


Abstract
The in vitro glucuronidation of a range of structurally diverse chemicals has been studied in hepatic and renal microsomes from human donors and the beagle dog. These studies were undertaken to improve on the limited knowledge of glucuronidation by the dog and to assess its suitability as a model species for pharmacokinetic studies. In general, the compounds studied were glucuronidated severalfold more rapidly (based on intrinsic clearance estimates) by DLM than by HLM. Intrinsic clearance values for human UGT1A1 and UGT2B7 substrates were an order of magnitude higher in DLM than in HLM (e.g., gemfibrozil: 31 μl/min/mg versus 3.0 μl/min/mg; ketoprofen: 2.4 μl/min/mg versus 0.2 μl/min/mg). There were also drug-specific differences. HLM readily glucuronidated propofol (2.4 μl/min/mg) whereas DLM appeared unable to glucuronidate this drug directly. Regioselective differences in morphine glucuronidation were also apparent. Human kidney microsomes catalyzed the glucuronidation of many xenobiotics, although glucuronidation of the endobiotic bilirubin was not detectable in this tissue. In direct contrast, dog kidney microsomes glucuronidated bilirubin only (no glucuronidation of all other xenobiotics was detected). These preliminary studies indicated significant differences in the glucuronidation of xenobiotics by microsomes from the livers and kidneys of human and dog and should be confirmed using a larger panel of tissues from individual dogs. Early knowledge of the relative rates of in vitro glucuronidation, the UGTs responsible for drug glucuronidation, and their tissue distribution in different species could assist the design and analysis of preclinical pharmacokinetic and safety evaluation studies.
Uridine diphosphate glucuronosyltransferases (UGTs1) are a family of microsomal enzymes that catalyze the conjugation of xenobiotic and endogenous substrates to a glucuronic acid moiety derived from the nucleotide sugar UDP-glucuronic acid in a nucleophilic substitution (SN2-like) reaction (Dutton, 1980). The transfer of this acidic group results in the formation of a glucuronide, which is more hydrophilic than the parent drug and can be excreted more readily in either bile or urine. This type of reaction has been designated phase II and can be the major route of clearance for compounds containing functional groups (hydroxyl, thiol, amine, or carbonyl) amenable to direct glucuronidation (Burchell and Coughtrie, 1989).
An understanding of the enzymology of the metabolic clearance of a drug, whether by phase I or phase II mechanisms, is pivotal to new drug development. Traditionally, rodents such as the rat and nonprimate species such as the dog have been used as animal models in studies aimed at evaluating the pharmacodynamics, metabolism, pharmacokinetics, and safety of new chemical entities. Therefore, it is surprising that few comparative studies have been performed that address quantitative and qualitative interspecies differences for drugs known to be extensively glucuronidated in human.
Perhaps the most comprehensive example of an interspecies comparison involving glucuronidation has been performed on amine substrates. TheN-glucuronidation of primary amines appears to occur in all common laboratory species (e.g., rat, dog, and nonhuman primate), although to markedly different degrees (reviewed by Chiu and Huskey, 1998).
Porter et al. (1975) were the first to demonstrate the formation of a quaternary ammonium-linked glucuronide in humans from tertiary amines, a process catalyzed by UGT1A4 (Green and Tephly, 1995). Early studies suggested that only humans and higher primates (chimpanzees) were able to glucuronidate tertiary amines, and most laboratory animals appeared unable to catalyze this pathway of metabolism (Hucker et al., 1978;Fischer et al., 1980). However, Lehman et al. (1983) have since demonstrated that rabbit can also form quaternary ammonium-linked glucuronides.
Another interspecies comparison by Sisenwine et al. (1982) demonstrated that stereoselective differences can occur in glucuronidation across species, using oxazepam as a model substrate. This group showed that in both human and dog, the S-enantiomer was glucuronidated preferentially by liver microsomes, whereas theR-enantiomer was glucuronidated in rhesus monkeys.
A preliminary in vitro study of phase I and II enzymes in the most commonly used drug metabolism species (Sharer et al., 1995) suggested that there may be differences in rates of glucuronidation by dog and human hepatic microsomes. Ethynylestradiol was glucuronidated by the dog at a rate 2-fold greater than by humans. However, this reaction was only studied at a single substrate concentration, and a more detailed kinetic evaluation would be required to make more valuable conclusions.
The aims of this study were to provide a more detailed understanding of in vitro glucuronidation catalyzed by dog tissue microsomes and to assess the suitability of the dog as a model drug metabolism species (in terms of glucuronidation) for human. In this study, in vitro kinetic parameters were determined in two of the major organs responsible for glucuronidation, the liver and kidney. Compounds known to be predominantly glucuronidated in human (Bertz and Granneman, 1997) were studied using human and canine tissues.
Materials and Methods
Chemicals.
Substrates, UDP-glucuronic acid, and other reagents used in the assays were purchased from Sigma (Gillingham, Dorset, UK), Aldrich (Gillingham, Dorset, UK), or BDH (Poole, Dorset, UK) and were of the highest grade available. [14C]UDPGA (293.6 mCi/mmol) and [3H]imipramine hydrochloride (48.7 Ci/mmol) were purchased from NEN DuPont (Stevenage, Hertfordshire, UK).
Microsomal Preparation from Liver and Kidney.
Pooled human liver microsomes were prepared by pooling microsomes from six human livers (male/female ratio 1:2, age range 27–62 years, one smoker) obtained from Keystone Skin Bank (Exton, PA) that exhibited representative UGT activity against prototypic substrates for the main hepatic human UGT isoforms and indicated the absence of poor metabolizers (Fig. 1). Microsomes prepared from a single dog liver were used for the detailed kinetic studies (Table 1). Kinetic data for selected key substrates were produced with a limited resource of two sets of pooled DLM [each prepared from five separate dog livers (In Vitro Technologies, Baltimore, MD)], which suggested low interanimal variability (Table 2).

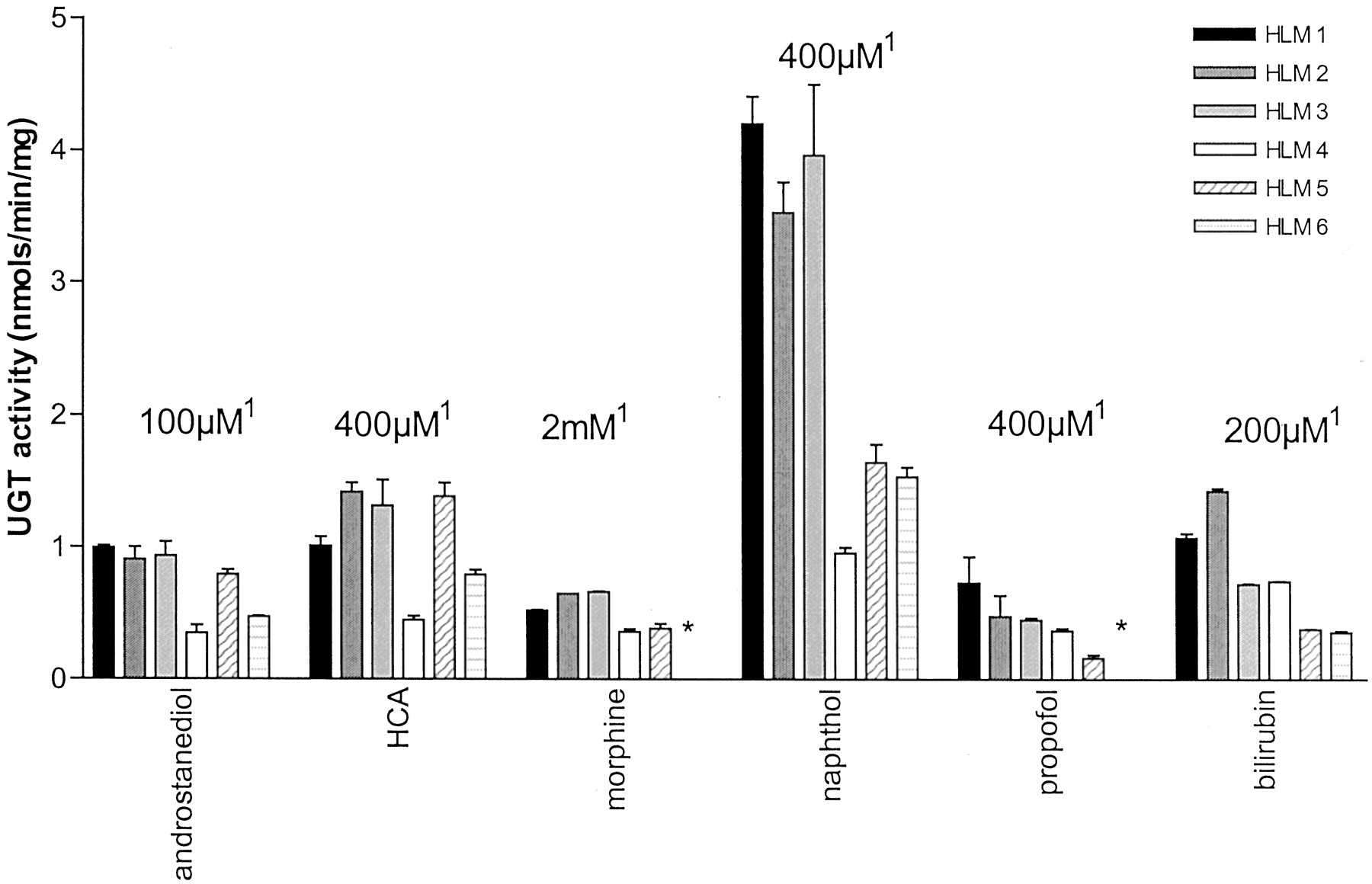
UGT activity from six human livers toward six probe substrates.
Screening of microsomes prepared from six human livers against the substrates androstanediol, hyodeoxycholic acid (HCA), morphine, naphthol, propofol, and bilirubin before pooling. ★, data not available; 1, substrate concentration used for screening.
Kinetic parameters for the glucuronidation of 16 compounds in human and dog liver microsomes
Reproducibility of kinetic parameters from microsomes prepared from beagle dogs
Human kidney samples were ethically obtained from pathological specimens from patients undergoing a nephrectomy. Nonpathological segments from these specimens were immediately frozen in liquid nitrogen and stored at −80°C. Dog kidney samples were removed from two healthy male beagle dogs (2–5 years old, weighing 10–15 kg) and were frozen in the same way as the human kidney samples. All microsomes were prepared using a method adapted from that of Coughtrie et al. (1987). Briefly, livers or kidneys were minced and homogenized in four volumes of ice-cold 0.25 M sucrose, 5 mM HEPES, pH 7.4. The resultant homogenates were then centrifuged for 15 min at 10,000g, and the supernatants were centrifuged at 100,000g for 60 min. Microsomal pellets were resuspended in 1 volume (ml) of ice-cold 0.25 M sucrose, 5 mM HEPES (pH 7.4) equal to the wet weight (in grams) of tissue. Protein in microsomal samples was determined using the method of Lowry et al. (1951), using bovine serum albumin as a standard. Aliquots were stored at −80°C.
The glucuronidation of 1-naphthol was used to determine the optimal activation for each microsomal preparation. Tissue was sonicated for 1-, 2-, 3-, 4-, and 5- × 5-s bursts with 1 min on ice between bursts. The resultant microsomal sonicates were used in UGT assays to determine the optimum level of sonication required for maximal 1-naphthol glucuronidation.
UGT Assays.
UGT assays (except bilirubin and imipramine) were performed as described previously (Ethell et al., 1998). Briefly, 100 mM Tris/Maleate buffer (pH 7.4) containing 5 mM MgCl2, 10 mM saccharic acid 1,4-lactone (present in all incubations), typically 500 μM substrate, 250 to 350 μg microsomal or cellular sonicate, and 2 mM UDPGA (0.1 μCi [14C]UDPGA/assay) were combined in a total volume of 100 μl. Incubations were run for 60 min and then terminated by the addition of 100 μl of methanol that had been prechilled to −20°C. The mixture was centrifuged for 10 min at 1000g. The resulting supernatant was then transferred to a high performance liquid chromatography vial, and 150 μl of this volume was directly injected onto gradient high performance liquid chromatograph system that used solid scintillant radioactive detection as described previously (Ethell et al., 1998).
Substrate solutions were prepared and diluted on the day of assay. Typically, the substrate concentrations used were 10, 50, 100, 250, 500, and 1000 μM. Kinetic parameters [Vmax, Km, and intrinsic clearance (CLint)] were calculated by fitting the experimental data (duplicate incubations at six substrate concentrations) to the Michaelis-Menten equation by a nonlinear least-squares regression method (Prism version 2, GraphPad Software, San Diego, CA). All data have been quoted to an appropriate level of accuracy.
A colorimetric assay was used to measure bilirubin glucuronidation (Heirwegh et al., 1972). Imipramine glucuronidation was determined using the method described by Coughtrie and Sharp (1991).
β-Glucuronidase Assay.
β-Glucuronidase assays were performed using a method adapted from that of Combie et al. (1982). Assays containing 100 mM potassium phosphate (pH 6.8) and 0.6 mM phenolphthalein glucuronide were preincubated for 2 min at 37°C before the addition of typically 200 to 500 μg of microsomal sonicate. Assay mixtures were incubated for 30 min at 37°C and then terminated by the addition of 1 ml of 200 mM glycine (pH 10.4). The absorbance of the resulting mixtures was measured at 540 nm using a negative control as a blank [an assay which had been quenched with 1 ml of 200 mM glycine (pH 10.4) before the addition of microsomal sonicate]. A standard curve was constructed by reading the absorbance (at 540 nm) of various concentrations of phenolphthalein dissolved in the assay mix. This curve was used to convert the absorbance readings produced from the assays to the amount of phenolphthalein produced.
Results
Glucuronidation by the Liver.
A total of 16 compounds including 12 drugs, 1-naphthol, and 3 endogenous substrates (androstanediol, bilirubin, and hyodeoxycholic acid) were studied in human and dog liver microsomes (HLM and DLM). The kinetic parameters Vmax andKm were determined, and the results are shown in Table 1. CLint, an indication of the inherent metabolic stability of the compounds studied, was calculated from Vmax/Km.
Kinetics for androstanediol, furosemide, gemfibrozil, and ketoprofen (these compounds were representative of those used in the main study) were confirmed in two separate pools of DLM, each composed of five separate dog livers (In Vitro Technologies). The kinetics of these substrates (Table 2) agrees with the kinetic parameters produced by the DLM used in the main study (Table 1), suggesting a low interanimal variability (variation in CLint < 30%).
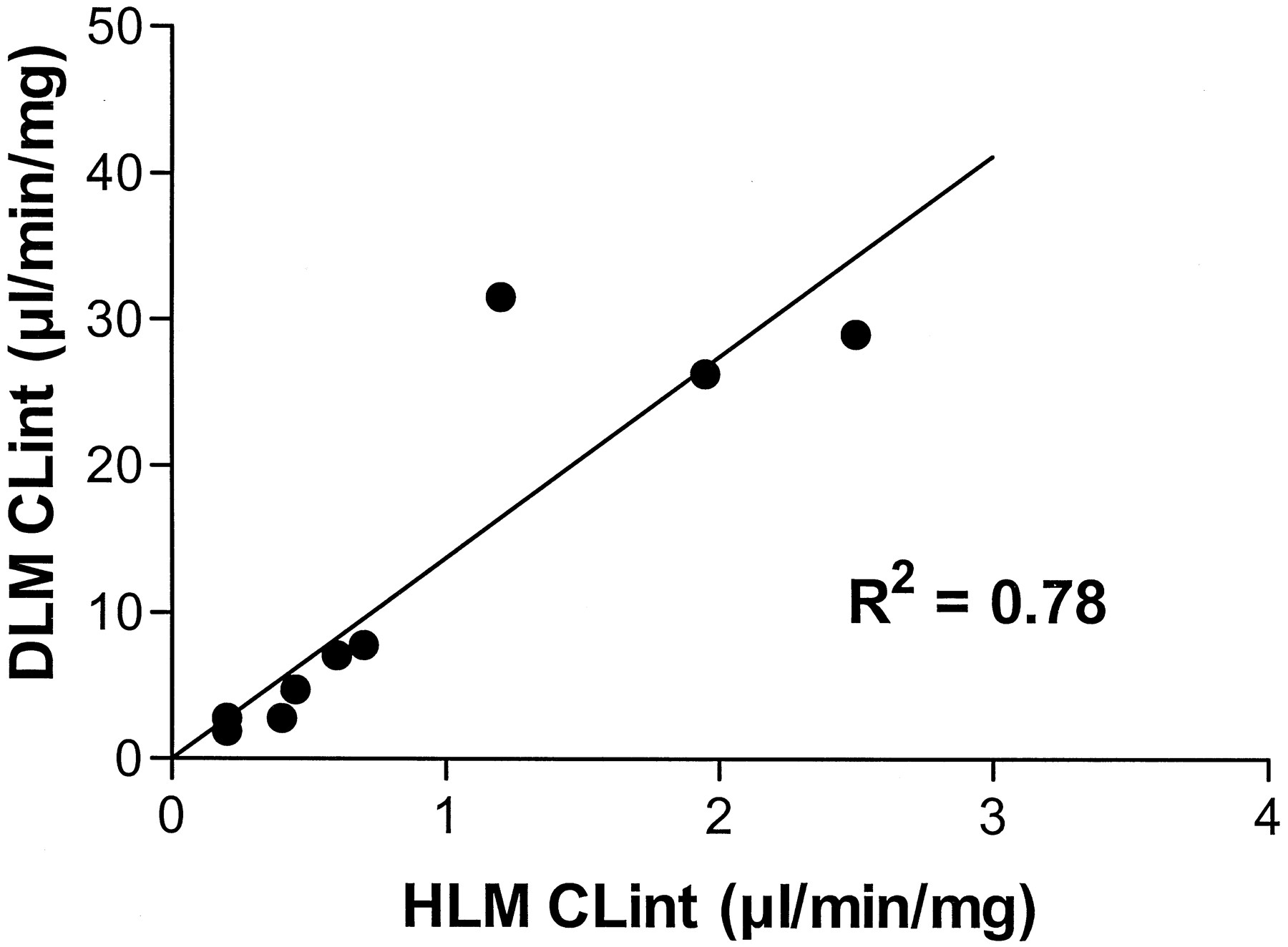
The maximum rate of glucuronidation of the compounds by DLM was at least 5-fold greater than in HLM, with the exception of the endobiotics bilirubin and hyodeoxycholic acid. In contrast, the affinity of HLM and DLM for compounds (as assessed by Km) did not display this simple trend but was compound specific. Hence, the CLint estimates for glucuronidation of drug and xenobiotic compounds by DLM were greater than by HLM (Fig.2), with the differences mainly reflecting Vmax differences. Figure 2 shows the relationship between glucuronidation by HLM and DLM of drugs and xenobiotics metabolized in human by UGT1A1 or UGT2B7.
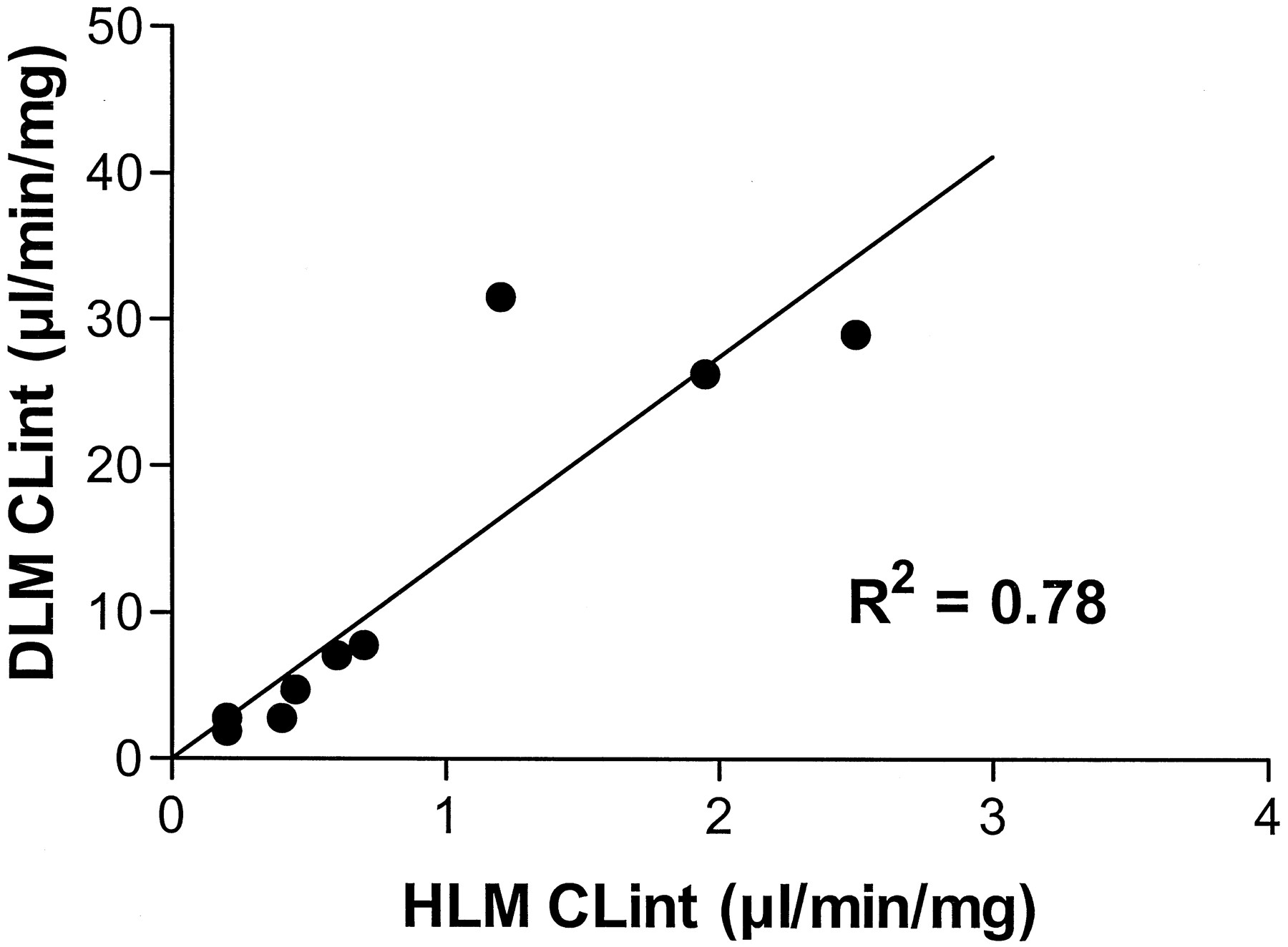
Comparison of CLint values for nine drugs obtained with HLM and DLM.
CLint values were determined for nine drugs (known to be substrates of human UGT1A1 or UGT2B7: codeine, ethynylestradiol, gemfibrozil, hydromorphone, ketoprofen, morphine, naloxone, naproxen, and valproic acid) in HLM and DLM. A correlation coefficient (R2) of 0.78 was determined.
Propofol was readily glucuronidated by HLM, but no glucuronidation of this compound was detected by DLM. However, the addition of 0.5 mM NADPH to incubations with propofol in DLM resulted in the formation of a glucuronide with a retention time earlier than the glucuronide produced by HLM (9.7 min versus 10.3 min, see Discussion). Morphine was glucuronidated by HLM, predominately at the 3-position (verified by authentic standards), but also by a significant amount at the 6-position (ratio 5:1). Glucuronidation of morphine was also extensively catalyzed by DLM at the 3-position (20 times greater than in HLM), but morphine glucuronidation at the 6-position was not readily detected by the methodology employed. This indicates both quantitative and qualitative differences in morphine glucuronidation by HLM and DLM. Glucuronidation of the tertiary amine imipramine was detected by DLM. However, the rate of glucuronidation was too low (0.01 nmol/min/mg) for accurate kinetics to be determined.
Glucuronidation by the Kidney.
The glucuronidation of 12 compounds was studied in human and dog kidney microsomes (HKM and DKM). The results are summarized in Table3. HKM catalyzed the glucuronidation of many structurally diverse compounds, including phenols, acids, amines, and steroids (Table 3). Table 3 shows that there was considerable interindividual variation for glucuronidation in human kidney (seeDiscussion). No glucuronidation of bilirubin was detectable in human kidney. In direct contrast, DKM catalyzed the glucuronidation of bilirubin but not the glucuronidation of all the other compounds studied. The Km was lower when using HKM compared with HLM for most compounds.
Kinetic parameters for the glucuronidation of 12 compounds in human and dog kidney microsomes
β-Glucuronidase Activity in Human and Dog Tissues.
The difference between the glucuronidation rates catalyzed by dog and human microsomes could be due to variation in β-glucuronidase activity between the two species. Therefore, β-glucuronidase activity was assayed in HLM, HKM, DLM, and DKM (Table4). HKM and DLM had similar levels of β-glucuronidase activity (approximately 35 pmol/min/mg), whereas a higher level of activity (130 pmol/min/mg) was detected in DKM. Addition of 10 mM saccharic acid 1,4-lactone (routinely included in all microsomal incubations) to β-glucuronidase assays of HLM, DLM, and DKM reduced activity by 79 to 83%. β-Glucuronidase activity was inhibited to a lesser extent (∼40%) by saccharic acid 1,4-lactone in HKM. These data indicate that the addition of this β-glucuronidase inhibitor to UGT assays of these microsomal preparations would significantly reduce hydrolysis of glucuronides formed.
β-Glucuronidase activity in tissue microsomal samples from human and dog
Therefore, the observed differences in levels of UGT activities between human and dog microsomes were not a consequence of differential hydrolysis of the glucuronides. Furthermore, the effect of addition of 10 mM saccharic acid 1,4-lactone was investigated directly by studying the glucuronidation of gemfibrozil and valproic acid in HLM, HKM, and DLM (Table 5). The addition of this β-glucuronidase inhibitor did not affect the rate of glucuronidation toward gemfibrozil or valproic acid by either human or dog microsomes, suggesting that endogenous β-glucuronidase had no significant contribution. Indeed, there was a slight decrease in gemfibrozil/valproic acid activity upon addition of saccharic acid 1,4-lactone.
The effect of saccharic acid 1,4-lactone on UDP-glucuronosyl transferase activity in tissue microsomes
Discussion
Many xenobiotics and endogenous compounds are metabolized by the phase II process of glucuronidation. Drugs that are mainly cleared by glucuronidation include nonsteroidal anti-inflammatory drugs, analgesics, antidepressants, and sedatives. Representatives from these therapeutic classes were investigated in this study. Dog is the most common secondary nonprimate species used in drug metabolism studies in the pharmaceutical industry. Therefore, the development of new drugs demands a knowledge of glucuronidation in the dog. The data generated in this study have indicated that the majority of the drugs studied exhibit greater in vitro CLint values in DLM compared with HLM. The higher CLint values in DLM suggest that many drugs undergoing direct glucuronidation may be cleared more rapidly by dog liver than by human liver, possibly due to a greater efficiency/capacity of glucuronidation.
Sharer et al. (1995) noted that the UGT1A1 substrate ethynylestradiol was glucuronidated severalfold more rapidly by dog liver than human liver. Information using human UGT recombinant cell lines has indicated that the compounds marked (a orb) in Table 1 are selectively glucuronidated by human UGT1A1 or UGT2B7 (Ebner et al., 1993; Coffman et al., 1997;Terrier et al., 1999). Preliminary findings in this lab using human UGTs expressed in Chinese hamster V79 cells have confirmed these results (data not shown). This subset of compounds was cleared at least 10-fold greater by DLM than by HLM (see Fig. 2). Therefore, we have considerably expanded the initial observation of Sharer et al. (1995). This information may be valuable to pharmaceutical companies using dog as a model species for pharmacokinetic analyses, and such detailed enzymology may prevent misleading pharmacokinetic studies in the dog. Indeed, substrates for human UGT1A1 and UGT2B7, which previously may have been discarded due to rapid glucuronidation in the dog, may in fact fulfill the desired pharmacokinetics in human (a screen with human recombinant UGT cell lines would determine if UGT1A1 or UGT2B7 were involved). It is important to state that the specific UGT isoforms responsible for the glucuronidation of compounds in DLM are unknown since no dog UGTs have been isolated to date.
In addition to this general species difference in hepatic glucuronidation capacity, an absolute difference in UGT activity was noted for propofol. No propofol glucuronidation was detected in DLM in direct contrast to HLM. The major metabolite of propofol in humans is the 1-glucuronide of the parent compound (Simons et al., 1992). This i.v. anesthetic is also known to be rapidly cleared (30–80 ml/min/kg) in dogs in vivo (Cockshott et al., 1992). The major metabolite of propofol in dog is a 4-hydroxylated derivative, which is then recovered as a glucuronide (Simons et al., 1991). Glucuronidation of propofol was observed when 0.5 mM NADPH was added to the assay mixture containing DLM. A glucuronide produced by DLM, which exhibited a retention time different from that produced by HLM (9.7 min versus 10.3 min), suggested that oxidation at the 4-position had occurred. The 4-hydroxy metabolite was subsequently glucuronidated, as predicted from the literature (Simons et al., 1991).
There was also a regioselective difference noted between HLM and DLM, illustrated by the glucuronidation of morphine. HLM were capable of forming both the 3- and 6-glucuronides of morphine, whereas only the 3-glucuronide was formed by DLM at significant levels. Previous enantiomeric differences in dog have been reported by Sisenwine et al. (1982) when using oxazepam as a model substrate. These pieces of evidence suggested that the route of glucuronidation in addition to the rate may be different between the dog and human. Although pooled HLM were used in the present study, which focused on interspecies variations in metabolism, future studies should include an examination of intersubject variability in human samples, in particular.
The liver is generally accepted as the main drug metabolic organ in human, but the kidney has also been shown to be capable of glucuronidating a wide range of drugs, including propofol, naproxen, ibuprofen, and octyl gallate (McGurk et al., 1998). The activities detected in the present study largely confirm these observations, and interindividual differences were also apparent between the three kidney samples studied. Since the method of tissue preparation was the same from these individuals, differences in glucuronidation may be attributable to either genetic or environmental effects (for example, administered xenobiotics or diet). The lowerKm in human kidney for drugs shown to be UGT substrates compared with human liver may reflect metabolism by a reduced number of different UGTs in kidney. This tissue distribution may be an important consideration for extrahepatic drug glucuronidation and target organ toxicity.
Comparison of glucuronidation by HKM with that of DKM showed major differences between these two species with respect to renal microsomal glucuronidation (Table 3). In direct contrast to the wide range of xenobiotics glucuronidated by HKM, DKM were only shown to glucuronidate bilirubin, confirming the earlier observations of Fevery et al. (1977). Therefore, DKM exhibited limited functional expression of renal UGTs. This may be attributable to the lack of expression of the relevant UGTs in DKM, or expression of these UGTs in dog kidney may be at such a low level that glucuronidation was below the level of detection. However, zonal expression of localized high concentrations of specific UGTs in dog kidney cannot be excluded from the present studies. This is another example of a marked difference in glucuronidation between human and dog that may well be attributed to differential expression of specific UGTs in the kidneys of these two species. This may have a bearing on the choice of the dog as a species for the toxicological assessment of some new chemical entities since target organ toxicity may be influenced by activation or detoxication in the kidney (Benet and Spahn, 1988).
This article has highlighted several significant differences in glucuronidation between human and dog and emphasizes again the importance of in vitro work with human tissues. Further studies using microsomes prepared from a larger panel of individual dogs would validate the preliminary findings of this report. The dog may be valuable as a second model species for compounds glucuronidated in human, providing the enzymological basis for quantitative differences in UGT activity is understood. Further analysis of the individual enzymes responsible for glucuronidation in the two species may provide a more detailed molecular insight. There is still much work to be done before glucuronidation in the dog is fully understood, and better diagnostic tools (e.g., the cloning of dog UGTs) will ultimately be required to achieve this goal.
Footnotes
-
Send reprint requests to: Brian Burchell, Department of Molecular and Cellular Pathology, Ninewells Hospital and Medical School, Dundee, DD1 9SY, Scotland. E-mail: b.burchell{at}dundee.ac.uk
-
This work was funded by AstraZeneca and The Wellcome Trust.
- Abbreviations used are::
- UGT
- uridine diphosphate glucuronosyltransferase
- HLM
- human liver microsomes
- HKM
- human kidney microsomes
- DLM
- dog liver microsomes
- DKM
- dog kidney microsomes
- UDPGA
- UDP glucuronic acid
- CLint
- intrinsic clearance
- Received April 13, 2000.
- Accepted October 5, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}