30 mM BSO on day 8 to replace their drinking water. Urine samples were collected 18-h post dose biweekly and stored frozen at −20°C. At the end of the 5-week experimental period, the animals were sacrificed and tissue samples from their liver and sternum were obtained and fixed in formalin for histological preparation and examination.
Histological Preparation.
Tissue samples were processed by American HistoLabs, Inc. (Gaithersburg, MD). Briefly, the bone was decalcified, paraffin-embedded, and stained with H & E. The liver slices were also paraffin-embedded and stained with H & E.
Patient Urine Samples.
Urine samples were obtained from a randomized patient population undergoing FBM therapy as approved by the University of Virginia Human Investigations Committee.
Identification of MCF-Glucuronide.
The urine was thawed at 37°C. For the rat urine, 200 μl of urine from a Sprague-Dawley rat dosed for a week at 50 mg/kg/day MCF, and for human urine, 1 ml of urine diluted 1:4 from a 900 mg/kg/day FBM patient was applied to an Oasis solid phase extraction cartridge that had been washed with 3 ml of CH3CN then 3 ml of 0.1% HOAc in water. The flow through was eluted, the column was washed with 2 ml of 0.1% HOAc in water, and the analyte was eluted with 2 ml of 10% CH3CN:90% (0.1%) HOAc. For the coelution experiments, 1 μl of 8.0 mM 2-phenyl-1,3-propanediol monocarbamate-α-d-glucuronic acid internal standard was added to 60 μl of the analyte eluent. The samples were analyzed by LC/ESI-MS/MS as described below.
LC/ESI-MS/MS.
LC/ESI-MS/MS analysis was performed on a Waters 2690 Separations Module with a Waters 486 Tunable Absorbance Detector. The LC was interfaced to a Finnigan MAT LCQ ion trap mass spectrometer with an ESI source. A 15-μl injection of each sample was separated on a Waters Symmetry 2.1 × 150 mm C8 reversed phase column and eluted isocratically at 200 μl/min with 33% CH3CN:67% (0.1%) HOAc. The column eluent was directed through a Waters 486 Tunable Absorbance Detector containing a 10-μl flow cell set at 214 nm for qualitative analysis. The flow was then directed into the ESI-MS for analytical analysis. A flow restrictor was used to split the flow 1:3, allowing one-fourth of the sample to enter the MS.
The values for the ESI were as follows: heated capillary temperature = 170°C; spray voltage = 4.9 kV; capillary voltage = 3.4 V; sheath gas flow rate = 35; auxiliary gas flow rate = 30. The data was collected in ms/ms full scan positive ion mode from 100 to 500 m/z. The ion trap was set to trap m/z = 372 with a 3-m/z isolation width, and fragmentation was achieved with 10% collision energy using helium as the collision gas. The automatic gain control was set at 7 × 107 ions and the maximum injection time was set at 1 s. The number of microscans was set at 1.
Preparation of the Urine Samples for Metabolite Quantification.
Urine sample preparation and metabolite quantification were performed using a modification of a previously described procedure (Thompson et al., 1999). Briefly, to 1 ml of human urine or FBM-dosed rat urine was added 100 μl of an internal standard solution containing 563 nmol of d4-FBM, 140 nmol of d2-3-carbamoyl-2-phenylpropionic acid (d2-CPPA), 54 nmol of d3-N-acetylcysteine alcohol, and 27.5 nmol of d3-N-acetylcysteine acid, and 2 μl of another internal standard solution containing 121 nmol of d4-MCF-glucuronide and 20 μl of 20% HOAc. To 50 μl of the monocarbamate-dosed rat urine was added an internal standard solution containing 42.5 nmol of d4-MCF, 140 nmol of d2-CPPA, 54 nmol of d3-N-acetyl-S-(2-phenylpropon-3-ol)-l-cysteine, and 27.5 nmol ofN-d3-acetyl-S-(2-phenypropanoic acid)-l-cysteine, and 2 μl of another internal standard solution containing 121 nmol of d4-MCF-glucuronide and 20 μl of 20% HOAc. The sample was applied to an Oasis SEP cartridge that had been equilibrated with 3 ml of CH3CN and then 3 ml of 0.1% HOAc in water. The sample was eluted and the column was washed with 500 μl of 0.1% HOAc in water. The analytes were eluted with 30% CH3CN:70% (0.1%) HOAc and saved for LC/ESI-MS analysis.
LC/ESI-MS Quantification.
The LC/ESI-MS conditions were identical with those described above for LC/ESI-MS/MS with the following exceptions. The data was collected in full scan positive ion mode from 190 to 390 m/z. The maximum injection time was set at 300 ms and the number of microscans was set at 2.
Quantitation of the respective analyte was achieved by integration of the mass chromatogram peaks using Rework, software accompanying the LCQ. Each of the analytes produced linear-response curves with respect to the corresponding deuterated standards within the concentration range examined. The ratio of the area under the analyte peak (expressed as counts per second) to area under the deuterated standard peak was determined. The ratio established a quantitative relationship between the amount of deuterated standard added and the amount of analyte present in the sample. Given that a known amount of deuterated standard was added, the absolute amount of metabolite in each sample was quantitatively determined.
Results
Quantification of FBM Metabolites in Patient and Sprague-Dawley Rat Urine.
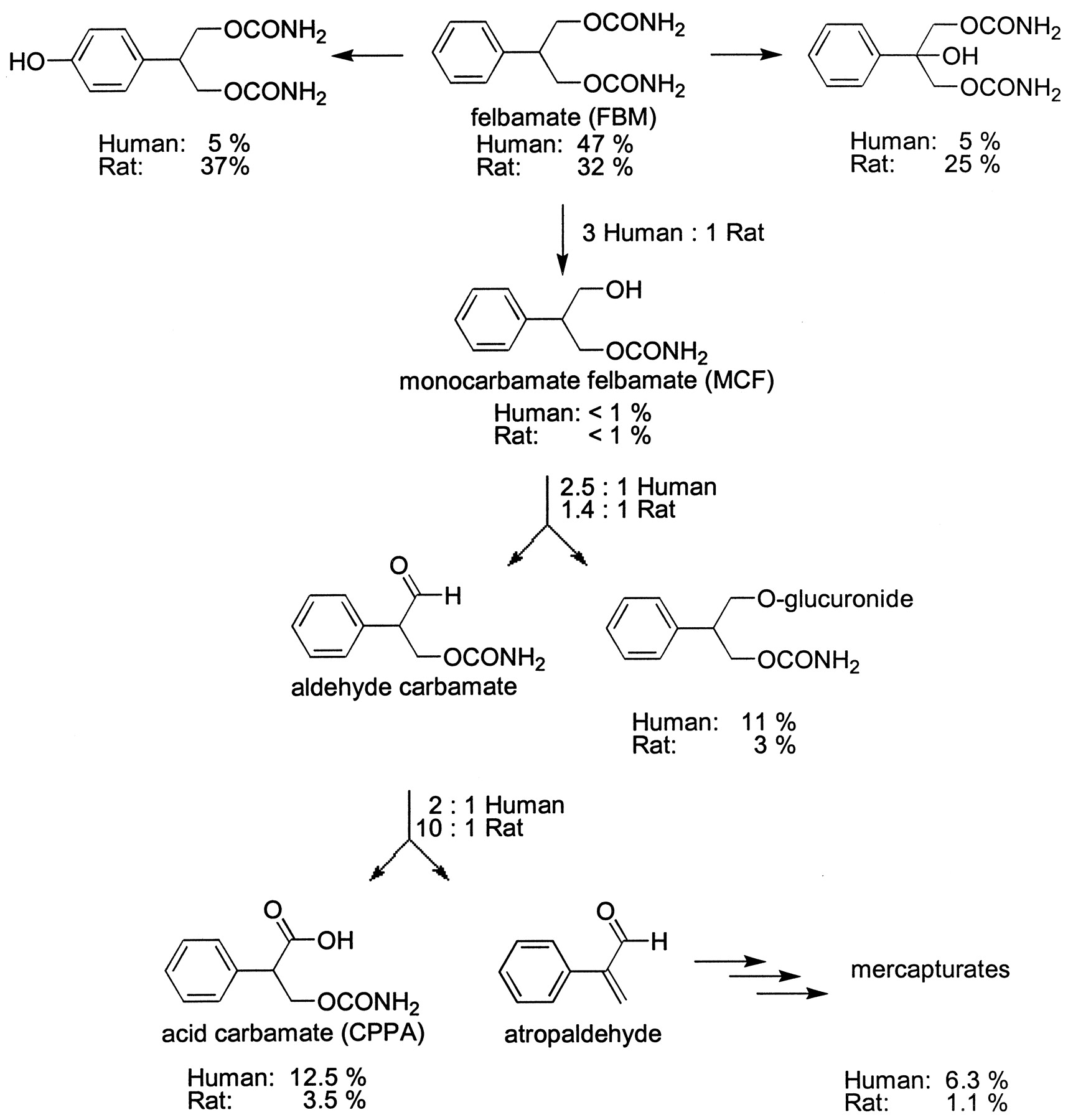
The quantification data for FBM metabolites in patient and Sprague-Dawley urine is given in Tables 1and 2 and summarized by Fig.1. The metabolites are reported as a percentage of FBM excreted to overcome dose and urine volume variability. For patients, the percentage of MCF-glucuronide excreted was calculated relative to the reported amount of CPPA excreted when patients were dosed with C14-FBM (Kucharczyk, 1995). Likewise, the percentage of mercapturates excreted was calculated to be 6.3% of the total dose, given that the ratio of CPPA/mercapturates is 2:1. Rats consistently excrete significantly fewer esterase-mediated FBM metabolites than humans, reflecting the characteristically poor esterase activity in rats. Additionally, the metabolite quantification in rats as compared with humans shows a statistically significant higher ratio of 3-carbamoyl-2-phenylpropionic acid (acid carbamate, CPPA) to mercapturates and a lower ratio of CPPA plus mercapturates to the newly identified MCF-glucuronide.
FBM-dosed Sprague-Dawley rats

Comparison of FBM metabolites excreted in urine for Sprague-Dawley rats versus humans.
Quantification of MCF Metabolites in Sprague-Dawley Rat Urine.
The quantification of MCF metabolites in Sprague-Dawley urine is summarized in Table 3. Again, the metabolites were reported as a percentage of MCF excreted to overcome dose and urine volume variability. MCF-dosed rats excrete 5-fold more MCF-glucuronide than the FBM-dosed rats (Fig.2).
MCF-dosed Sprague-Dawley rats

Comparison of FBM and MCF metabolism in Sprague-Dawley rats.
Long-Term Treatment of GSH-Depleted Rats.
In an exhaustive effort to demonstrate FBM toxicity as a result of atropaldehyde production and GSH depletion, rats were treated with either FBM or MCF over the course of several weeks. GSH depletion was monitored as a reduction in the excretion of mercapturates. In all of the GSH-depleted animals, mercapturate excretion was half that amount excreted in control animals. However, even after long-term treatment neither liver toxicity, marked by neutrophil infiltration, nor bone marrow toxicity, marked by an increase in adipocytes and loss of blood cells precursors, was observed.
Identification and Quantification of 2-Phenyl-1,3-propanediol monocarbamate-α-d-glucuronic Acid in Rat and Human Urine.
The synthesis of MCF-glucuronide was readily achieved by coupling 2-phenyl-1,3-propanediol monocarbamate with acetobromo-α-d-glucuronic acid methyl ester and subsequent saponification. A synthetic standard of the proposed metabolite allowed for the characterization of this metabolite by a coelution experiment. Under the LC/MS/MS conditions applied, the synthetic standard eluted at 5.6 min with a parentm/z = 372 and daughter ions havingm/z = 354 corresponding to the loss of water and m/z = 196 corresponding to MCF. Under collisionally activated dissociation, the glucuronic acid portion of the molecule is a neutral loss and therefore not observed.
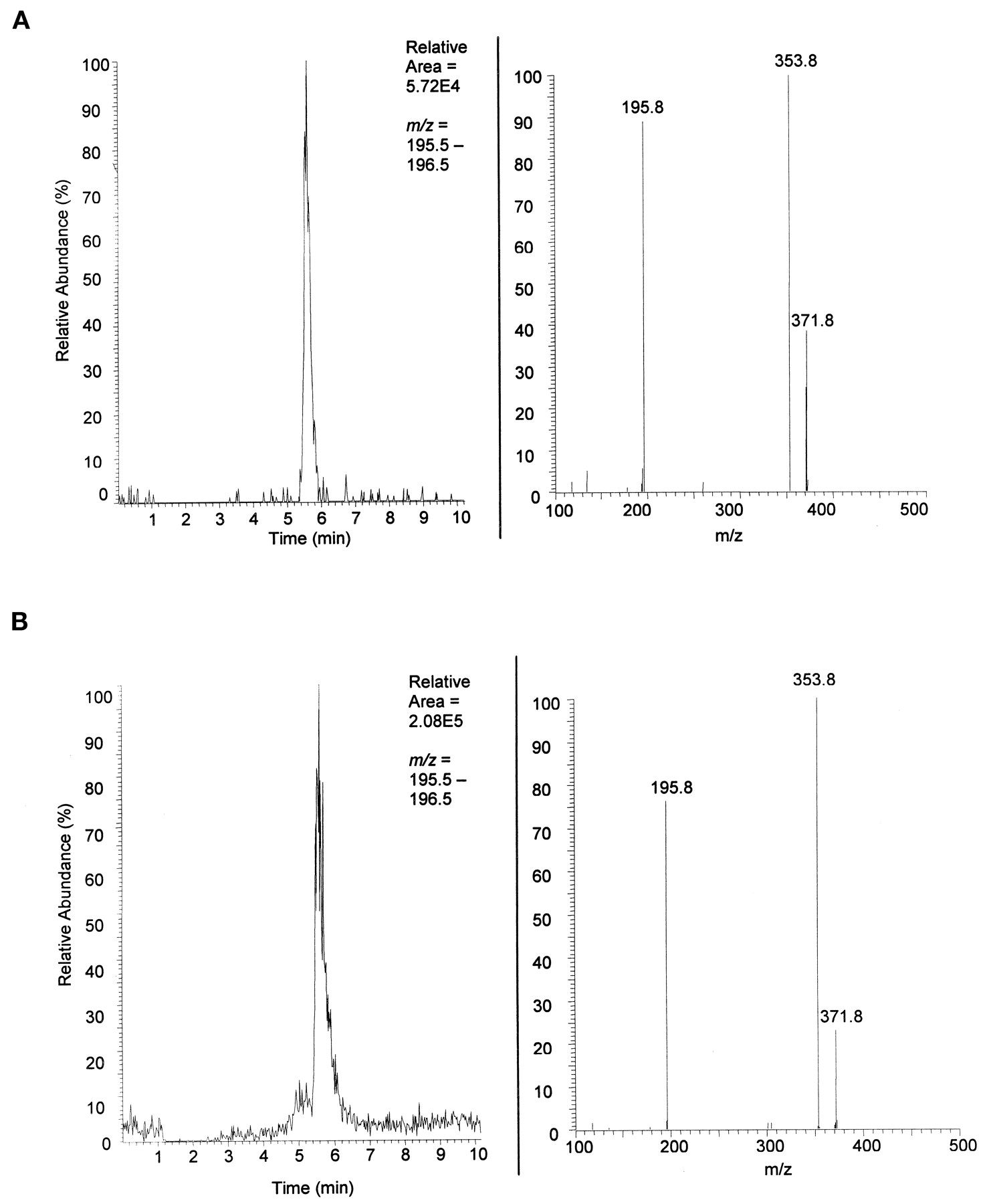
Urine from rats treated with 50 mg/kg/day MCF was collected and purified by solid phase extraction. The extracted material was loaded onto a reversed phase column, and the proposed analyte eluted at 5.6 min. The parent m/z = 372 of the peak at 5.6 min yielded daughter ions of m/z = 354 andm/z = 196 (Fig.3A). To complete the positive identification of the new metabolite as the glucuronide conjugate of 2-phenyl-1,3-propanediol monocarbamate, the synthetic standard was added to the solid phase extracted mixture and loaded onto the reversed phase column. The elution was monitored by ESI-MS/MS. The synthetic standard and the analyte of interest coeluted at 5.6 min yielding daughters with m/z = 354 andm/z = 196, Fig. 3B. The coelution experiment positively identified the new metabolite in rat urine post 2-phenyl-1,3-propanediol monocarbamate treatment as 2-phenyl-1,3-propanediol monocarbamate-α-d-glucuronic acid.

Identification of MCF-glucuronide in MCF-dosed Sprague-Dawley rat urine.
A, rat urine. Top left shows the LC chromatograph with mass filter m/z = 196, characteristic daughter ion of parent MCF-glucuronide. Top right shows the MS/MS spectrum [resident time (RT) = 5.60 on left] of parent m/z = 372 and daughters m/z = 354 (loss of water) and m/z = 196 (MCF). Neutral loss of mass 176 is characteristic of glucuronide conjugates. B, rat urine + synthetic standard. Top left shows the LC chromatograph with mass filter m/z = 196. The increase in area under the curve and same retention as shown in Fig. 3 indicate coelution of the synthetic standard with the unknown metabolite in patient urine. Top right shows the characteristic MS/MS spectrum (RT = 5.62 on left) of parent m/z = 372 and daughtersm/z = 354 (loss of water) andm/z = 196 (MCF).
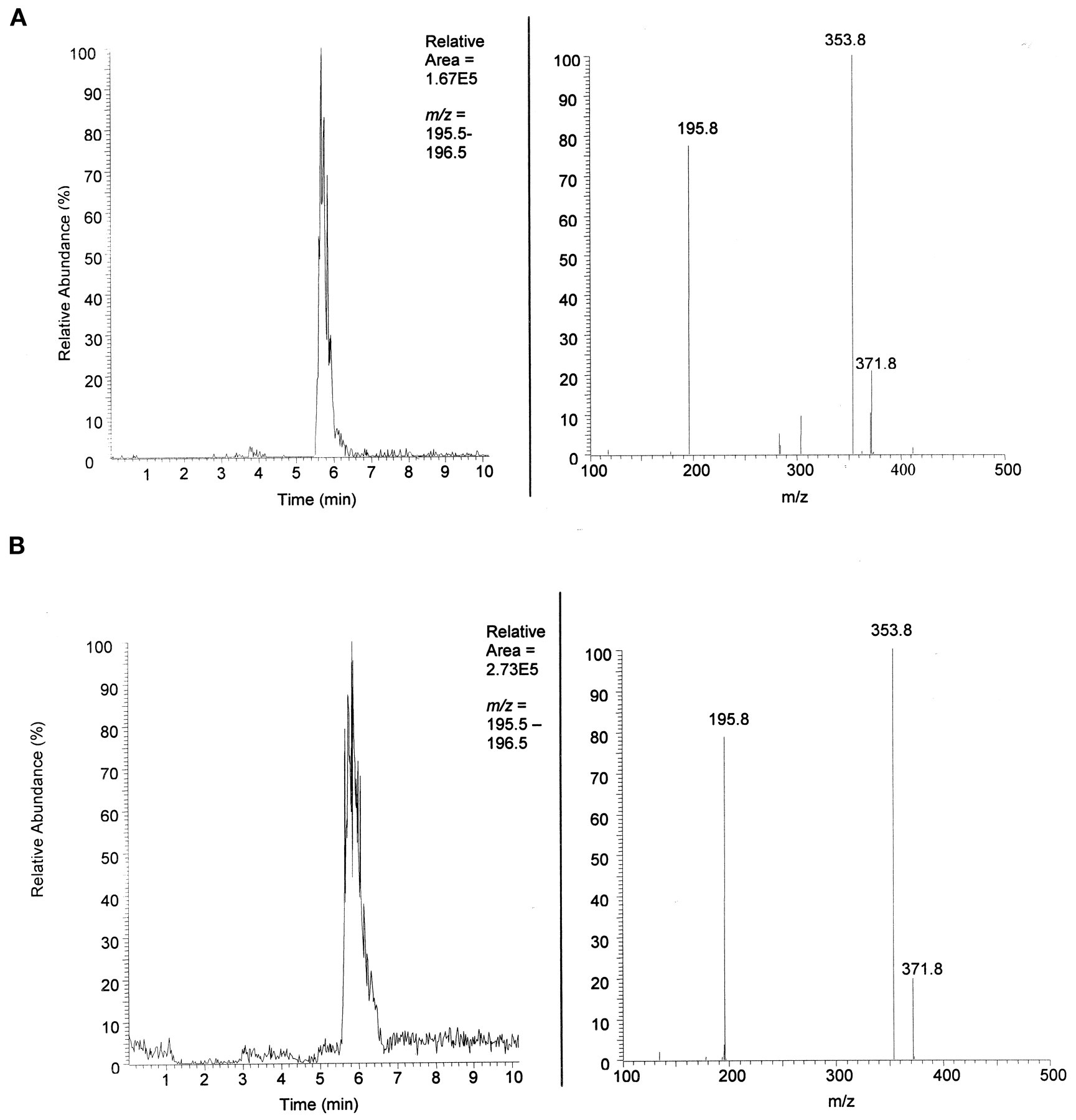
The identification of MCF-glucuronide in rat urine suggested its formation in patients being treated with FBM. Similar coelution experiments were completed with the urine of patients treated with FBM. The data for the extracted human urine is shown in Fig.4A. The data for extracted human urine plus synthetic standard is shown in Fig. 4B. Both the extracted patient urine and extracted patient urine spiked with synthetic standard elute at about 5.6 min and show the characteristic parentm/z = 372 with characteristic daughter ionsm/z = 354 and m/z= 196.

Identification of MCF-glucuronide in FBM-treated patient urine.
A, human urine. Top left shows the LC chromatograph with mass filter m/z = 196, characteristic daughter ion of parent MCF-glucuronide. Top right shows the MS/MS spectrum (RT = 5.61 on left) of parent m/z = 372 and daughtersm/z = 354 (loss of water) andm/z = 196 (MCF). Neutral loss of mass 176 is characteristic of glucuronide conjugates. B, human urine + synthetic standard. Top left shows the LC chromatograph with mass filterm/z = 196. The increase in area under the curve and same retention as shown in Fig. 3 indicate coelution of the synthetic standard with the unknown metabolite in patient urine. Top right shows the characteristic MS/MS spectrum (RT = 5.76 on left) of parent m/z = 372 and daughtersm/z = 354 (loss of water) andm/z = 196 (MCF).
Given that uridine diphosphoglucuronosyltransferase (UGT) activity varies between species and within a species population (Clarke et al., 1997), and that conjugation of MCF with UDP-glucuronic acid contributes to the elimination of an atropaldehyde precursor, we quantitated the amount of MCF-glucuronide in the urine of rats dosed with FBM or MCF and patients being treated with FBM. The amount of FBM or MCF, 3-carbamoyl-2-phenylpropionic acid,N-acetyl-S-(2-phenylpropan-3-ol)-l-cysteine, and N-acetyl-S-(2-phenylpropanoic acid)-l-cysteine as well as MCF-glucuronide was quantified using LC/ESI-MS isotope dilution. The results of the metabolite quantification in rats are shown in Table 1 (humans), Table2 (FBM-dosed rats), and Table 3 (MCF-dosed rats). MCF-glucuronide is the major metabolite of rats dosed with MCF, representing 80% of the total metabolites excreted (MCF, CPPA, mercapturates, and MCF-glucuronide).
To address the concern that patients with poor UGT activity may produce relatively more atropaldehyde and represent potential at risk individuals for FBM toxicity, we quantified the amount of MCF-glucuronide reported as a ratio of CPPA plus mercapturates to MCF-glucuronide (Fig. 5). The average amount of CPPA plus mercapturates to MCF-glucuronide in FBM patients was 2.5 ± 1. One patient produced a relatively high ratio of CPPA plus mercapturates to MCF-glucuronide of 8.6. The high ratio corresponds to a relatively small amount of MCF-glucuronide formed. However, the patient did not have reduced levels of GSH when measured as a ratio of CPPA to mercapturates (Thompson et al., 1999).

The bar graph represents a patient's ratio of acid carbamate plus mercapturates to MCF-glucuronide.
The “outlier” at 8.6 had a normal ratio of acid carbamate to mercapturates and is not considered at risk for FBM toxicity.
Quantification of MCF-Glucuronide in FBM- or MCF-Treated Gunn Rats.
Gunn rats have been genetically altered to be deficient in UGT enzymes and are generally considered a good model for UGT deficiency. We expected that the Gunn rats would produce less MCF-glucuronide and model the FBM toxicities observed in humans. The quantification of metabolites in FBM- or MCF-dosed Gunn rats is reported in Tables4 and 5. Surprisingly, the Gunn rat did not form reduced amounts of MCF-glucuronide.
FBM-dosed Gunn rats
MCF-dosed Gunn rats
Discussion
The species differences in the bioactivation of FBM are significant and could contribute to an explanation of selective toxicity in humans as compared with rats. The major metabolites of FBM in rats are p-hydroxyfelbamate (37%) and 2-hydroxyfelbamate (25%), (Kucharczyk, 1995) (Fig. 1). A significant portion of the parent drug is also excreted as unchanged drug (32%), (Kucharczyk, 1995). The major metabolites of FBM in humans are MCF-glucuronide (11%), CPPA (13%), and atropaldehyde excreted as mercapturates (6%). As in rats, a significant amount of the FBM is excreted as unchanged drug (47%), (Kucharczyk, 1995). Given the hypothesis that atropaldehyde is the toxic metabolite in FBM bioactivation, a decrease in its production should protect from FBM toxicity.
Our data (Fig. 1) indicate that rats demonstrate a protective metabolism of FBM generating less atropaldehyde (1%). The minor formation of atropaldehyde in rats results from a number of factors including: 1) significant contribution from P450-mediated hydroxylation of the parent drug, 2) relatively poor esterase activity, and 3) relatively high aldehyde dehydrogenase activity, as compared with humans. We do not believe that alcohol dehydrogenase plays a major role in species-selective bioactivation of FBM because MCF was found to be <1% total metabolites in rats and humans dosed with FBM (Kucharczyk, 1995). Even at high doses of FBM, the small amount of atropaldehyde generated in rats could easily be detoxified with the GSH available in the liver.
In humans, atropaldehyde constitutes ∼6% of FBM metabolites. The relatively high production of atropaldehyde in humans results from a decrease in the P450-mediated metabolic pathways (Kucharczyk, 1995). Relatively higher esterase activity and lower aldehyde dehydrogenase activity serve to further promote atropaldehyde production (Fig.1). The apparent lower aldehyde dehydrogenase activity in humans is not a result of the substrate exceeding the Kmfor the enzyme in vivo (C.M. Dieckhaus, unpublished data). The resultant 5-fold increase in atropaldehyde production coupled with the relative high therapeutic dosage (1–6 g/day) contributes importantly to the appearance of the observed idiosyncratic reactions. Typically, atropaldehyde undergoes detoxification by conjugation with GSH. In a small percentage of the population, GSH may become depleted allowing, by our hypothesis, atropaldehyde to induce the observed FBM-associated toxicities. It is also possible that GSH conjugation may be catalyzed by GSH transferases, and the known polymorphisms in GSH transferases may also promote depressed atropaldehyde detoxification.
In an attempt to generate high levels of atropaldehyde in rats and elicit a toxic response, rats were dosed with the esterase-mediated metabolite, MCF. Even after 2 months of high dose treatment (350 mg/kg/day) in GSH-depleted rats, we were not able to demonstrate a toxic response, as monitored by neutrophil infiltration in the liver and an increase in adipocytes and a loss of blood cell precursors in the bone marrow. This virtual lack of toxicity is thought to be the result of alternative, protective metabolic pathways as evidenced by the occurrence of the newly identified metabolite, MCF-glucuronide. The potential importance of the glucuronide metabolite stems from the fact that conjugation of MCF with UDP-glucuronic acid contributes to the elimination of an atropaldehyde precursor metabolite. About 80% of the MCF administered to Sprague-Dawley rats was conjugated and not, therefore, available to undergo oxidation to the corresponding aldehyde, which has been shown to produce atropaldehyde.
Additional experiments were carried out in Gunn rats. The Gunn rat has been characterized to be deficient in bilirubin and phenol UGT isozymes and is well accepted as an animal model for UGT deficiencies (Chowdhury et al., 1993; Kren et al., 1999). Continuing with our efforts to produce high levels of atropaldehyde in an animal model, we dosed Gunn rats with FBM or MCF. Given the UGT deficiencies, we proposed that the Gunn rat would form relatively small amounts of MCF-glucuronide. The reduction in MCF-glucuronide formation would result in higher levels of MCF available to undergo oxidation to the aldehyde and subsequent elimination to atropaldehyde. Similar experiments demonstrated a 110-fold greater hepatotoxic response to acetaminophen-dosed Gunn rats as compared with Wistar rat controls (Morais and Wells, 1988). Interestingly, quantification of MCF-glucuronide in FBM- or MCF-dosed Gunn rats demonstrated no reduction in the amount of MCF-glucuronide formed (Tables 4 and 5). The results are most likely an effect of enzyme isoform specificity. The broad classification of UGT enzymes is broken down into the UGT1 and UGT2 family of enzymes (Burchell et al., 1995). Our results indicate that MCF is not conjugated through the bilirubin or phenol isozymes (UGT1) and explains the lack of toxicity in the Gunn rat. Similar results were obtained when Gunn rats were dosed with 2-ethylhexanoic acid (Hamdoune et al., 1995), 2-arylpropionic acids (Magdalou et al., 1990), and 3′-azido-3′-deoxythymidine (AZT), (Haumont et al., 1990). We hypothesize that the UGT 2 family of enzymes catalyzes the glucuronidation of MCF based on structural similarity (Iwersen and Schmoldt, 1998).
Given the known variability of UGT activity between species and within the population (Clarke et al., 1997), we considered that poor UGT activity could result in an increased risk for FBM toxicity. To study the metabolic disposition of MCF to form either the glucuronide conjugate or become oxidized to the corresponding aldehyde, we quantified the amount of MCF-glucuronide, CPPA, and mercapturates in the urine of rats dosed with MCF or FBM and in patients undergoing FBM therapy. The metabolic disposition in rats and humans appears similar. In FBM-dosed rats, the disposition is 1.4 ± 0.2 (CPPA + mercapturates)/(MCF-glucuronide) and in patients the disposition is 2.5 ± 1.0 (CPPA + mercapturates)/(MCF-glucuronide). Although the two are not statistically different, the disposition is different in MCF-dosed rats. In MCF-dosed rats, the disposition is 0.3 ± 0.0 (CPPA + mercapturates)/(MCF-glucuronide). This difference may occur due to the differences in distribution and enzyme exposure between the parent drug and its metabolite, or the lack of competing substrates for UDP-glucuronic acid in MCF-dosed rats. For example, FBM undergoes P450-mediated p-hydroxylation forming thep-hydroxyfelbamate metabolite that also undergoes glucuronidation (Kucharczyk, 1995). Studies with acetaminophen have demonstrated that the amount of UDP-glucuronic acid is rate-limiting in the amount of glucuronide formed (Hjelle, 1986). The average FBM dose is within a range whereby the amount of UDP-glucuronic acid may be depleted. The formation of greater amounts of the MCF-glucuronide conjugate in MCF-dosed rats may, therefore, may be a factor of UDP-glucuronic acid availability.
The amount of MCF-glucuronide formed in patients being treated with FBM represents about 11% of all FBM metabolites. Given that CPPA represents 12% of the total FBM metabolites (Admusmalli, 1993), we would expect that a patient with a reduced ability to form MCF-glucuronide would produce elevated amounts of atropaldehyde, possibly by as much as a factor of 2. Two diseases, Crigler-Najjar and Gilbert's Syndrome, are characterized by a deficiency in UGT activity (Burchell et al., 1987). Whereas Crigler-Najjar is relatively uncommon, Gilbert's syndrome is present in 6% of the population (Owens et al., 1996). To determine whether poor UGT activity correlates with an increase in atropaldehyde production in humans, we quantified the relative amounts of MCF-glucuronide as a ratio of (CPPA + mercapturates)/(MCF-glucuronide). An individual with apparently poor UGT activity would be expected to have a relatively high ratio of CPPA plus mercapturates to MCF-glucuronide. Heterogeneity of UGT activity within a population undergoing acetaminophen therapy was found to be 3-fold (Miners et al., 1984).
Of the first 34 patients screened, one patient presented as forming relatively low levels of MCF-glucuronide, seen at 8.6 in Fig.5. Although the patient was forming relatively low levels of MCF-glucuronide, his ratio of acid to carbamate to mercapturates was well within the normal range as previously defined (Thompson et al., 1999) at 2.25 (Table 2). These results indicate that although a patient with poor UGT activity may produce relatively more atropaldehyde, the at risk assessment should still be based on monitoring the levels of GSH available to react/detoxify atropaldehyde, and supports the continued use of the at risk FBM metabolite patient monitoring developed previously in our laboratory (Thompson et al., 1999). Future experiments will determine the isoform(s) of aldehyde dehydrogenase that oxidize aldehyde carbamate to CPPA and the GST isoform(s) that may aid in the conjugation of GSH with atropaldehyde. At that time, the heterogeneity of aldehyde dehydrogenase and GST will be addressed in detail.
If idiosyncratic reactions are the result of the bioactivation of a compound to a reactive metabolite, reactive metabolite/protein-adduct formation, and a subsequent immunological response, species differences in bioactivation deserves attention. A mechanistic approach to understanding the species differences in FBM bioactivation has allowed us to uncover metabolic differences thought to partially account for the species-selective toxicity observed in humans. We have shown that large doses of FBM do not produce toxic levels of atropaldehyde in rats, and hypothesize that this is due to an increase in P-450-mediated hydroxylations of the parent compound, poor esterase activity, and relatively high aldehyde dehydrogenase activity relative to humans. Directed attempts to overcome the species differences in metabolism by dosing rats with the esterase-mediated metabolite MCF resulted in the protective phase II conjugation to form MCF-glucuronide. In addition, Gunn rats did not produce less MCF-glucuronide, also illustrating the protective metabolism in rats. Although a patient with poor UGT activity may produce relatively more atropaldehyde, the at risk assessment should still be based on monitoring the levels of GSH available to react/detoxify atropaldehyde.
An understanding of the differences in species metabolism/bioactivation of compounds such as FBM may serve as a basis for developing a model that is capable of predicting species-selective toxicities for other therapeutically useful compounds. In addition to differences in bioactivation, variance in immunological response to hapten formation is equally important in predicting idiosyncratic reactions and critical in the development of animal models for predicting idiosyncratic adverse drug reactions. Taken as a whole, these issues may aid in the development of other clinically useful agents.
Acknowledgments
We thank Donald J. Innes, M.D., David J. Deck, M.D., and Milton Brown, M.D., Ph.D. (University of Virginia Hospital) for their guidance and advice regarding the tissue pathology. We also thank Charles D. Thompson, Ph.D., for useful conversations concerning the animal metabolite experiments, and Angela Bretz for assistance in processing the rat urine samples. Appreciation is also extended to Warren Kline Bolton, Ph.D. (University of Virginia Nephrology), for use of the metabolic animal cages.
Footnotes
-
Send reprint requests to: Timothy L. Macdonald, Ph.D., University of Virginia, Department of Chemistry, McCormick Road, Charlottesville, VA 22901. E-mail: tlm{at}virginia.edu
-
↵1 Presented in part at the 29th Annual Gordon Research Conference on Drug Metabolism, Plymouth, New Hampshire, July 4–8, 1999.
-
This work was supported by Carter-Wallace, Inc. and National Institutes of Health Cell and Molecular Pharmacology Training Grant T32GM07055 (C.M.D.).
- Abbreviations used are::
- FBM
- felbamate (2-phenyl-1,3-propanediol dicarbamate)
- MCF
- monocarbamate felbamate (2-phenyl-1,3-propanediol monocarbamate)
- MCF-glucuronide
- 2-phenyl-1,3-propanediol monocarbamate-α-d-glucuronic acid
- d4-MCF-glucuronide
- 6-[3-(Aminocarbonyloxy)-2-phenyl-1,1,3,3-tetradeuteriopropoxy]-3,4,5-trihydroxyperhydro-2H-pyran-2-carboxylic acid
- UGT
- uridine diphosphoglucuronosyltransferase
- LC/ESI-MS
- liquid chromatography/electrospray ionization-mass spectrometry
- LC/ESI-MS/MS
- liquid chromatography/electrospray ionization-tandem mass spectrometry
- BSO
- buthionine-[S,R]-sulfoximine
- CPPA
- 3-carbamoyl-2-phenylpropionic acid
- RT
- retention time
- Received December 21, 1999.
- Accepted April 13, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}