


Abstract
The biotransformation of prasugrel to R-138727 (2-[1-2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid) involves rapid deesterification to R-95913 (2-[2-oxo-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone) followed by cytochrome P450 (P450)-mediated formation of R-138727, the metabolite responsible for platelet aggregation. For identification of the P450s responsible for the formation of the active metabolite, the current studies were conducted with R-95913 as the substrate. Incubations required supplementation with reduced glutathione. Hyperbolic kinetics (Km 21–30 μM), consistent with a single enzyme predominating, were observed after incubations with human liver microsomes. Correlation analyses revealed a strong relationship between R-138727 formation and CYP3A-mediated midazolam 1′-hydroxylation (r2 = 0.98; p < 0.001) in a bank of characterized human liver microsomal samples. The human lymphoblast-expressed enzymes capable of forming R-138727, in rank order of rates, were CYP3A4>CYP2B6>CYP2C19≈CYP2C9>CYP2D6. A monoclonal antibody to CYP2B6 and the CYP3A inhibitor ketoconazole substantially inhibited R-138727 formation, whereas inhibitors of CYP2C9 (sulfaphenazole) and CYP2C19 (omeprazole) did not. Scaling of in vitro intrinsic clearance values from expressed enzymes to the whole liver using a relative abundance approach indicated that either CYP3A4 alone or CYP3A4 and CYP2B6 are the major contributors to R-138727 formation. R-95913 and R-138727 were also examined for their ability to inhibit metabolism mediated by five P450s. R-138727 did not inhibit the P450s tested. In vitro, R-95913 inhibited CYP2C9, CYP2C19, CYP2D6, and CYP3A, with Ki values ranging from 7.2 μM to 82 μM, but did not inhibit CYP1A2. These Ki values exceed circulating concentrations in humans by 3.8- to 43-fold. Therefore, neither R-95913 nor R-138727 is expected to substantially inhibit the P450-mediated metabolism of coadministered drugs.
Prasugrel (CS-747, LY640315; Fig. 1) is a novel thienopyridine antiplatelet prodrug that is administered orally as a racemic mixture. It is rapidly metabolized in vivo to a potent ADP-receptor antagonist, designated R-138727 (Fig. 1), which binds irreversibly to P2Y12 receptors, causing inhibition of platelet aggregation that persists for the life of the platelets (Kurihara et al., 2005; Niitsu et al., 2005). Prasugrel is rapidly hydrolyzed by carboxylesterases to yield the thiolactone, R-95913 (Fig. 1), which circulates in human plasma, whereas prasugrel is not detected in human plasma (Farid et al., 2005b). Further metabolism of R-95913 yields a ring-opened form, R-138727 (Fig. 1) (Sugidachi et al., 2000, 2001), which has two chiral centers and is actually a mixture of four stereoisomers that vary in potency of inhibition at P2Y12 receptors (Kazui et al., 2001). Other drugs of the thienopyridine class, ticlopidine and clopidogrel, also require metabolic activation.
The active metabolites (both ring-opened forms similar to R-138727) of clopidogrel (Savi et al., 2000) and ticlopidine (Yoneda et al., 2004) have recently been identified. Some information as to the biotransformation routes of these two drugs is available, although only the surrogate endpoint of parent drug loss, rather than the more informative endpoint of metabolite formation, has been measured. For ticlopidine, the cytochromes P450 (P450s) most active in metabolizing the parent drug are CYP3A4 and CYP2C19 (Dalvie and O'Connell, 2004). For clopidogrel, CYP1A2 has been found to be involved in the metabolism of parent drug in rat microsomes (Savi et al., 1994), but in a more recent study, the CYP3A enzymes were identified as the major enzymes involved in metabolism of parent drug in rat microsomes and a panel of expressed human P450s (Clarke and Waskell, 2003).
For prasugrel, direct measurement of R-138727 in preliminary in vitro studies identified CYP3A4 and CYP2B6 as potential mediators of active metabolite formation (Kazui et al., 2000, 2001). The current studies were performed at a more relevant range of concentrations, used a validated LC/MS/MS analytical method in which the unstable free thiol-containing R-138727 was stabilized by derivatization with 2-bromo-3′-methoxyacetophenone (BMAP), and used a scaling exercise to estimate the contributions of the relevant enzymes to R-138727 formation in vivo. The results of the current studies confirmed the roles of CYP3A and CYP2B6, and identified additional contributors to R-138727 formation.

Prasugrel and the biotransformation pathway to the inactive intermediate R-95913, followed by conversion to active R-138727.
For drugs such as the thienopyridine prodrugs, which require metabolism to form active compounds, identification of the enzymes responsible for biotransformation to the active metabolite helps determine whether therapeutic levels of active drug will be consistently achieved. Clopidogrel is the current standard of care for a variety of cardiovascular indications and has been administered to many thousands of patients, but there has been recent debate as to whether interpatient variability in CYP3A is related to the phenomenon of clopidogrel resistance and whether coadministration of CYP3A substrates with clopidogrel affects the formation of the active metabolite of clopidogrel (Clarke and Waskell, 2003; Lau et al., 2003; Saw et al., 2003; Wienbergen et al., 2003; Lau et al., 2004; Mitsios et al., 2004). The ability to measure the formation of the active metabolite of prasugrel allowed the results of the current in vitro studies to be used to 1) evaluate possible variability in the pharmacokinetics of the major circulating metabolites of prasugrel, and 2) actually predict, rather than clarify retrospectively, the likelihood of potential metabolism-mediated drug-drug interactions that could affect the achievement of therapeutic levels of active drug. In addition, R-95913 and R-138727 were evaluated in vitro for their ability to inhibit the P450s for which drug-drug interactions are of most concern: CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A.
Materials and Methods
Materials. R-95913, R-138727, and 2H4-R-138727 were obtained from UBE Industries (Yamaguchi, Japan). Testosterone, 6β-hydroxytestosterone, 16-epiestriol, l-ascorbic acid, α-hydroxytriazolam, Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid), phenytoin, NADPH, reduced glutathione (GSH), ketoconazole, sulfaphenazole, phenacetin, acetaminophen, and BMAP were purchased from Sigma-Aldrich (St. Louis, MO). Midazolam was obtained from Hoffmann-LaRoche (Nutley, NJ). The 4′-hydroxy metabolite of diclofenac was obtained from BD Gentest (Woburn, MA). Deuterated acetaminophen was obtained from C/D/N Isotopes (Pointe-Claire, QC, Canada). Bufuralol, 1′-hydroxybufuralol, S-mephenytoin, 4′-hydroxy S-mephenytoin, and 1′-hydroxymidazolam were obtained from Ultrafine (Manchester, UK). Omeprazole was obtained from Sigma/RBI (Natick, MA). Inhibitory monoclonal antibody (mAb) to CYP2B6 (in ascites fluid) was obtained from Invitrogen (Carlsbad, CA). Control mouse ascites fluid was obtained from MP Biomedicals (Irvine, CA).
Microsomes. Human liver samples designated HLA through HLT were obtained from the Medical College of Wisconsin (Milwaukee, WI), Medical College of Virginia (Richmond, VA), or Indiana University School of Medicine (Indianapolis, IN), under protocols approved by the appropriate committee for the conduct of human research. Hepatic microsomes were prepared by differential centrifugation (van der Hoeven and Coon, 1974) and characterized for relative levels of P450s through the use of form-selective catalytic activities or immunoquantification (Ekins et al., 1998; Ring et al., 2001). A mixture of equal microsomal protein concentrations from HLB, HLH, HLM, and HLP was used in the studies examining the ability of R-95913 and R-138727 to inhibit various P450-mediated reactions. Microsomes prepared from a human β-lymphoblastoid cell line engineered to express the cDNA for the individual P450s were obtained from BD Gentest.
Examination of R-138727 Formation. Metabolism of R-95913 to R-138727 was examined in vitro in incubations (200 μl) conducted under linear rate of formation conditions at 37°C. The final incubations contained varying concentrations of R-95913, 0.125 mg/ml human hepatic microsomal protein, 5 mM GSH, and 1 mM NADPH in 100 mM sodium phosphate buffer (pH 7.4). After 3-min preincubations at 37°C, reactions were initiated with NADPH. The mixtures were incubated for 15 min at 37°C. Reactions were stopped and samples treated as described under Analytical Conditions (below).
For estimation of apparent enzyme kinetic parameters for the formation of R-138727 by human liver microsomes, concentrations of R-95913 ranged from 0.5 to 75 μM. For correlation analyses and incubations with selective chemical inhibitors or inhibitory mAb, the concentration of R-95913 (20 μM) approximated the Km for R-138727 formation. When P450 inhibitors were added to incubations, concentrations were: 10 μM sulfaphenazole (CYP2C9-selective; Newton et al., 1995; Bourrié et al., 1996), 10 μM omeprazole (CYP2C19-selective; Ko et al., 1997), and 2 μM ketoconazole (CYP3A-selective; Newton et al., 1995; Bourrié et al., 1996). When CYP2B6 mAbs in ascites were included, modifications to the procedure were required. Microsomes with mAb (up to 8 μl), in 100 mM sodium phosphate buffer (pH 7.4) with 5 mM GSH, were preincubated for 5 min at 37°C. Reactions (a total volume of 200 μl) were initiated with addition of a solution of 1 mM NADPH plus 20 μM R-95913. Reactions were stopped at the appropriate times and treated as described under Analytical Conditions (below).
Microsomes prepared from human β-lymphoblastoid cells engineered to express human P450s and P450 reductase were examined for their ability to form R-138727 from R-95913. Incubations (200 μl) contained 5 mM GSH, 2 mM NADPH, and 20 μM R-95913 in 100 mM sodium phosphate buffer (pH 7.4). After 3-min preincubations, microsomes (0.125 mg/ml) from cells expressing CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 were added to start reactions, which continued for 15 min at 37°C. Apparent enzyme kinetic parameters were estimated for the formation of R-138727 by microsomes from β-lymphoblastoid cells expressing CYP2B6, CYP2C9, CYP2C19, and CYP3A4 under linear rate of formation conditions (0.125 mg/ml protein and 30 min for CYP2B6, CYP2C9, and CYP2C19; 0.125 mg/ml and 20 min for CYP3A4). Concentrations of R-95913 in these experiments ranged from 1 to 100 μM.
Inhibition of P450-Selective Catalytic Activities by R-95913 and R-138727. All incubations were conducted under linear rate conditions and contained 100 mM sodium phosphate buffer at pH 7.4. A Ki value was determined when a preliminary experiment indicated substantial inhibition (50%) of one of the P450s by a metabolite of prasugrel.
The O-deethylation of phenacetin (acetaminophen formation) was used as a marker of CYP1A2 activity (Ring et al., 2001). Incubations contained 0.5 mg/ml microsomal protein, 1 mM NADPH, 12.5 μM phenacetin, and 0, 0.5, 2.5, 10, 50, and 100 μM R-95913 or R-138727 as potential inhibitors.
The biotransformation of diclofenac to 4′-hydroxydiclofenac was used as the marker activity for CYP2C9 (Ring et al., 2001). Incubations contained 0, 10, 50, 100, and 200 μM R-95913 and 2.5, 5, 10, 25, and 50 μM diclofenac when R-95913 was the inhibitor. When R-138727 was the inhibitor, 0, 0.5, 2.5, 10, 50, and 200 μM R-138727 were incubated with a concentration of diclofenac approximating the Km (2.5 μM) for the diclofenac 4′-hydroxylation. Additional components were 0.25 mg/ml microsomal protein and 1 mM NADPH.
CYP2C19 activity was studied with the formation of 4′-hydroxy S-mephenytoin from S-mephenytoin as the form-selective activity (Ring et al., 2001). Incubations contained 0.5 mg/ml microsomal protein, 1 mM NADPH, and at least a single concentration (5 μM) of S-mephenytoin. When R-95913 was the inhibitor, R-95913 concentrations were 0, 0.5, 2.5, 10, and 20 μM, and S-mephenytoin concentrations were 5, 10, 25, 50, and 100 μM. When R-138727 was the inhibitor, R-138727 concentrations were 0, 0.5, 2.5, 10, 50, and 100 μM.
The biotransformation of bufuralol to 1′-hydroxybufuralol was used as the marker activity for CYP2D6 (Ring et al., 1996). Incubations contained 0, 10, 50, 100, and 200 μM R-95913 and 5, 10, 25, 50, and 100 μM bufuralol when R-95913 was the inhibitor. When R-138727 was the inhibitor, 0, 0.7, 3.3, 13, 67, and 267 μM R-138727 were incubated with a concentration of bufuralol approximating the Km (5 μM) for the bufuralol 1′-hydroxylation. Additional components were 0.1 mg/ml microsomal protein and 1 mM NADPH.
The biotransformation of midazolam to 1′-hydroxymidazolam was used as one of the marker activities for CYP3A (Ring et al., 2001). Incubations contained 0, 10, 20, 40, and 60 μM R-95913 and 2.5, 5, 10, 25, and 50 μM midazolam when R-95913 was the inhibitor. When R-138727 was the inhibitor, 0, 0.5, 2.5, 10, 50, and 200 μM R-138727 were incubated with a concentration of midazolam approximating the Km (2.5 μM) for the midazolam 1′-hydroxylation. Additional components were 0.5 mg/ml microsomal protein and 1 mM NADPH.
The biotransformation of testosterone to 6β-hydroxytestosterone was used as the second marker activity for CYP3A. Incubation conditions were as described previously (Fayer et al., 2001). Incubations contained 0, 2.5, 100, 150, and 200 μM R-95913; 5, 25, 50, 150, and 250 μM testosterone; 0.375 mg/ml microsomal protein; and 1 mM NADPH. Coelution of R-138727 with 6β-hydroxytestosterone prevented inclusion of R-138727 as an inhibitor of testosterone 6β-hydroxylation.
Analytical Conditions. A variation of the method of Pang et al. (2005) was used for the analyses of samples containing R-138727 (Table 1). R-138727 was stabilized by derivatization with BMAP (10 mM; in excess of the combined R-138727 and GSH concentrations), which was considered to have proceeded to completion in all samples.
LC/MS/MS analytical methods
Conditions for LC/MS/MS analyses of samples containing acetaminophen, 4′-hydroxy S-mephenytoin, and 1′-hydroxymidazolam are also found in Table 1. HPLC sample analyses for 4′-hydroxydiclofenac (Ring et al., 2001) and 1′-hydroxybufuralol (Ring et al., 1996) formation were described previously. All LC/MS/MS analyses were conducted with a Sciex API 3000 (Applied Biosystems/MDS Sciex, Foster City, CA) equipped with a Shimadzu (Kyoto, Japan) LC10 AD pump, a Shimadzu SCL 10 AD VP controller, and a HTS PAL Leap Autosampler (CTC Analytics, Zwingen, Switzerland). For analysis of 6β-hydroxytestosterone formation, HPLC with UV detection was as described under “Analytical Method A” from Fayer et al. (2001) when R-95913 was the inhibitor.
Calculations. Enzyme kinetics of the formation of R-138727 from R-95913 were initially evaluated by visual examination of Eadie-Hofstee plots to assess whether one or more enzymes were involved (Enzyme Kinetics Module version 1.1 for SigmaPlot; SPSS Inc., Chicago, IL). Nonlinear regression analysis (Ring et al., 2001) was used to fit the Michaelis-Menten model [Segel (1975); eq. 1 to the formation rate data (WinNonlin, Version 3.1, Pharsight, Mountain View, California)] to determine apparent Km and Vmax parameters. 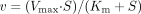 Intrinsic clearance (CLint) was determined with eq. 2:
Intrinsic clearance (CLint) was determined with eq. 2: 
Using eqs. 3 and 4, relative contributions of various P450s to total P450-mediated clearance were determined by scaling up CLint determined for expressed P450s based on abundance of P450s in the human liver (Hijazi and Boulieu, 2002): 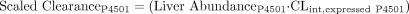
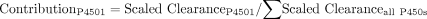
Correlation analyses were performed (JMP, Version 4.0.2; SAS Institute, Cary, NC) between the rates of R-138727 formation during incubation with 20 μM R-95913 and enzymatic activities or immunoquantified levels of various P450s in a human liver microsomal bank of up to 20 samples as described previously (Ekins et al., 1998; Ring et al., 2001). The form-selective catalytic activities or immunoquantified levels for CYP1A2 (phenacetin O-deethylation), CYP2A6 (coumarin 7-hydroxylation), CYP2B6 (immunoquantified levels), CYP2C8 (taxol 6-hydroxylation), CYP2C9 (diclofenac 4′-hydroxylation), CYP2C19 (S-mephenytoin 4′-hydroxylation), CYP2D6 (bufuralol 1′-hydroxylation), CYP2E1 (chlorzoxazone 6-hydroxylation), and CYP3A (midazolam 1′-hydroxylation) were used as possible coregressors.
For inhibition studies, the apparent kinetic parameters Vmax, Km, and Ki and the standard errors of the parameter estimates were determined using conventional relationships for reversible inhibition (Segel, 1975). The predicted in vivo inhibition by R-95913 of the catalytic activities of the examined P450s was calculated as follows (Segel, 1975; Ring et al., 2002): 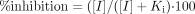 The Ki terms entered into this formula are those generated in this study. The inhibitor concentration (I) was the highest individual Cmax value observed for R-95913 in a clinical study after a 60-mg loading dose of prasugrel: 616 ng/ml [1.9 μM; N. Farid, C. Payne, and K. Winters (Eli Lilly and Co.), unpublished observations].
The Ki terms entered into this formula are those generated in this study. The inhibitor concentration (I) was the highest individual Cmax value observed for R-95913 in a clinical study after a 60-mg loading dose of prasugrel: 616 ng/ml [1.9 μM; N. Farid, C. Payne, and K. Winters (Eli Lilly and Co.), unpublished observations].
Results
Identification of the Enzymes Responsible for the Formation of R-138727 from R-95913. The rates of R-138727 formation over a range of R-95913 concentrations were determined in incubations containing human liver microsomal samples HLC, HLG, and HLH, each of which possessed a full complement of P450s (Ekins et al., 1998; Ring et al., 2001). Eadie-Hofstee plots (not shown) were monophasic for all three samples, consistent with a single enzyme predominating in the formation of R-138727. Samples HLC, HLG, and HLH exhibited apparent Km values of 21, 30, and 26 μM, respectively, for the formation of R-138727 (Table 2). The corresponding CLint values in these microsomal samples, calculated from eq. 2, ranged from 5.8 to 12 μl/min/mg protein (Table 2).
Kinetic parameters for the formation of R-138727 by human liver microsomal samples and expressed P450s
The conversion rates of R-95913 to R-138727 were determined in triplicate incubations as indicated under Materials and Methods. Values for Km and Vmax are reported as parameter estimate ± the standard error of the parameter estimate.
When the R-138727 formation rates were analyzed for correlation with the rates of form-selective catalytic activities or immunoquantified levels of the P450s (Table 3), the only regressor that exhibited statistical significance in relation to R-138727 formation was the activity associated with CYP3A, 1′-hydroxymidazolam formation (r2 = 0.98, p < 0.001), suggesting that CYP3A predominates in R-138727 formation.
Rates of formation of R-138727 from R-95913 by a bank of human liver microsomal samples
The ability of cDNA-expressed CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 to form R-138727 was evaluated after incubations containing 20 μM R-95913. In rank order of rates, CYP3A4>CYP2B6> CYP2C19≈CYP2C9 >CYP2D6 were all able to form R-138727 (Fig. 2). Michaelis-Menten kinetic parameters were determined [using eq. 1 for the four most active of these enzymes under linear rate conditions (Table 2)]. As demonstrated by the low Km value, 2.3 μM, R-95913 had the highest affinity for CYP2B6. The Km value for R-95913 interaction with CYP3A4 (21 μM) was the highest of the Km values for the four P450s and similar to the Km value observed for human liver microsomal samples (21 μM to ∼30 μM). The corresponding Vmax for CYP3A4-mediated R-138727 formation from R-95913 was higher than for the other three P450s. Considering the Km and Vmax for CYP2B6 and CYP3A4, CYP2B6 appears to be a high affinity, low capacity enzyme for R-138727 formation, whereas CYP3A4 appears to be a low affinity, high capacity enzyme.
The CLint values from the in vitro expressed P450 system were then scaled according to the relative abundances of each P450 in human liver microsomes determined in five different studies (Shimada et al., 1994, 1999; Lasker et al., 1998; Rodrigues, 1999; Hijazi and Boulieu, 2002), and the potential contributions from the respective enzymes to the clearance of R-95913 via the R-138727 formation route were estimated (Table 4). Overall, the projected contribution of CYP2B6 varied 18-fold, from 2% to 36%, depending on the source of the relative abundance data (Table 4). Estimates of the contributions of CYP2C9 (14% to 19%) and CYP2C19 (8% to 11%) varied only slightly and indicated relatively minor involvement in overall R-138727 formation by these enzymes (Table 4). The contribution of CYP3A4 was consistently projected to be the highest of the four enzymes, ranging from 38 to 70% (Table 4).
Scaling of in vitro CLint to project contributions of CYP2B6, CYP2C9, CYP2C19, and CYP3A to in vivo clearance of R-95913 via R-138727
Specific inhibitors of CYP2B6 (8 μl of inhibitory mAb), CYP2C9 (10 μM sulfaphenazole), CYP2C19 (10 μM omeprazole), and CYP3A4 (2 μM ketoconazole) were examined to determine their effects on the formation of R-138727 from R-95913 (20 μM) (Fig. 3). The effects of adding up to 8 μl of the CYP2B6 mAb to incubations were evaluated in comparison to control incubations containing the same volume of vehicle (ascites fluid). Although a range of volumes of mAbs were examined, maximal inhibition occurred in the presence of 8 μl of CYP2B6 mAb, yielding a consistent 48% to 52% inhibition in the four microsomal samples tested (Fig. 3). Sulfaphenazole produced only minor (12–32%) inhibition (Fig. 3). The effects of omeprazole varied from 29% inhibition to a 36% increase relative to controls without omeprazole (Fig. 3). Ketoconazole inhibited R-138727 formation activity 33% to at least 86% (Fig. 3). The maximal inhibition (≥86%), observed for HLL, was calculated based on the lower limit of quantitation for R-138727, although the level of R-138727 formed was actually below the lower limit of quantitation.
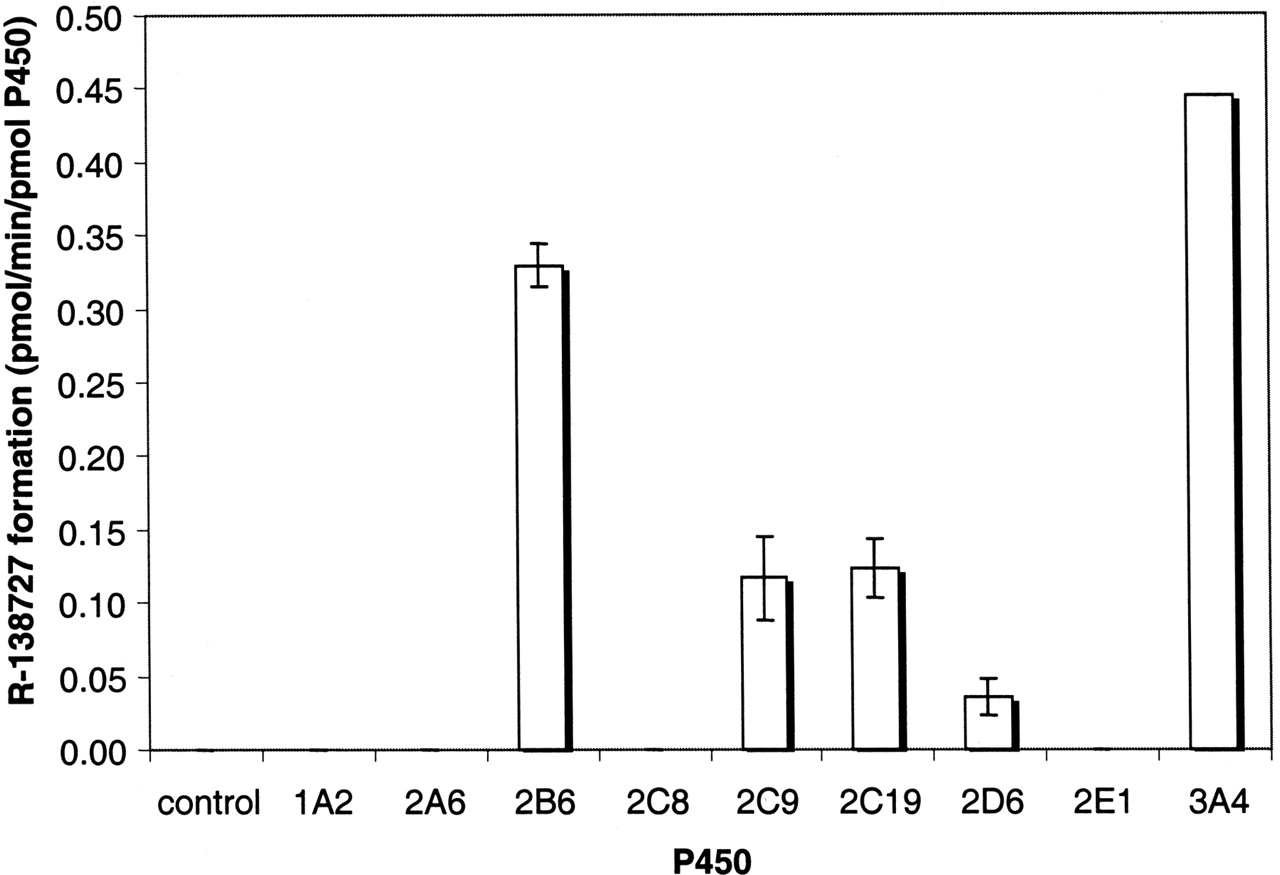
Formation of R-138727 by human lymphoblast-expressed P450s. Results are presented as means of triplicate results ± S.D. (except for CYP3A4, where the mean of duplicate results is shown).
Inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A by R-95913 and R-138727. In vitro, R-95913 was found to inhibit CYP2C9, CYP2C19, CYP2D6, and CYP3A. The inhibition of CYP2C9-mediated diclofenac 4′-hydroxylation, CYP2C19-mediated S-mephenytoin 4′-hydroxylation, and CYP3A-mediated midazolam 1′-hydroxylation by R-95913 best fit the competitive inhibition model, yielding respective apparent Ki values of 82 μM, 7.2 μM, and 13 μM (Table 5). The noncompetitive inhibition model was the best fit to the data for inhibition of CYP2D6-mediated bufuralol 1′-hydroxylation and CYP3A-mediated testosterone 6β-hydroxylation by R-95913, yielding apparent Ki values of 41 μM and 56 μM, respectively (Table 5). Using these Ki values and a plasma concentration of 1.9 μM (as described under Materials and Methods) in eq. 5 yielded estimates of predicted in vivo inhibition ranging from 2% to 21% (Table 5).
In vitro inhibition of P450 form-selective catalytic activities and predicted in vivo inhibition of P450s by R-95913
The ability of R-95913 to inhibit CYP1A2-mediated phenacetin O-deethylation and of R-138727 to inhibit all the P450s was examined in incubations containing substrate concentrations approximating the Km values for the respective reactions. Inhibition at the highest concentration of inhibitor (200 μMto267 μM) did not exceed 50% (data not shown). Thus, inhibition with these substrate/inhibitor combinations was deemed nonsubstantial, and full Ki determinations were not performed. Due to interference in the HPLC chromatograms, R-138727 could not be tested as an inhibitor of testosterone 6β-hydroxylation.
Discussion
After oral administration to humans, prasugrel is not detected in plasma because of rapid deesterification to R-95913 (Farid et al., 2005b). The active metabolite of prasugrel, R-138727, is then generated from R-95913 via P450s. In the current study, the P450s involved in the biotransformation of R-95913 to R-138727 were identified and the relative contributions of those enzymes to R-138727 formation were projected. The second series of experiments explored the potential for R-95913 and R-138727 to cause drug-drug interactions resulting from inhibition of the major P450s involved in drug metabolism. This information, collectively, can potentially be used to evaluate 1) possible pharmacokinetic variability in the formation of prasugrel metabolites within the human population; 2) pharmacokinetic/pharmacodynamic relationships for prasugrel metabolites; and 3) the effects of coadministered drugs on R-138727 formation and the effects of the circulating metabolites of prasugrel on the metabolism of coadministered drugs.

Effects of an inhibitory mAb to CYP2B6 and chemical inhibitors selective for CYP2C9 (sulfaphenazole), CYP2C19 (omeprazole), and CYP3A (ketoconazole) on R-138727 formation by four human liver microsomal samples. The CYP2B6 inhibition study was conducted with human liver samples HLB, HLD, HLG, and HLP, whereas the CYP2C9, CYP2C19, and CYP3A inhibition study was conducted with human liver samples HLB, HLD, HLG, and HLL.
Inclusion of GSH in incubations of R-95913 was required, consistent with in vitro studies in which the active thiol metabolites of clopidogrel (Savi et al., 2000; Pereillo et al., 2002) and ticlopidine (Yoneda et al., 2004) have been detected. To measure the chemically unstable, free thiol-containing R-138727, stabilization of R-138727 in the reduced form also required postincubation derivatization with 10 mM BMAP. The ratio of GSH in the incubations to BMAP in the stop solution was optimized and the derivatization was found to be relatively specific. Glutathione/BMAP mixed disulfides were observed, but in relatively small amounts. In addition, low levels of glutathione/R-138727 mixed disulfides were observed, but R-138727 homodimers were never detected. Therefore, with the addition of excess BMAP, the derivatization of R-138727 with BMAP was considered to have proceeded to completion in all samples.
The formation of R-138727 by five of nine expressed P450s established that multiple P450s may contribute to R-138727 formation in vivo. Upon gauging the relative efficiency of this conversion by four of these P450s with the kinetic parameter CLint, CYP2B6 was the most efficient enzyme involved, followed by CYP2C19, CYP3A, and CYP2C9. However, to account for the varying quantities of the relevant P450s in the liver, an exercise was conducted in which each CLint estimate was adjusted using the relative hepatic abundances of the P450s (Table 4). Because the P450 contents reported in the literature vary extensively, multiple determinations were made, and a range of potential in vivo contributions was then projected (Table 4). The CYP2B6 levels reported in the literature varied 39-fold, with the low estimate (1 pmol/mg) from Shimada et al. (1994), and the high (39 pmol/mg) estimate from Rodrigues (1999). CYP2B6 expression has been shown to vary with genotype, sex, and ethnicity (Lamba et al., 2003), but a more likely explanation for the extensive variability in reported CYP2B6 abundance has to do with the antibodies used for protein detection (reviewed in Stresser and Kupfer, 1999). CYP2B6 antibody sensitivity and specificity have improved in recent years, with later studies (for instance, Code et al., 1997; and Ekins et al., 1998) indicating measurable CYP2B6 in all samples, and higher maximal and average levels than reported in earlier studies. As the wide-ranging estimates of CYP2B6 contribution to R-138727 formation (2% to 36%; Table 4) show, the ability to robustly measure CYP2B6 greatly influences the projected in vivo outcome, and, in the case presented here, use of the higher abundance value for CYP2B6 (determinations B and E; Table 4) resulted in a compensatory reduction in the projected contributions for CYP3A. In fact, in determination E (Table 4), the projected contributions from CYP2B6 and CYP3A are similar. With CYP2B6 being the more efficient catalyst of the R-138727 reaction (CLint = 0.170 versus 0.065; Table 2), there may be cases in which individuals possess high levels of CYP2B6 versus CYP3A, and CYP2B6-mediated formation of R-138727 predominates. The inherent uncertainty in these scaling exercises is a major reason why a multi-pronged in vitro approach is used for a comprehensive picture of metabolism and drug-drug interaction potential (Ring and Wrighton, 2000).
In the earliest reports of the relative abundances of the P450s in the human liver (Shimada et al., 1994, 1999), the individual contributions of CYP2C9 and CYP2C19 were not reported. Instead, a total CYP2C content was indicated. Therefore, in two of the five determinations of the contributions of CYP2C9 and CYP2C19 to the formation of R-138727, the total CYP2C content was adjusted, using the results of Lasker et al. (1998), to reflect the content of the individual CYP2C enzymes (Table 4). Even after these adjustments, CYP2C9 and CYP2C19 abundance estimates varied only 1.9-fold, much less than the reported CYP2B6 abundances. The reported average CYP3A abundances were consistently high and also varied only 1.9-fold. Upon scale-up, the ranges of projected contributions from each enzyme varied 19-fold for CYP2B6, but only 1.4- to 1.8-fold for CYP2C9, CYP2C19, and CYP3A. Contributions to R-138727 formation from CYP2C9 and CYP2C19 were projected to be relatively minor in all cases, whereas substantial CYP3A contributions were consistently predicted.
Inhibition of R-138727 formation by the chemical inhibitors selective for CYP2C9 and CYP2C19 was minimal and consistent with the results of the expressed enzymes experiment and the abundance-scaling exercises. Inhibition by the mAb to CYP2B6 was consistently near 50% (Fig. 3) across the four microsomal samples tested, confirming a substantial involvement of CYP2B6 in R-138727 formation. In addition, ketoconazole, the selective CYP3A inhibitor, inhibited R-138727 substantially (more than approximately 50%) for three of the four microsomal samples tested, confirming the involvement of CYP3A in R-138727 formation. In summary, the overall results for the formation of R-138727 indicate substantial contributions of CYP3A and CYP2B6, with CYP3A predominating in most cases and CYP2B6 predominating in certain individuals.
The inhibitory potentials of the major circulating metabolites of prasugrel, R-95913 and R-138727, toward CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A were also measured. Inhibition by R-138727 did not exceed the ≥50% threshold for Ki determinations for any of the form-selective activities in vitro. Consequently, R-138727 would not be predicted to be an inhibitor in vivo. Conversely, R-95913, in vitro, inhibited the catalytic activities for all the P450s tested except CYP1A2. To put these results into perspective, a conservative estimate of an R-95913 concentration in human plasma (1.9 μM, the highest individual Cmax achieved after a 60-mg prasugrel loading dose) was used to relate the in vitro Ki values to inhibition in vivo. Inhibition of CYP2C19, the enzyme most potently affected in vitro, was predicted to affect metabolism of other CYP2C19 substrates in vivo by only 21%. In addition, because CYP3A contributes to the clearance of so many drugs, potential inhibition of CYP3A-mediated metabolism by R-95913 was a concern. However, CYP3A was predicted to be affected only a maximum of 13% by R-95913 in vivo. CYP2C9 and CYP2D6 were predicted not to be affected by R-95913 in vivo. Thus, neither R-95913 nor R-138727 is expected to have a clinically meaningful impact on the P450-mediated metabolic clearance of other drugs.
These studies demonstrate that R-138727, the active metabolite of prasugrel, is produced from R-95913 by multiple P450s. CYP3A appears to be the enzyme most predominant in R-138727 formation, based on the high degree of correlation between R-138727 formation activity and midazolam-1′-hydroxylation activity, the substantial inhibition of R-138727 formation by ketoconazole in human liver microsomal samples, and the consistently high (38–70%) contribution predicted from the abundance scaling exercise. CYP2B6 may also contribute substantially to R-138727 formation in some individuals, as evidenced by consistent substantial inhibition of R-138727 formation in the presence of mAb to CYP2B6 and the moderate to low contribution predicted from the abundance scaling exercise. The scale-up exercises reinforced the finding that CYP3A and possibly CYP2B6 play substantial roles in R-138727 formation. In addition, CYP2C9, CYP2C19, and CYP2D6 were capable of forming R-138727 and, as estimated by scaling, may contribute as much as 10% to 20% to R-138727 formation. Therefore, in vivo, it was predicted that, should any single P450 involved in R-138727 formation be absent or inhibited by a coadministered drug, other P450s would remain capable of forming R-138727. Thus, therapeutic failure due to lack of formation of the active metabolite of prasugrel would be unlikely. This hypothesis was proven in a clinical drug-drug interaction study in which the potent CYP3A inhibitor ketoconazole was coadministered with prasugrel but affected neither the exposure to active R-138727 over 24 h nor the antiplatelet effects of prasugrel (Farid et al., 2005a). Furthermore, predictions of in vivo inhibition potential indicate that neither R-95913 nor R-138727 is expected to substantially inhibit the CYP1A2-, CYP2C9-, CYP2C19-, CYP2D6-, or CYP3A-mediated metabolism of coadministered drugs. In total, this series of in vitro studies indicates that drug-drug interaction liability with prasugrel therapy appears minimal.
Footnotes
-
A portion of this work was presented at the 7th International ISSX Meeting, Vancouver, BC, Canada, 2004 and was published in abstract form in Drug Metab Rev36 (Suppl 1): 342.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.007989.
-
ABBREVIATIONS: prasugrel, (±)-2-[2-acetyloxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone hydrochloride; R-95913, 2-[2-oxo-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone; R-138727, 2-[1-2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid; P450, cytochrome P450; LC/MS/MS, liquid chromatography/tandem mass spectrometry; GSH, reduced glutathione; BMAP, 2-bromo-3′-methoxyacetophenone; mAb, monoclonal antibody; HPLC, high-performance liquid chromatography; CLint, intrinsic clearance.
- Received October 31, 2005.
- Accepted January 11, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}