


Abstract
A new metabolic scheme of flutamide is proposed in this article. Some patients treated with flutamide, a nonsteroidal antiandrogen, have developed severe hepatic dysfunction. Toxic metabolites have been proposed to be responsible for these negative effects. In this study, the qualitative aspects of the in vitro metabolism of flutamide in liver microsomes from human, dog, pig, and rat were evaluated. A direct comparison of the flutamide metabolism in liver and prostate microsomes from pig was made, and the in vivo metabolism of flutamide was investigated in urine from orally treated prostate cancer patients. Liquid chromatography/tandem mass spectrometry was used for analysis. The mass spectrometer was equipped with an electrospray interface and operated in the negative ion mode. In liver microsomes from pig, dog, and rat, extensive hydroxylation of flutamide occurred. One, two, or three hydroxy groups were attached, and isomeric forms were detected for both monohydroxylated and trihydroxylated drug. In pig liver microsomes, isomers of a third metabolite, hydroxylated 4-nitro-3-(trifluoromethyl)-aniline, were also found after incubation with either flutamide or 2-hydroxyflutamide. In human liver microsomes, the pharmacologically active 2-hydroxyflutamide was the only metabolite detected. Several phase I metabolites as well as four intact phase II metabolites could be recovered from the urine samples. For the first time in humans, glucuronic acid conjugates of hydroxylated 4-nitro-3-(trifluoromethyl)-aniline, and mono- and dihydroxylated flutamide were identified, together with hydroxylated 4-nitro-3-(trifluoromethyl)-aniline conjugated with sulfate. In addition, one mercapturic acid conjugate of hydroxylated flutamide, probably formed from flutamide via a reactive intermediate, was detected.
Flutamide was the first pure nonsteroidal antiandrogen compound discovered. After oral administration, flutamide is readily metabolized to its active metabolite, 2-hydroxyflutamide, which appears to be mainly responsible for the pharmacological effect (Brogden and Chrisp, 1991; Farthing et al., 1994). It acts as a competitive antagonist that blocks the growth-stimulating effects of androgens (testosterone and dihydrotestosterone) at the androgen receptor in the prostate (Brogden and Chrisp, 1991; Labrie, 1993) and has been widely used in the treatment of prostate cancer since the 1970s. Cancer itself represents the second leading cause of death in men, and prostate cancer alone ranks number five in overall causes of death among men in the United States (Chan et al., 2004).
Some patients treated with flutamide orally have developed severe hepatic dysfunction at doses above the recommended dose, i.e., 250 mg three times daily, and minor hepatoxicity has been documented with oral doses as low as 250 to 375 mg/day (Ibanez et al., 2005). Because the dysfunction seems to be dose-dependent and the liver has been reported to recover after flutamide withdrawal (Aizawa et al., 2003), an improved characterization of the flutamide metabolism may contribute to the understanding of this safety issue. Both flutamide and potentially formed toxic metabolites have been proposed to be responsible for these negative effects, possibly due to affected mitochondrial respiration (Fau et al., 1994; Aizawa et al., 2003; Takashima et al., 2003; Thole et al., 2004).
Phase I metabolites have been identified in vitro (Shet et al., 1997) and in vivo in plasma and urine from humans (Katchen and Buxbaum, 1975). It has been shown that CYP1A2 is the dominant enzyme forming 2-hydroxyflutamide, but CYP1A1 and CYP1B1 may also be involved (Shet et al., 1997). The formation of 4-nitro-3-(trifluoromethyl)-aniline (Flu-1), a metabolite produced by carboxyesterase, has been suggested to be directly involved in the toxicity (Aizawa et al., 2003). Katchen and Buxbaum (1975), who treated urine from flutamide patients with glucuronidase and sulfatase, were the first to propose conjugation reactions, and later, a sulfate-conjugated metabolite was recovered in urine from rats (Asakawa et al., 1995b). However, to our knowledge, intact phase II metabolites of flutamide have not been previously identified in humans. A schematic presentation of the phase I metabolites already identified, together with their short names, is provided in Fig. 1.

In this study, liquid chromatography (LC) electrospray ionization (ESI) tandem mass spectrometry (MS/MS) was the analytical technique used, and the mass spectral data obtained for the metabolites were compared with those of the available synthetic standards. Detection by mass spectrometric methods allows low detection limits and high selectivity, and by the use of tandem MS, structural information about the compounds can be obtained. A complementary technique to determine the elemental composition of drugs and their metabolites is accurate mass determination by high resolution MS. Because a smaller experimental mass error results in a smaller set of possible elemental compositions, accurate mass determination is very useful in structural evaluation of drug metabolites.
The aim of the present study was, first, to qualitatively evaluate the in vitro metabolism of flutamide in liver microsomes from the four species dog, human, pig, and rat and to compare the qualitative aspects of metabolism of flutamide in pig liver and prostate microsomes. Second, the in vivo metabolism of flutamide was investigated by identification of metabolites in urine from prostate cancer patients treated with oral doses of flutamide, and a new metabolic scheme is proposed.
Materials and Methods
Chemicals. Sigma (Steinheim, Germany) supplied the synthetic standards of flutamide and 4-nitro-3-(trifluoromethyl)-aniline, MIKROMOL GmbH (Luckenwalde, Germany) supplied the synthetic standard of 2-hydroxyflutamide, and β-glucuronidase 1585665 from Escherichia coli K12 was purchased from Roche (Bromma, Sweden). NADPH regenerating system solutions A and B (No. 451220 and 451200, respectively) were purchased from BD Bioscience (Stockholm, Sweden). The water was purified using a Milli-Q water purification system (Millipore, Bedford, MA). All other chemicals were of analytical grade or better and used without further purification.
Microsomal Origin, Preparation, and Incubation. Liver microsomes from human (male), dog (male), and rat (male) were purchased from In Vitro Technologies Inc. (Baltimore, MD). Pig (male) liver and prostate microsomes were prepared as described elsewhere (Andersson et al., 1985; Norlin, 2002).
Incubations of liver microsomes from humans, dogs, pigs, and rats were performed at 37°C for 120 min. Microsomes from pig liver and prostate were incubated at 37°C for 80 min. All experiments were started by preincubation of Tris buffer (50 mM, pH 7.4) with microsomes (1 mg/ml) and flutamide or 2-hydroxyflutamide (5 μM in methanol) at 37°C. The reaction was initiated by the addition of NADPH regenerating systems (50 μl of solution A and 10 μl of solution B, total volume 1 ml). The incubations were terminated by the addition of 2 volumes of ice-cold methanol.
Sample Preparation.Microsomes. After termination of the incubation, 1 ml was removed and centrifuged at 3600 rpm for 10 min, and the supernatant was transferred to a fresh test tube and evaporated to dryness under nitrogen at 60°C. The residue was dissolved in 100 μl of 0.1% formic acid/acetonitrile (75:25) and transferred to an LC vial before analysis.
Prostate Cancer Patient Urine. After individual approval, urine from three prostate cancer patients was collected before curative brachytherapy. The patients were treated with oral 250-mg (b.i.d.) doses of flutamide and were at the steady-state plasma level of 2-hydroxyflutamide (the active metabolite). The patients had not taken any other drugs considered likely to interfere with the analysis.
Before the analysis of phase I metabolites, the urine was treated with β-glucuronidase. To 3 ml of urine, 1 ml of potassium phosphate buffer (0.1 M, pH 6.1) was added and pH was measured to 6.6. β-Glucuronidase was added (75 μl), and the test tubes were placed in a water bath for 1 h (50°C, horizontal shaking). After centrifugation (3600 rpm, 10 min), the supernatant was transferred to LC vials. The urine was treated in the same way except for the addition of β-glucuronidase in the search for conjugates with glutathione, sulfate, and glucuronic acid.
Instrumentation.Q-Tof Mass Spectrometry. A Jasco (Tokyo, Japan) PU-980 was used to pump the mobile phase consisting of 0.1% formic acid in water/acetonitrile (50:50), flow rate 200 μl/min, through the guard column (ODS-C18, 4 mm × 2.0 mm; Scandinavian GeneTech, Västra Frölunda, Sweden) and the analytical column (Zorbax Eclipse XDB-C18, 5 μm, 2.1 mm × 50 mm; Agilent, Partille, Sweden).
The mass spectrometer used for the accurate mass measurements was an ESI hybrid quadrupole/time of flight instrument (Q-Tof Model I, upgraded with a 3.6-GHz time-to-digital converter card; Micromass, Manchester, UK) equipped with an orthogonal electrospray source (Z spray) operated in the negative ion mode. The source block and desolvation temperatures were 120 and 350°C, respectively, the capillary and cone voltage were -3000 and -40 V, respectively, and argon was used for collision-induced dissociation (CID; the inlet pressure was set to 20 psi).
Calibration was performed every day by a constant infusion of a sodium iodide solution (2 mg/ml in 2-propanol/water 50:50) at 1 μl/min. The argon gas was turned on and the collision energy was set to 4 eV when data were collected for 1 min from m/z 100 to m/z 800. Spectra were combined and smoothed. To make sure that no dead time distortion was present, the TOF constants, resolution, and number of pushes correction factor (Np multiplier), were first set to zero. Then the procedure was repeated with the TOF constants set to 5000 and 0.7 for resolution and Np multiplier, respectively (m/z values for both centered spectra should be the same). According to the recommendations from the manufacturer, a polynomial of order four was used for the calibration.
The collision energy was set to 35 eV when CID mass spectra of synthetic standards of flutamide and 2-hydroxyflutamide dissolved in 0.1% formic acid in water/methanol 50:50 (concentrations 18 and 17 μM, respectively) were obtained by the Q-Tof mass spectrometer. The samples were introduced to the system manually through a 20-μl injection loop (Rheodyne 7125; Rheodyne, Cotati, CA). The internal lock mass solution used was a mixture of sodium iodide, d-tryptophan, and acetylsalicylic acid (m/z 276.7987, 1.11 mM; m/z 203.0821, 10.6 mM; and m/z 179.0345, 74.4 mM, respectively), infused at a flow rate of 20 μl/min through a T-connection, where it was mixed with the LC mobile phase (see above). The software Masslynx version 3.4 (Micromass) combined the spectra, and the internal calibrant peak (lock mass) was chosen to be as close as possible to the m/z of the sample peak of interest. The software allowed the use of one lock mass only. The dead time distortion was evaluated as above.
Triple Quadrupole Mass Spectrometry. The system described below was used for the analysis of microsome and urine samples throughout this study.
Solvents in the liquid phase were 0.1% formic acid in water (A) and acetonitrile (B), and the gradient program was: 0 to 2 min, 25% B; 2 to 10 min, 25 to 90% B; 10 to 12 min, 90% B; 12 to 14 min, 90 to 25% B. The mobile phase was delivered at a flow rate of 200 μl/min by a Surveyor MS pump (Thermo Electron Corp., San Jose, CA). The guard and analytical columns used were the same as in the Q-Tof section. A sample volume of 10 μl was injected by an HTC PAL auto sampler (CTC Analytics AG, Zwingen, Switzerland). MS detection was achieved by a Finnigan TSQ Quantum Ultra mass spectrometer (Thermo Electron Corp.) equipped with an electrospray interface. Instrument control, data acquisition, and data processing were carried out with the Xcalibur software, version 1.3 (Thermo Electron Corp.). The MS parameters were optimized for sensitivity automatically during direct infusion of a flutamide solution in methanol. This solution was mixed with the LC flow through a connecting T. The instrument was operated in the negative ion mode. The total ion chromatogram of LC/MS was obtained by scanning the first quadrupole from m/z 50 to 500 in a 1-s scan time. In CID, the deprotonated molecules ([M - H]-) were transmitted through the first quadrupole into the collision cell that contained argon gas at 1.5 mTorr pressure. The last quadrupole was scanned from m/z 50 to 500 in 1.0 s, resulting in a product ion chromatogram. During analysis, the sheath and auxiliary gas was nitrogen at 30 and 2 mTorr pressure, respectively, the capillary temperature was 250°C, the spray voltage was -3000 V, and the collision energy was 22 V unless otherwise stated.
The Search for Metabolites. The search for flutamide metabolites was performed manually by interpretation of mass spectra and chromatograms. Mass spectral information of samples, blank samples, and synthetic standard solutions of flutamide, 2-hydroxyflutamide, and 4-nitro-3-(trifluoromethyl)-aniline were compared by looking at peaks in the total ion chromatogram and known mass shifts; for example, +16 for hydroxylation and +176 for glucuronidation. Neutral loss scans of 80 and 176 were used to recognize conjugations by sulfate and glucuronic acid, respectively. In the search for glutathione conjugates, neutral loss scans of 75, 129, and 146 were used.
Results
Accurate Mass Determination of MS/MS Fragments from Flutamide and 2-Hydroxyflutamide Standards. The fragments produced from the flutamide and 2-hydroxyflutamide standards are summarized in Tables 1 and 2, respectively. Sternal and Nugara (2001) identified fragments corresponding to B and E in positive fast atom bombardment, but the other fragments have not been reported in the literature before. Fragment C (I) was a common fragment for the two compounds but was produced to a greater extent from 2-hydroxyflutamide. In flutamide, D is the dominating fragment. The accurate mass data indicate that fragment D was produced by a net loss of C3H7NO, which corresponds to one propyl radical and one NO radical. The latter one could be formed from the aromatic nitro group (Madhududanan, 1996). This fragment (m/z 202) could not be found in 2-hydroxyflutamide, possibly because it is less likely that the hydroxy propyl group is lost compared with the propyl group. The loss of NO2 and HF can be observed in both substances, resulting in fragments B and K, and F and L, respectively.
Accurate mass measurement of the fragments after CID of a flutamide standard solution (18 μM) For LC and MS settings, see Materials and Methods.
Accurate mass measurement of the fragments produced after CID of a 2-hydroxyflutamide standard solution (17 μM) For LC and MS settings, see Materials and Methods.

LC-ESI-MS/MS of m/z 205. Top, spectrum from a rat liver microsome sample after incubation with flutamide. Bottom, spectrum from the synthetic standard of 4-nitro-3-(trifluoromethyl)-aniline, 10 μg/ml. For more information, see Materials and Methods.
In Vitro Metabolism in Liver Microsomes from Dog, Human, Pig, and Rat. 2-Hydroxyflutamide was the only metabolite found in the liver microsomes from all four species. Interestingly, in liver microsomes from the three animals, either two or three isoforms of hydroxyflutamide were detected (Table 3). These isomers have not previously been reported.
Metabolites found in liver microsomes from four species, human, pig, dog, and rat The microsomal fractions were incubated with flutamide for 120 min before termination. Fore more information, see Materials and Methods.
In rat liver microsomes M1, Flu-1, three isomeric forms of hydroxyflutamide, and two isomers of M3 were detected. The chromatographic peak for m/z 205 ([M - H]- of Flu-1) eluted at 7.1 min. Both the retention time and fragmentation pattern, produced after collision-induced dissociation, were consistent with findings from the synthetic standard of 4-nitro-3-(trifluoromethyl)-aniline (Fig. 2). Fragmentation of the parent ion [M - H]-m/z 291, which corresponds to the mass-to-charge ratio of flutamide plus one hydroxylation, results in the major fragment m/z 205. This means that the hydroxy group is positioned at the aliphatic side chain of flutamide. There were two peaks in the extracted product ion chromatogram for m/z 205 (at 6.0 and 7.1 min in Fig. 3), indicating two isomeric forms of aliphatic hydroxyflutamide. Interestingly, our findings also suggest aromatic hydroxylation on the intact flutamide structure in rat liver microsomes, resulting in the fragment m/z 221 (at 8.1 min in Fig. 3). The fragment m/z 205 visible in Fig. 3 is probably due to overlap with the more abundant peak at 7.1 min (metabolite with aliphatic hydroxylation). In total, three isomers of monohydroxylated flutamide could be separated chromatographically at 6.0, 7.1, and 8.1 min, respectively. The peak that eluted at 7.1 min was consistent in retention time and mass spectrum with the synthetic standard of 2-hydroxyflutamide. Because the mass spectral information after CID indicated that the metabolite at 8.1 min was aromatically hydroxylated flutamide, the peak at 6.0 min probably represented 3-hydroxyflutamide. For m/z 307 ([M - H]- of M1), the major fragment after CID was m/z 205, indicating that both the hydroxy groups were positioned on the aliphatic side chain of flutamide (Fig. 4A). In the case of m/z 323 ([M - H]- of M3), there were two peaks in the chromatogram (at 1.8 and 5.4 min), and the two main fragments observed after CID were m/z 205 and m/z 221, respectively, suggesting that either all three hydroxy groups can be aliphatic or two of them aliphatic and the third aromatic (results not shown).

Sample from rat liver microsomes incubated with flutamide for 120 min. Left, extracted ion chromatograms for m/z 291, m/z 221, and m/z 205 after LC-ESI-MS/MS of m/z 291. Right, spectra from the chromatographic peaks at 8.1 and 7.1 min, respectively. For more information, see Materials and Methods.
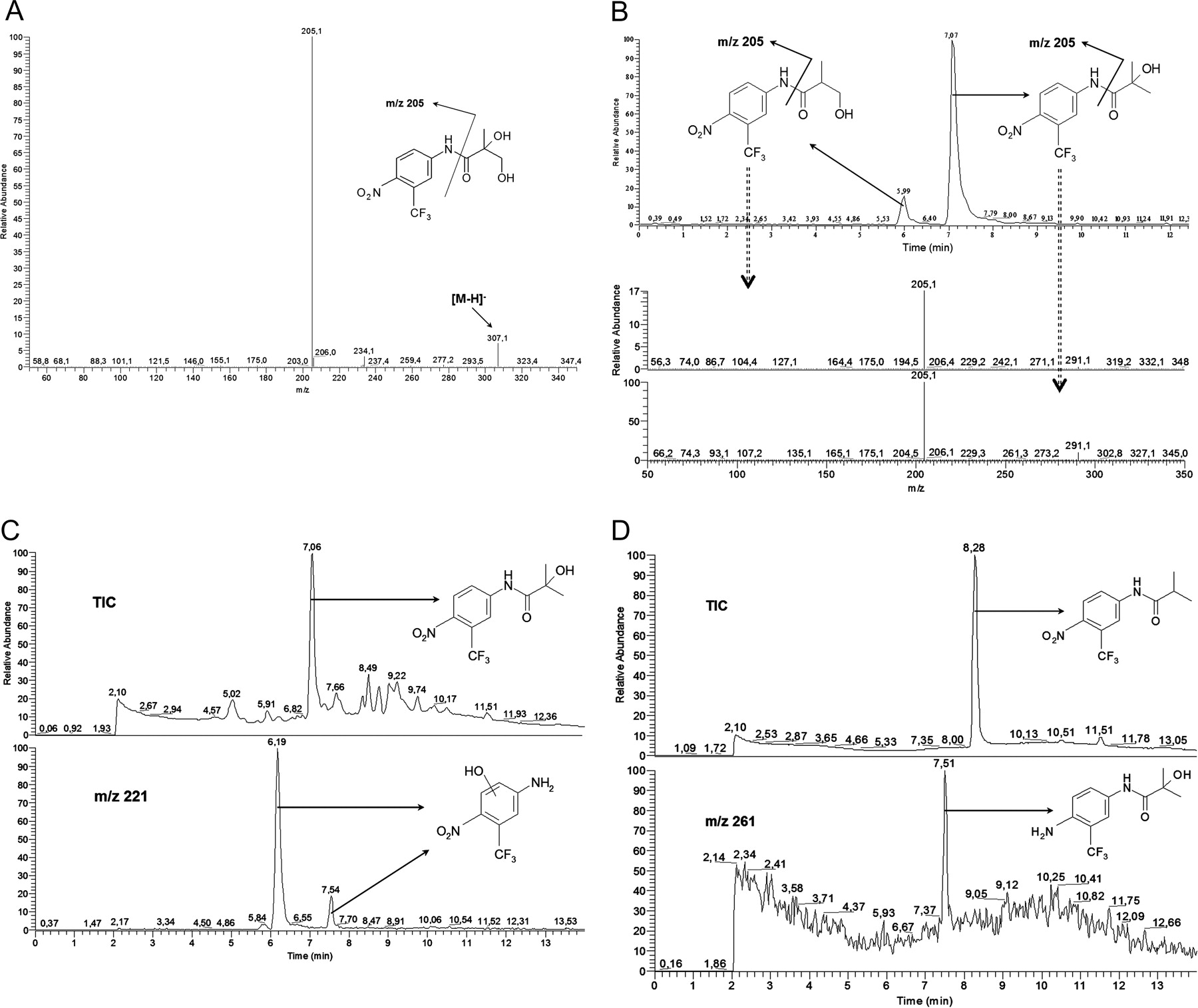
Dog liver microsomes were similar to rat liver microsomes regarding the qualitative aspects of flutamide metabolism. For instance, Flu-1, M1, M3, and hydroxyflutamide (aliphatic and aromatic) were observed in both species. According to the retention time, the aliphatic hydroxyflutamide metabolite was consistent with 2-hydroxyflutamide (results not shown). In pig liver microsome fractions incubated with flutamide, Flu-1, M1, and aliphatic M3 were also detected. Additionally, two isomers of aliphatic hydroxyflutamide were observed. Both the peaks for m/z 291 in the chromatogram resulted in the major fragment m/z 205 (Fig. 4B), indicating that hydroxylation can occur on either the tertiary or primary aliphatic carbon (cf. Figure 4A).
In liver microsomes from humans, the only metabolite found was 2-hydroxyflutamide, as previously reported (Shet et al., 1997). However, the two minor metabolites that were detected but not identified by Shet et al. (1997) could not be detected in this study.
Comparison between the Pig Liver and Prostate in Vitro Metabolism. The in vitro metabolism in the microsomes from pig liver and prostate was compared after incubation of either flutamide or 2-hydroxyflutamide. In liver microsomes, three mass-to-charge ratios matching known flutamide metabolites were found after flutamide incubation. These were m/z 291, 307, and 221, corresponding to [M - H]- for hydroxyflutamide, M1, and Flu-3, respectively. There were two peaks in the extracted ion chromatogram for m/z 291, at 6.0 and 7.1 min, indicating isomeric forms of hydroxyflutamide. In the extracted ion chromatogram for m/z 221, corresponding to [M - H]- for Flu-3, there were two peaks at 6.2 and 7.5 min, respectively. To our knowledge, this is the first report of isomeric forms of the metabolite called Flu-3 (cf. Fig. 1). Hydroxylated Flu-1 might be a more correct nomenclature for the metabolites formed, but to reduce the confusion, we will continue to call the metabolite Flu-3 throughout this paper, without any conclusion of the exact position of the hydroxy group. Peaks corresponding to [M - H]- for M1 and Flu-3 were also discovered after incubation with 2-hydroxyflutamide, which indicated that these two metabolites could be formed by further metabolism of 2-hydroxyflutamide. There were, again, two peaks in the extracted ion chromatogram for m/z 221 at 6.2 and 7.5 min, respectively, indicating two isomeric forms of Flu-3 (Fig. 4C).
Metabolism in the pig prostate microsomes was also investigated by incubation with flutamide or 2-hydroxyflutamide. After flutamide incubation, m/z 261, indicating [M - H]- of Flu-5, eluted at 7.5 min (Fig. 4D), and one isomer of hydroxylated flutamide was detected, for which the retention time was consistent with 2-hydroxyflutamide. Chromatographic peaks at 6.2 and 7.5 min for m/z 221 again indicated two isomers of the Flu-3 metabolite after incubation with 2-hydroxyflutamide (results not shown).
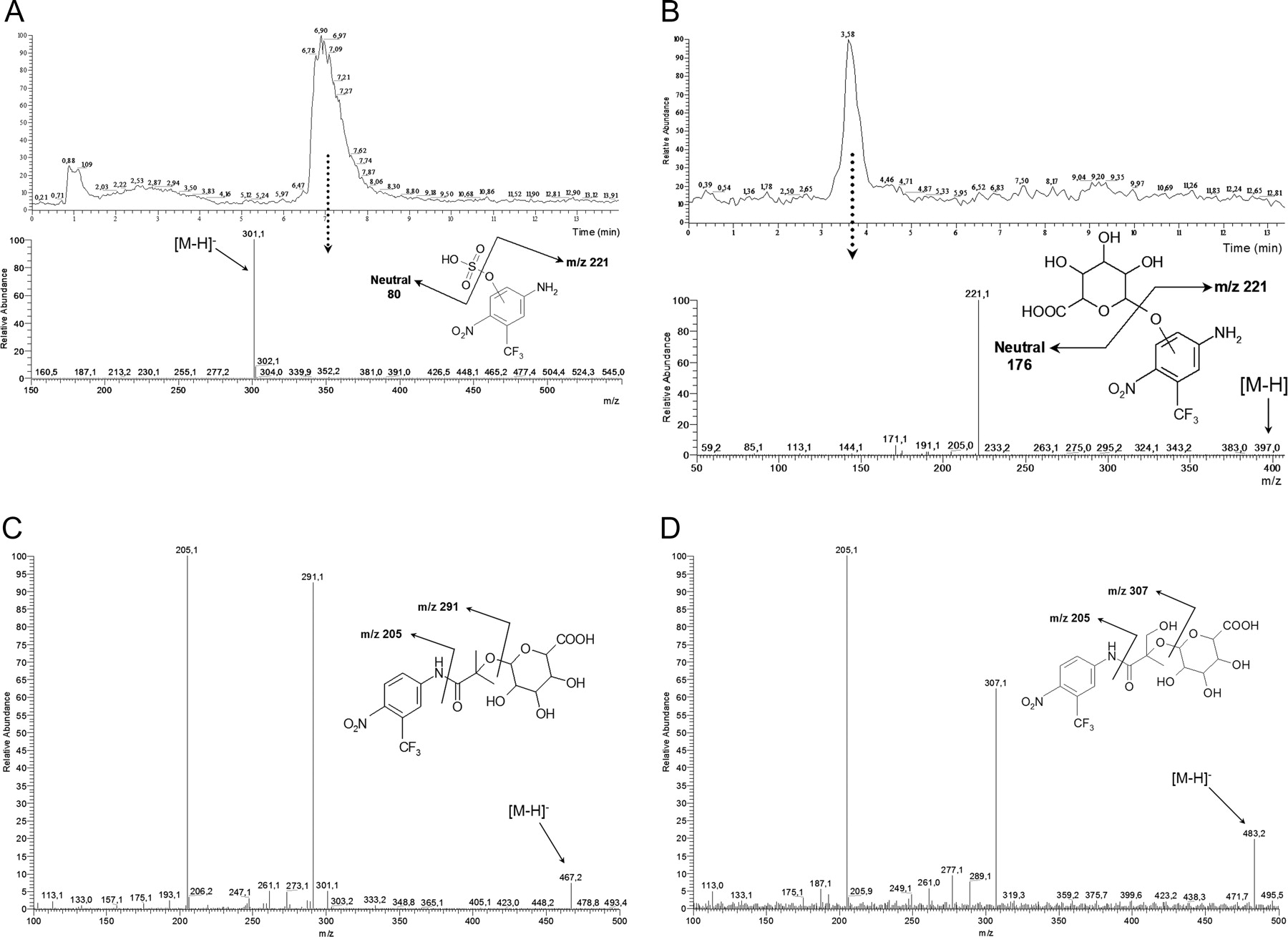
Urine from Patients Treated with Flutamide. In the present study, several phase I and phase II metabolites were recovered in urine from prostate cancer patients treated with oral 250-mg doses (b.i.d.) of flutamide (Fig. 5). After hydrolysis with β-glucuronidase, a peak corresponding to m/z 221 ([M - H]- of Flu-3) eluted at 5.9 min. Neutral loss scans of 80 and 176 of an untreated urine sample indicated that Flu-3 could be conjugated with either sulfate or glucuronic acid, resulting in m/z 301 or m/z 397, respectively. CID of the precursor ions of these phase II metabolites resulted in m/z 221, i.e., [M - H]- for Flu-3 (Fig. 6, A and B).

A, LC-ESI-MS/MS spectrum after CID of m/z 307, corresponding to [M - H]- of dihydroxylated flutamide. The sample injected was from rat liver microsomes incubated with flutamide for 120 min. The peak eluted at 5.0 min. B, LC-ESI-MS/MS of m/z 291, corresponding to [M - H]- of hydroxylated flutamide. Pig liver microsome sample after incubation with flutamide for 120 min. Top: extracted ion chromatogram for m/z 205. Bottom: spectra for the two chromatographic peaks. C, pig liver microsomes incubated with 2-hydroxyflutamide for 80 min. Top: total ion chromatogram after full MS scan m/z 50 to 500. Bottom: extracted ion chromatogram for [M - H]-m/z 221. D, pig prostate microsomes incubated with flutamide for 80 min. Top: total ion chromatogram after full MS scan m/z 50 to 500. Bottom: extracted ion chromatogram for [M - H]-m/z 261. For more information, see Materials and Methods.
Flutamide was hydroxylated in one, two, or three positions according to peaks of m/z 291 ([M - H]- of hydroxyflutamide) at 7.1 min, m/z 307 ([M - H]- of M1) at 5.1 min, and m/z 323 ([M - H]- of M3) at 2.0 min (data not shown). Addition of β-glucuronidase to the urine indicated glucuronidation of the mono- and dihydroxylated forms, which was confirmed by neutral loss scan and MS/MS of urine samples that had not been treated with β-glucuronidase. Chromatographic peaks for m/z 483 ([M - H]- of M1 glucuronide) and m/z 467 ([M - H]- of hydroxyflutamide glucuronide) were observed at 2.5 and 5.6 min, respectively. The main fragments of the phase II metabolites were the same as those found in the phase I metabolites: m/z 307 and 205, or m/z 291 and 205, respectively (Fig. 6, C and D). Other ions of interest in the spectra are m/z 175 and m/z 113, derived from monodehydrated glucuronic acid. Trihydroxylated flutamide (M3) did not appear to be conjugated with glucuronic acid.
An experiment with a neutral loss scan of 129 on urine that had not been treated with β-glucuronidase gave a peak at 6.2 min with a precursor ion of m/z 452. CID of m/z 452 resulted in the spectrum in Fig. 7. These results indicate the presence of a mercapturic acid conjugate of hydroxylated flutamide. The fragment m/z 205 shows that the hydroxylation was aliphatic.
Flutamide, [M - H]-m/z 275, could not be recovered in any of the urinary samples. Figure 5 provides a proposed metabolic pathway for flutamide and its active metabolite 2-hydroxyflutamide in humans. It includes these novel conjugates detected directly by LC-ESI-MS/MS.

Possible metabolic pathway of flutamide in humans. Novel metabolites identified in the present study are indicated in bold. Of special interest is the mercapturic acid conjugate formed from flutamide because the suggested reactive intermediate might be responsible for the reported hepatoxicity.
Discussion
LC-ESI-MS/MS is a powerful analytical tool in metabolite identification studies. The selectivity of the technique is high and the limit of detection low compared with many other techniques. For the analysis of the microsome and urine samples, we used a triple quadrupole mass spectrometer that can identify families of structurally related metabolites by using, for example, neutral loss and precursor ion scans. Metabolites are derivatives of the parent drug and, as such, it can be assumed that many of the metabolites show the same fragment ions as the parent drug or characteristic neutral losses (Kostiainen et al., 2003). To identify drug metabolites by mass spectral information, the fragmentation of a synthetic standard of the drug is advantageous. We had access to synthetic standard substances of flutamide, 2-hydroxyflutamide, and 4-nitro-3-(trifluoromethyl)-aniline (Flu-1), for which the retention times and fragmentation patterns could be used for the determination of metabolites in the in vitro and in vivo samples. The fragments produced from the standard were then used to draw conclusions about the structure of the metabolites that are similar in their chemical structure and thus fragmented in a similar manner. A positive ion fast atom bombardment mass spectrum of flutamide can be found in the literature (Sternal and Nugara, 2001), but chemical entities tend to fragment differently, depending on the technique and analytical conditions applied. In this study, a hybrid quadrupole/time of flight instrument with an electrospray ion source was used for accurate mass measurements to assist structural assignment of the fragment ions of flutamide and 2-hydroxyflutamide standards. The fragments were produced by collision-induced dissociation, and all fragments except for two could be determined within 3 mDa mass difference. In the cases of the larger mass difference, the peak intensities were low in combination with a larger difference between the peak measured and the lock mass value.
The pharmacologically active 2-hydroxyflutamide has been reported as the major plasma metabolite of flutamide in rats (Asakawa et al., 1995a), dogs (Farthing et al., 1994), and humans (Katchen and Buxbaum, 1975). This finding was also supported in our study since 2-hydroxyflutamide was the only metabolite found in the liver microsomes from all four species. Because 2-hydroxyflutamide is responsible for the antiandrogen effect, it may be concluded that these three animal species are relevant models, at least from a metabolic perspective. Furthermore, there are reports concluding that there are similarities between pig and human metabolizing systems (Anzenbacher et al., 1998, 2002; Myers et al., 2001; Soucek et al., 2001). The hydroxylation has, until now, only been observed on the second (tertiary) aliphatic carbon, which might be considered an uncommon metabolic process (Schulz et al., 1988). However, this study shows that hydroxylation also can occur on the primary aliphatic carbon as well as on the aromatic ring. The three isomers of the monohydroxyflutamide metabolite were separated by the liquid chromatography system used. In the case of trihydroxylated flutamide, two isomers were identified in this study. In one of the metabolites, all three hydroxy groups were aliphatic, and in the other isomer, two were aliphatic and one aromatic.
It has been reported that the prostate tissue contains the gene transcripts of a broad array of drug- and steroid-metabolizing cytochromes P450 (Finnstrom et al., 2001). However, in the comparison of the qualitative metabolism in pig prostate and liver, it is clear that the metabolism was more extensive regarding the number of metabolites formed in the liver than in the prostate. An interesting observation was that the Flu-3 metabolite existed in two isomers, both of which could be found in liver and prostate microsomes after incubation with flutamide and in liver microsomes after incubation with 2-hydroxyflutamide. Until now, the 2-amino-5-nitro-4-(trifluoromethyl)phenol was the only known isomer of the Flu-3 metabolite. The reduction product Flu-5 was only identified in pig prostate microsomes, possibly due to cytosolic contamination in the microsome preparation. Gao et al. (2006) recently identified a metabolite of the compound S4 (a novel nonsteroidal selective androgen receptor modulator) formed by reduction as the main metabolite in the presence of human liver cytosol.

A, urine sample from a human male patient analyzed with full neutral loss scan of 80 mass units. Top: total ion chromatogram. Bottom: the spectrum from the peak at 7 min. B, urine sample from a male patient treated with flutamide analyzed by LC-ESI-MS/MS of m/z 397. Top: total ion chromatogram. Bottom: the spectrum from the peak at 3.6 min. C, LC-ESI-MS/MS spectrum in negative ion mode from a urine sample from a patient treated with flutamide. The parent ion is [M - H]-m/z 467, the weight of the deprotonated molecule hydroxyflutamide + 176, indicating a glucuronic acid conjugate of hydroxyflutamide. D, LC-ESI-MS/MS spectrum in negative ion mode from a urine sample from a patient treated with flutamide. The parent ion is [M - H]-m/z 483, the weight of the deprotonated molecule M1 + 176, indicating a glucuronic acid conjugate of M1. The collision energy was 22 V in A and 30 V in B to D. For more information, see Materials and Methods.
In urine from prostate cancer patients treated with 250 mg of flutamide orally (b.i.d.), four intact phase II metabolites could be directly identified by the use of LC-ESI-MS/MS in the negative ion mode. Previous studies have used indirect techniques to indicate the formation of phase II metabolites in humans. In 1975, Katchen and Buxbaum published data in which plasma, urine, and feces were analyzed after administration of tritium-labeled flutamide to three men. They discovered that Flu-3 was the major urine metabolite and that treatment of the urine with glucuronidase and sulfatase increased the extraction recovery of the total amount of tritium-labeled compounds, indicating the presence of conjugated metabolites (Katchen and Buxbaum, 1975). After administration of flutamide to rats, Flu-3 sulfate has been reported to be the major metabolite in urine, and glucuronic acid conjugates have been detected in bile (Asakawa et al., 1995b). In this study, intact sulfate and glucuronic acid conjugates of Flu-3, together with glucuronides of both mono- and dihydroxylated flutamide, were identified in human urine for the first time. One metabolic product of a glutathione conjugate, mercapturic acid conjugate of hydroxylated flutamide, was detected in the urine samples in the present study. It has been reported that hepatocytes produce electrophilic metabolites that often are subjected to glutathione conjugation (Robert et al., 2005). Our suggestion is that the mercapturic acid conjugate is produced from flutamide through a reactive intermediate (Fig. 5), which might be responsible for the toxic effects observed in numerous articles (Fau et al., 1994; Aizawa et al., 2003; Takashima et al., 2003).
In the metabolic scheme presented by Katchen and Buxbaum (1975), they suggest that Flu-6 plays a major role as an intermediate and that the isobutyryl chain is removed by stepwise oxidation rather than hydrolysis (Katchen and Buxbaum, 1975; Katchen et al., 1976). On the contrary, our findings of end-stage conjugates (see Fig. 5) clearly suggest that the isobutyryl chain does not have to be removed before conjugation and renal excretion. Instead, it is more likely that the major part of the flutamide drug is metabolized into 2-hydroxyflutamide, which may be conjugated with glucuronic acid, or further hydroxylated and then conjugated with glucuronic acid, or hydrolyzed to Flu-1 which, after hydroxylation to Flu-3, can be conjugated with either sulfate or glucuronic acid. Some part of the flutamide drug appears to be converted into a reactive intermediate that can be detoxified through glutathione conjugation. Furthermore, in a recent publication by Gao et al. (2006), it was concluded that the flutamide metabolism was well characterized, and they showed a metabolic scheme in which the only metabolites were 2-hydroxyflutamide and Flu-1, further metabolized into Flu-3. We disagree that the metabolism of flutamide was well characterized before the present publication. As an example, we were able to clearly demonstrate that conjugation reactions play an important role in the flutamide metabolism.

Urine sample from a human male patient analyzed with LC-ESI-MS/MS of m/z 452. Fragments correspond to a mercapturic acid conjugate of hydroxylated flutamide.
In conclusion, the present study for the first time detected five different intact conjugates derived from flutamide in human urine, and a number of isomers of phase I metabolites were detected in liver microsomes from the three animal species studied. A similar metabolic pattern was established in microsomes from pig liver and prostate when incubated with flutamide or 2-hydroxyflutamide, and the metabolism was more extensive in the liver than in the prostate in pigs, as could be expected. Liver microsomes from all four species studied formed the pharmacologically active main metabolite 2-hydroxyflutamide, which we suggest is then further metabolized before excretion (see the proposed metabolic scheme in Fig. 5). We were also able to show the formation of a mercapturic acid conjugate formed from flutamide in human urine and suggest that the reported liver effects of flutamide are due to the molecular mechanism caused by a reactive intermediate.
Footnotes
-
B.L. and H.L. have an owner interest in LIDDS AB, which develops a product for the treatment of prostate cancer (information at www.pulsinvest.se).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008516.
-
ABBREVIATIONS: LC, liquid chromatography; MS, mass spectrometry; MS/MS, tandem mass spectrometry; ESI, electrospray ionization; Q-Tof, hybrid quadrupole/time of flight; CID, collision-induced dissociation.
-
↵1 Current affiliation: The Pharmacy callcenter, Uppsala, Sweden.
- Received November 26, 2005.
- Accepted March 14, 2006.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}