


Abstract
This study demonstrates the stereoselective metabolism of the optical isomers of omeprazole in human liver microsomes. The intrinsic clearance (CLint) of the formation of the hydroxy metabolite from S-omeprazole was 10-fold lower than that from R-omeprazole. However, the CLint value for the sulfone and 5-O-desmethyl metabolites fromS-omeprazole was higher than that fromR-omeprazole. The sum of the CLint of the formation of all three metabolites was 14.6 and 42.5 μl/min/mg protein for S- and R-omeprazole, respectively. This indicates that S-omeprazole is cleared more slowly than R-omeprazole in vivo. The stereoselective metabolism of the optical isomers is mediated primarily by cytochrome P450 (CYP) 2C19, as indicated by studies using cDNA-expressed enzymes. This is the result of a considerably higher CLint of the 5-hydroxy metabolite formation forR- than for S-omeprazole. ForS-omeprazole, CYP2C19 is more important for 5-O-desmethyl formation than for 5-hydroxylation. Predictions of the CLint using data from cDNA-expressed enzymes suggest that CYP2C19 is responsible for 40 and 87% of the total CLint of S- andR-omeprazole, respectively, in human liver microsomes. According to experiments using cDNA-expressed enzymes, the sulfoxidation of both optical isomers is metabolized by a single isoform, CYP3A4. The CLint of the sulfone formation by CYP3A4 is 10-fold higher for S-omeprazole than forR-omeprazole, which may contribute to their stereoselective disposition. The results of this study show that both CYP2C19 and CYP3A4 exhibit a stereoselective metabolism of omeprazole. CYP2C19 favors 5-hydroxylation of the pyridine group ofR-omeprazole, whereas the same enzyme mainly 5-O-demethylates S-omeprazole in the benzimidazole group. Sulfoxidation mediated by CYP3A4 highly favors theS-form.
Omeprazole, lansoprazole, pantoprazole, and rabeprazole are all drugs of a class referred to as proton pump inhibitors (PPIs1, Fig.1). They act to regulate acid production in the stomach and are used to treat various acid-related gastrointestinal disorders. Chemically, they have in common a pyridinylsulfinylbenzimidazole backbone but have different substitution patterns. The in vitro metabolism of these compounds has been reported previously, and the main routes of metabolism, i.e., sulfoxidation and hydroxylation, have been shown to be mediated via cytochrome P450 (CYP) 3A4 and CYP2C19, respectively, in studies that have used therapeutically relevant concentrations (Andersson et al., 1993b; Chiba et al., 1993; Simon, 1995; Karam et al., 1996; Pearce et al., 1996; VandenBranden et al., 1996). The major metabolites of omeprazole found in plasma are hydroxyomeprazole and omeprazole sulfone (Fig. 2) (Andersson, 1996), which agrees with in vitro findings (Andersson et al., 1993b). In addition to hydroxyomeprazole and omeprazole sulfone, a minor metabolite, 5-O-desmethylomeprazole (Fig. 2), was identified in human liver microsomal incubations (Andersson et al., 1993a). As for the other PPIs, hydroxylation of omeprazole is done mainly by CYP2C19, which is also reflected in the reduced omeprazole plasma clearance observed in poor S-mephenytoin hydroxylators in vivo. The formation of sulfone is mediated almost exclusively by CYP3A4 (Andersson et al., 1998). However, in vitro results suggest that this pathway contributes less to omeprazole elimination than does the formation of the hydroxy metabolite, as the formation intrinsic clearance (CLint) of the sulfone is four times lower than that of hydroxyomeprazole in human liver microsomes (Andersson et al., 1993b). This is also supported by clinical studies (Andersson, 1996). The minor metabolite 5-O-desmethylomeprazole was suggested to be formed mainly by CYP2C19 (Andersson et al., 1993b). According to these results, the overall metabolism of omeprazole depends more on the activity of CYP2C19 than on that of CYP3A4.

Structures of PPIs discussed.

Structural formulae of S- and R-omeprazole and the three identified metabolites.
The sulfur of the sulfoxide functionality is a chiral center but no in vitro metabolism studies on the individual optical isomers have yet been reported for the PPIs. However, the in vitro metabolism of the optical isomers of a closely related compound (H 259/31, Fig. 1), which was selected to study the influence on the metabolic pathways by the substitution pattern in comparison with racemic omeprazole, has recently been described by this laboratory (Äbelö et al., 2000). The results of these studies show a clear stereoselectivity in the in vitro metabolism of the optical isomers. The total CLint for the S-form of H 259/31 was higher than for the R-form, and the preferential site of metabolism was the benzimidazole moiety. Although the in vitro metabolism of the optical isomers of the PPIs listed above has not been described yet, there are a few reports on the in vivo pharmacokinetics of the individual optical isomers obtained by chiral analysis of plasma after administration of the racemates of omeprazole (Cairns et al., 1995; Tybring et al., 1997), lansoprazole (Katsuki et al., 1996), and pantoprazole (Tanaka et al., 1997) in humans. In these studies, the stereoselective disposition is evident in extensive metabolizers (EM) as the area under the plasma concentration versus time curves (AUCs) are higher for the S-forms of omeprazole and pantoprazole than for the respective R-forms. The opposite was reported for lansoprazole as the AUC of the R-form of lansoprazole is higher than that of the S-form.
The aim of this study was to investigate the in vitro metabolism of the individual optical isomers to explain the findings of stereoselective pharmacokinetic disposition in vivo. The potential differences in the enzyme kinetics of the formation of the three metabolites (omeprazole sulfone, hydroxyomeprazole, and 5-O-desmethylomeprazole) from the individual optical isomers of omeprazole in human liver microsomes and the involvement of specific CYP isoforms were investigated.
Materials and Methods
Chemicals.
The following compounds were synthesized at the Department of Medicinal Chemistry at AstraZeneca R&D Mölndal, Sweden: theS-form of omeprazole [S-omeprazole; (S)-5-methoxy-2[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole) sodium], the R-form of omeprazole [R-omeprazole, (R)-5-methoxy-2[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole) sodium], sulfone (5-methoxy-2[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfonyl]-1H-benzimidazole), hydroxyomeprazole (5-methoxy-2[[(4-methoxy-3-methyl-5-hydroxymethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole), 5-O-desmethylomeprazole (5-hydroxy-2[[(4-methoxy-3,5-dimethyl-2-pyridinyl)-methyl]sulfinyl]-1H-benzimidazole), and the internal standard (4,6-dimethyl-2[[(4-methoxy-2-pyridinyl)methyl]sulfinyl]-1H-benzimidazole). The trivial names of the metabolites are used throughout this report. The molecular structures are shown in Fig. 2. The following chemicals (of analytical grade) were all obtained from Merck (Darmstadt, Germany): MnCl2, MgCl2, NaH2PO4, NH4OH (aq), Na2CO3, Tris(hydroxymethyl-aminomethane), hydrochloric acid (fuming 37%), 1-butanol, and dimethyl sulfoxide (DMSO). Methanol and methylene chloride were supplied by Rathburn Chemicals Ltd (Walkerburn, Scotland). NADP and isocitrate dehydrogenase (ICDH) were obtained from Boehringer Mannheim (Bromma, Sweden) and isocitrate was purchased from Fluka (Stockholm, Sweden). Water was passed through a MilliQ filter.
Human Liver Microsomes and cDNA-Expressed P450s.
Human liver samples (excess material removed during surgery on the liver) were obtained from the Department of Surgery 1, Sahlgrenska Hospital, Göteborg, Sweden. Small cubes (1 cm3) were frozen in liquid nitrogen and stored at −70°C until preparation of liver microsomes. Microsomes were prepared according to the method of Ernster et al. (1962), and the microsomal protein concentration was measured according to Lowry et al. (1951) using BSA as standard. Human liver microsomes from three different liver samples were used in the determination of the kinetic parameters (Vmax,Km).
Microsomes from human lymphoblastoid cell lines expressing CYP1A2, CYP2C9, CYP3A4, CYP2D6, CYP2A6, CYP2E1, CYP2C8, CYP2B6, and CYP2C19 were obtained from Gentest Corp. (Woburn, MA). The microsomal preparations were stored at −70°C until use.
HPLC Analysis of Omeprazole Metabolism.
The metabolites of S-omeprazole and R-omeprazole were determined by the method of Andersson et al. (1993a), with a few modifications. The incubated samples were extracted with dichloromethane/butanol (99:1). The mobile phase consisted of dichloromethane mixed with 3.5% methanol and 0.175% aqueous ammonia solution (25% by volume) or dichloromethane mixed with 1.0% methanol, 5.0% 2-propanol, and 0.175% aqueous ammonia solution (25% by volume) and was delivered at a flow rate of 1.5 ml/min at ambient temperature. The injection volume was 110 μl. The eluate was monitored at 302 nm. The HPLC system consisted of a pump (model 480; Gynkotek, Munich, Germany), an autosampler (model CMA/200; Carnegie Medicine AB, Stockholm, Sweden), a UV detector (model 975 Jasco; Japan Spectroscopic Co., Tokyo, Japan), and a model Chrom Jet integrator (Spectra-Physics Inc., San Jose, CA). The instrument was fitted with a Superspher SI-60 4-μm separation column (125 mm × 4 mm i.d.; E. Merck, Darmstadt, Germany) and a Brownlee Aquapore Silica, 7-μm guard column (15 mm × 3 mm i.d.; Applied Biosystems, Foster City, CA). The quantitation of the sulfone-, the hydroxy-, and the 5-O-desmethyl metabolites was done by comparison with standard samples (containing known amounts of the synthetic racemic metabolites and microsomes but no NADPH) by using the peak area ratio method. The samples were analyzed by an achiral method, and it was assumed that the metabolites formed retained the optical configuration of the parent.
Enzyme Kinetics.
Linear conditions for the formation of metabolites were evaluated with respect to protein content and incubation time for the human liver microsomes. The optimum conditions selected for the kinetic study were a 16-min incubation period at a protein concentration of 1.0 mg/ml at 37°C. Reactions were initiated by the addition of substrate after a preincubation period of 1 min. The reactions were stopped by an addition of 2 ml of methylene chloride 1-butanol (99:1). Seven different concentrations, ranging from 5 to 200 μM, were used in theVmax/Kmdetermination. The substrates were dissolved in 10% DMSO. The final concentration of DMSO in the incubation mixture was always 0.5%. This concentration of DMSO inhibits the formation of omeprazole sulfone, hydroxyomeprazole, and 5-O-desmethylomeprazole by 16, 17, and 9%, respectively (Andersson et al., 1993b), but was used to ensure complete dissolution of the test compounds. Controls were prepared as above at each substrate concentration, but the extraction solvent was added before the substrate and the controls were not incubated. All experiments, incubated samples as well as controls, were performed in triplicate.
Enzyme kinetic parameters were obtained by nonlinear regression analysis (WinNonlin; SCI, Inc., Apex, NC). Homoscedastic weighting was used in all regression analyses. The Michaelis-Menten equation,V = (Vmax · C)/Km+ C, was fitted to the formation rates of the metabolites versus substrate concentrations. V is the velocity of the reaction at substrate concentration C, Vmax is the maximum velocity, and Km is the substrate concentration at which the reaction velocity is 50% ofVmax. Intrinsic clearance of the in vitro incubation was calculated as CLint =Vmax/Km.
cDNA-Expressed Human P450s.
The linearity of the formation rate of the metabolites ofS-omeprazole and R-omeprazole was established with respect to protein concentration and time. Based on these results, the kinetic studies were performed at a protein concentration of 1.0 mg/ml at 37°C over periods of 60 min (2D6, 2A6, 2C9, and 3A4) and 30 min (2C19). Five different substrate concentrations, ranging from 20 to 500 μM (2D6, 2A6, and 3A4), 10 to 200 μM (2C9), and 5 to 200 μM (2C19), were used in these experiments. Owing to difficulties in the evaluation of the chromatograms of the sulfone metabolite at high concentrations of R-omeprazole, the formation rate of this metabolite by CYP3A4 was studied only in the concentration range of 20 to 100 μM. cDNA-expressed CYP2C8 was evaluated at 5 μM and CYP2E1, 1A1, and 2B6 at 5 and 200 μM. Microsomes from lymphoblastoid cells transfected with vector only were used as control and did not metabolize the optical isomers of omeprazole.
Prediction of Blood Clearance Using Data from Human Liver Microsomes.
The venous equilibration model was used in an attempt to predict in vivo metabolic clearance. The sum of the CLintfor the formation of the three metabolites was used in the calculation. The parameter values used for scaling the in vitro metabolism data were 21 g/kg for liver weight, 20 ml/min/kg for hepatic blood flow, and 45 mg of protein/g of liver for microsomal yield (Houston, 1994). The degree of binding to plasma proteins was assumed to be equal to that in the microsomal suspension. The blood to plasma partition constant in humans for racemic omeprazole is 0.6 (Regårdh et al., 1985); this value was used for the estimation of the blood clearance forS-omeprazole (data from Hassan-Alin et al., 2000).
Prediction of CLint in Human Liver Microsomes Using Data from cDNA-Expressed Enzymes.
In an attempt to calculate CLint values in human liver microsomes by using the enzyme kinetic parameters estimated from the experiments performed with cDNA-expressed enzymes, we used the information from Shimada et al. (1994) and Yamazaki et al. (1997) on the average content of each P450 isoform in a microsomal preparation (Tables 1 and2).
Prediction of CLint in human liver microsomes from enzyme kinetic parameters from cDNA-expressed enzymes for S-omeprazole
Prediction of CLint in human liver microsomes from enzyme kinetic parameters from cDNA-expressed enzymes for R-omeprazole
Results
The formation of the three main metabolites (the sulfone, hydroxy, and 5-O-desmethyl metabolites) from S- andR-omeprazole was studied using human liver microsomes. For the individual metabolites, the initial formation rates were calculated and used in the estimates of parameters according to the Michaelis-Menten equation. The mean values of the computer-derived Michaelis-Menten parameters and calculated CLintvalues for the formation of metabolites from S- andR-omeprazole are presented in Table3. The CLint values show that the three metabolites formed from S-omeprazole are equally important for its elimination, whereas the hydroxy metabolite dominates the elimination of R-omeprazole. The sum of the formation CLint of all three metabolites was 14.6 and 42.5 μl/min/mg of protein for S- andR-omeprazole, respectively, indicating stereoselectivity in the metabolism, and that S-omeprazole is cleared more slowly than R-omeprazole. Representative saturation curves for the formation of the metabolites from one of the three liver microsome preparations are shown in Fig. 3. TheKm values evaluated for the formation of the hydroxy metabolite seem to be lower for R-omeprazole than for S-omeprazole, whereas no significant differences can be seen between S- and R-omeprazole for the formation of the other metabolites. Although the results from cDNA-expressed enzymes indicate that more than one enzyme is involved in the formation of the hydroxy and 5-O-desmethyl metabolites, no improvement in the regression was obtained when a biphasic model describing a two-enzyme system was used in the liver microsome study. The mean Km values for 5-O-desmethyl and hydroxy formation may thus be overestimated owing to the use of the simple model. However, the evaluated Km values are probably more a reflection of the property of the high-affinity enzymes in human liver microsomes, as they are close to those obtained with expressed enzymes exhibiting low Km values.
Mean enzyme kinetic parameters ± S.D. of the formation of metabolites from S-omeprazole and R-omeprazole in human liver microsomes
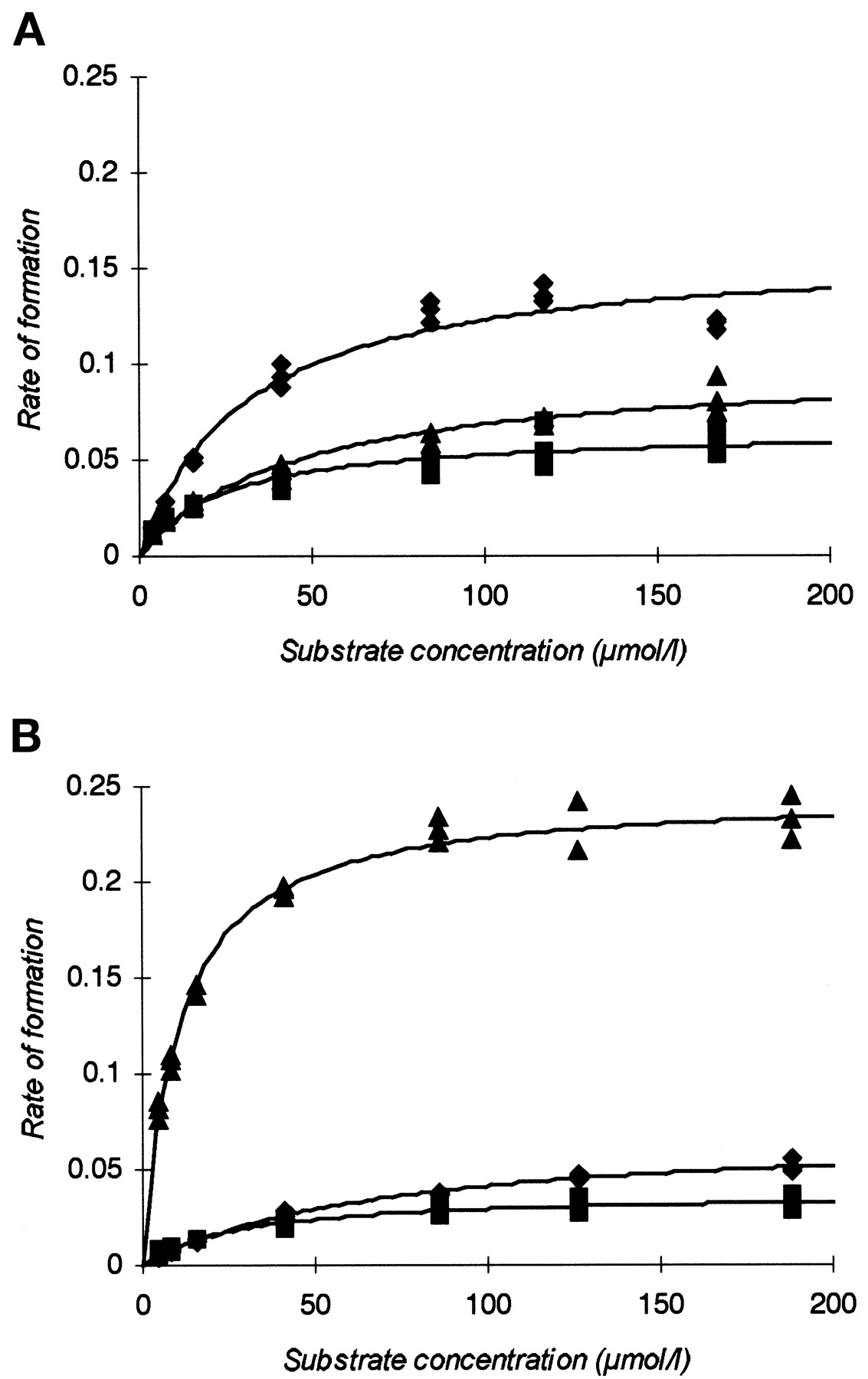
Representative plots of the formation of sulfone (-♦-), hydroxy (-▴-), and 5-O-desmethyl (-▪-) from S- omeprazole (A) and R-omeprazole (B) in human liver microsomes from one liver preparation.
Symbols are experimentally determined values, whereas solid lines are the computer-generated curves of best fit.
The enzyme kinetics of the metabolism of S- andR-omeprazole was studied in microsomes from lymphoblastoid cells over-expressing CYPs 1A2, 2C9, 3A4, 2D6, 2A6, 2E1, 2C8, 2B6, and 2C19. The derived Km andVmax values, as well as the calculated CLint values, of the formation of the metabolites are shown in Tables 4 and5. CYP2C19 catalyzes the formation of the hydroxy and 5-O-desmethyl metabolites from both optical isomers. The CLint representing the 5-O-desmethylation by CYP2C19 is higher forS-omeprazole than for R-omeprazole owing to its lower Km and higherVmax values. The CLint for hydroxylation by CYP2C19 is higher for the R-form, mainly due to a considerably higherVmax value as compared with theS-form. Incubations with cDNA-expressed CYP2C9 indicate minor catalytic involvement in the formation of the hydroxy and 5-O-desmethyl metabolites from the S-form, whereas no metabolism of the R-form by this enzyme was detected. According to the CLint values, CYP3A4 mainly supports the sulfone metabolite formation fromS-omeprazole, whereas hydroxylation and 5-O-desmethylation are minor pathways via this enzyme. ForR-omeprazole, the CLint values for the formation of the hydroxy and sulfone metabolites via CYP3A4 are similar, although the total contribution of CYP3A4 metabolism is much lower than for S-omeprazole. CYP2A6 formed the hydroxy metabolite from S-omeprazole. The highKm value does indicate a very low affinity, however, suggesting that CYP2A6 is a relatively unimportant enzyme in the metabolism of S-omeprazole in vivo. In addition, a large S.E. of the estimated Km andVmax values was observed. No metabolites were formed from R-omeprazole by CYP2A6. Incubations of cDNA-expressed CYP2D6 resulted in hydroxy and 5-O-desmethyl formation for S- and R-omeprazole. However, the reactions were not saturable over the concentration range used, andKm and Vmaxvalues could not be estimated. Instead, the CLintwas estimated using linear regression of the formation rate at unsaturated enzyme reactions. Incubation of the optical isomers with CYPs1A2, 2C8, 2B6, and 2E1 did not result in any metabolites.
Kinetic parameters ± S.E. of the formation of metabolites from S-omeprazole in microsomes prepared from a human lymphoblastoid cell line
Kinetic parameters ± S.E. of the formation of metabolites from R-omeprazole in microsomes prepared from a human lymphoblastoid cell line
An estimate of the in vivo hepatic clearance was calculated from in vitro enzyme kinetic parameters (Vmax/Km) from microsomes from three separate livers (Table 3) for the formation of the three metabolites using the well stirred model. The values forS- and R-omeprazole were calculated to be 572 and 935 ml/min, respectively. In vivo, the corresponding blood clearance for S-omeprazole was measured to be 602 ml/min (Hassan-Alin et al., 2000). No data are available for R-omeprazole.
The enzyme kinetic data from expressed enzymes (Tables 4 and 5) were used to predict the hepatic microsomal CLintgiven in Tables 1 and 2. The CLint values obtained suggest that sulfone formation is a major elimination pathway for S-omeprazole, followed by hydroxy and 5-O-desmethyl formation. The percentage of CLint calculated for each isoenzyme suggests CYP3A4 to be the dominant enzyme (57%), followed by CYP2C19 (40%). The calculations indicated a minor contribution of CYP2C9 to the metabolism of S-omeprazole, whereas the contributions of CYP2A6 and 2D6 were negligible. For R-omeprazole, the hydroxy pathway is the dominant elimination pathway. According to the calculations in Table 2, CYP2C19 and CYP3A4 contribute to 87 and 13%, respectively, of the total metabolism of the R-form. Thus, CYP2C19 followed by CYP3A4 were identified as the most important enzymes in the overall metabolism of the optical isomers of omeprazole. As indicated by Shimada et al. (1994) and Yamazaki et al. (1997), however, the interindividual variation in specific CYP isoform content is considerable. As measurements of the individual CYP isoenzymes were made in a limited number of individuals, the predicted microsomal CLint from expressed enzymes should be interpreted with care.
The sum of the predicted CLint values for the individual enzymes (Tables 1 and 2) and the three metabolites should reflect the CLint for the same metabolites measured in liver microsomes (Table 3). However, the total microsomal CLint, calculated from expressed enzymes, forS-omeprazole was calculated to be 38.2 μl/min/mg, which is 2.6-fold higher than the CLint measured in our studies using liver microsomes (i.e., 14.6 μl/min/mg). On the other hand, the microsomal CLint calculated from expressed enzymes for R-omeprazole was 35.6 μl/min/mg, which is 84% of that measured in liver microsomes (i.e., 42.5 μl/min/mg).
Discussion
The optical isomers of omeprazole show a clear difference in their metabolism by human liver microsomes. The sum of the CLint values for the three metabolites is considerably lower for S-omeprazole than forR-omeprazole. The slower metabolism ofS-omeprazole is indeed reflected in a 2-fold higher AUC in vivo for S-omeprazole than for R-omeprazole when given orally as individual optical isomers to EM (Andersson et al., 2000). The hepatic clearance for S-omeprazole calculated from the in vitro data on human liver microsomes (572 ml/min) is close to the blood clearance measured in vivo after an i.v. dose (602 ml/min) (Hassan-Alin et al., 2000), indicating that calculation of the CLint using metabolite formation in human liver microsomes can give a good prediction of the in vivo situation. The results also suggest that hepatic metabolism is a major determinant of the overall clearance in vivo. An attempt to predict metabolic clearance in human liver microsomes from cDNA-expressed enzymes did not predict the CLint measured in liver microsomes, indicating that the values obtained in microsomes overexpressing single enzymes may not be easily scaled up to the in vivo situation. In fact, these calculations suggest that the sums of the CLint values for the formation of the three metabolites from S- and R-omeprazole would be almost equal, i.e., that there would be no stereoselectivity displayed in the overall metabolism of the two compounds. However, as discussed by Ito et al. (1998), expressed enzymes are a valuable aid in predicting the relative importance of the individual enzymes for each metabolite.
The two major enzymes involved in omeprazole metabolism are CYP3A4, which forms the sulfone, and CYP2C19, which forms the major part of the 5-O-desmethyl and hydroxy metabolites. In human liver microsomes, the CLint for the two CYP2C19-dependent routes, 5-O-desmethyl and hydroxy formation, from the S-form is 73% of the total and from theR-form is 98% of the total (data from Table 3). These in vitro results support the in vivo findings reported by Tybring et al. (1997). When given as the racemate, the mean AUC forR-omeprazole was 7.5-fold higher in poor metabolizers (PM) (i.e., subjects lacking CYP2C19 activity) than in EM, whereas the difference between PM and EM for S-omeprazole was 3.1-fold. These in vivo results suggest that CYP2C19 is responsible for about 70% of the metabolism of S-omeprazole and about 90% of that of R-omeprazole. These data further support the contention that R-omeprazole is metabolized to a higher extent than S-omeprazole by CYP2C19.
Our findings that the contributions of CYP2C9 and 2D6 are of minor importance for the formation of the three metabolites of the optical isomers of omeprazole are analogous to results reported from in vitro studies on racemic PPIs. Studies using expressed enzymes, isoenzyme-specific inhibitors, and/or antibodies have shown minor or negligible contributions of CYP2C9 and 2D6 to the overall metabolic clearance of omeprazole, pantoprazole, lansoprazole, and rabeprazole (Andersson et al., 1993b; Simon, 1995; Karam et al., 1996; Pearce et al., 1996; VandenBranden et al., 1996).
The estimated Km values for hydroxy metabolite formation from the incubations with cDNA-expressed enzymes indicate that the optical isomers of omeprazole have a considerably higher affinity to CYP2C19 (Km= 4–6 μM) than to CYP3A4 (Km= 316–367 μM). TheKm values for the hydroxy metabolite formation obtained in human liver microsomes (31.7 and 7.93 forS- and R-omeprazole, respectively) most likely reflect CYP2C19-dependent metabolism. In previous studies on racemic omeprazole, where data were evaluated using a biphasic model, Andersson et al. (1993b) calculated two Km values ranging from 3.4 to 16 and 81 to 354 μM, respectively. Similar values for omeprazole hydroxylation were also estimated by Chiba et al. (1993)(Km1 = 6.0 μM andKm2 = 106 μM). Thus, theKm values obtained for the hydroxy metabolite formation from the optical isomers of omeprazole in our study are closer to the high-affinity Kmvalues determined in those studies than to the low-affinity components. As indicated by the CLintvalues calculated from expressed enzymes, the hydroxylation ofR-omeprazole in human liver microsomes is probably dominated by CYP2C19. Expressed enzymes exhibit 2-fold higherVmax and 80-fold lowerKm values for CYP2C19 than for CYP3A4; this is a strong indicator of the major role of CYP2C19 in the hydroxylation of R-omeprazole. As predicted from the results from expressed enzymes, the metabolic clearance of S-omeprazole via the hydroxy route is much less important than that ofR-omeprazole. Hydroxylation of S-omeprazole by CYP3A4 may be equally important as that via CYP2C19 for its elimination because of the relatively high Vmax and the high abundance of CYP3A4 in liver microsomes.
The formation of 5-O-desmethyl is a relatively more important route of metabolism for S- thanR-omeprazole in human liver microsomes. Expressed enzymes indicate that this is a result of a higher affinity and capacity of CYP2C19 for the S- than for the R-form.
The sulfoxidation of the optical isomers of omeprazole is catalyzed by a single enzyme, CYP3A4, as indicated by the results of incubations with cDNA-expressed enzymes. Owing to the higherVmax value obtained, the formation CLint of the sulfone from S-omeprazole is considerably higher than that when formed fromR-omeprazole, as indicated both by human liver microsomes and expressed CYP3A4.
A major finding in our study is that the stereoselective disposition of the optical isomers of omeprazole is considerable, not only via CYP2C19 but also via CYP3A4. This is also reflected in the in vivo data on the pharmacokinetics in PM subjects reported for omeprazole (Tybring et al., 1997) and pantoprazole (Tanaka et al., 1997). In PM subjects with a deficiency of metabolism by CYP2C19, the AUCs of theS-forms are significantly lower than those of theR-forms. [Note: the absolute configuration of the optical isomers of pantoprazole has not been published but, by analogy to other PPIs and as discussed previously (Äbelö et al., 2000), it is highly probable that (+)-pantoprazole is of theR-configuration.] However, in the EM subjects, the higher clearance via CYP2C19 switches the stereoselectivity from favoring metabolism of the S-forms, as in the PM, to theR-forms for both omeprazole and pantoprazole. In the case of lansoprazole, no in vivo data on the pharmacokinetics of the individual optical isomers after administration of the racemic compound to PM subjects are available. However, such data have been reported in a study including six nongenotyped subjects. In that study, the AUC of the S-form after oral administration of racemic lansoprazole was significantly lower than that of the R-form. Assuming that these data reflect the pharmacokinetics in EM subjects (EM constitute 77–87% of the Oriental population; Goldstein et al., 1997), the results indicate that the stereoselective metabolism of lansoprazole is opposite to that of omeprazole and pantoprazole in EM subjects, where the metabolism of the R-forms is favored (Katsuki et al., 1996). In vitro data on the metabolism of lansoprazole using human liver microsomes clearly show that the clearance depends mainly on CYP2C19 (Pearce et al., 1996); it can thus be concluded that structural features of the closely related PPIs will determine whether the S- or the R-forms will be the substrates preferred by CYP2C19.
For omeprazole and pantoprazole, the pyridine substituent is the favored target of metabolism and, as discussed above, theS-form is cleared more slowly than the R-form. For lansoprazole, on the other hand, the hydroxylation occurs in the benzimidazole group, and the clearance of the S-form is favored over that of the R-form. By analogy, our data on expressed CYP2C19 indicate that 5-O-demethylation of the benzimidazole is highly favored for S- as compared withR-omeprazole. This was also found in our in vitro studies on the compound H 259/31 (Fig. 1; Äbelö et al., 2000), as theS-form is more prone to metabolism and as the CYP2C19-mediated hydroxylation occurs mainly in the benzimidazole group. Thus, our present data on the optical isomers of omeprazole and previously published data on the metabolism of other PPIs indicate that the metabolic disposition via CYP2C19 is regio- and stereoselective.
These findings are most probably of importance when studying the structure-activity relationships of CYP active sites and their substrates. Thus, if omeprazole is used in structure-activity models of the site of CYP2C19, the S-form should be docked to effectuate 5-O-demethylation of the benzimidazole group, whereas the R-form should be docked to allow for hydroxylation of the 5-methylpyridine group. However, several papers have appeared (Ibeanu et al., 1996; Lewis and Lake, 1998; Lewis et al., 1998) in which the chirality of the sulfoxide apparently has been overlooked. First, in the studies by Lewis et al. (Lewis and Lake, 1998; Lewis et al., 1998), their model of omeprazole itself includes a planar rather than a tetrahedral configuration of the sulfoxide functionality. Second, the optimum binding for hydroxylation of the 5-methylpyridine group of omeprazole by CYP2C19 is suggested to occur via a hydrogen bond between His74 and the achiral benzimidazole nitrogen rather than the chiral sulfinyl group. Obviously, the binding via the achiral benzimidazole nitrogen is less likely to result in the stereo- and regioselective outcome of the reaction that was demonstrated in our studies. Consequently, the regioselective metabolism of S- and R-omeprazole by CYP2C19 indicates that the chirality of the sulfinyl group should be accounted for when developing active site models for the catalysis of the metabolism of omeprazole.
In conclusion, our studies in human liver microsomes and in cDNA-expressed enzymes show a significant stereoselectivity in the metabolism of the optical isomers of omeprazole, the CLint for the S-form being approximately three times lower than that of the R-form. This is mainly explained by the considerably lower CLint for the formation of 5-hydroxy fromS-omeprazole than from R-omeprazole via CYP2C19. We also conclude that S-omeprazole is favored for the 5-O-demethylation of the benzimidazole group by CYP2C19 and of the sulfoxidation by CYP3A4. However, the total metabolic clearance of S-omeprazole is lower than that ofR-omeprazole, which explains the higher plasma levels of theS-form in vivo when given as the racemate.
Acknowledgments
We thank Dr. Tommy Andersson for valuable input during the preparation of the manuscript.
Footnotes
-
Send reprint requests to: Angela Äbelö, AstraZeneca R&D Mölndal, S-431 83 Mölndal, Sweden. E-mail:angela.abelo{at}astrazeneca.com
- Abbreviations used are::
- PPIs
- proton pump inhibitors
- CYP
- cytochrome P450
- DMSO
- dimethyl sulfoxide
- CLint
- intrinsic clearance
- EM
- extensive metabolizers
- PM
- poor metabolizers
- S-omeprazole
- theS-form of omeprazole
- R-omeprazole
- theR-form of omeprazole
- AUC
- area under the plasma concentration versus time curve
- Received February 17, 2000.
- Accepted May 9, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}