


Abstract
Human liver microsomes are a reagent commonly used to predict human hepatic clearance of new chemical entities via phase 1 metabolism. Another common metabolic pathway, glucuronidation, can also be observed in human liver microsomes, although the scalability of this process has not been validated. In fact, several groups have demonstrated that clearance estimated from liver microsomes with UDP-glucuronic acid typically underpredicts the actual in vivo clearance more than 10-fold for compounds that are predominantly glucuronidated. In contrast, clearance predicted using human hepatocytes, for these same compounds, provides a more accurate assessment of in vivo clearance. We sought to characterize the kinetics of glucuronidation of the selective UGT2B7 substrate AZT (3′-azido-3′-deoxythymidine), a selective UGT2B7 substrate, in human liver microsomes (HLMs), recombinant UGT2B7, and human hepatocytes. Apparent Km values in these three preparations were 760, 490, and 87 μM, with apparent Vmax values highest in hepatocytes. The IC50 for ibuprofen against AZT glucuronidation, when run at its Km concentration in HLMs and hepatocytes, was 975 and 170 μM, respectively. Since incubation conditions have been shown to modulate glucuronidation rates, AZT glucuronidation was performed in various physiological and nonphysiological buffer systems, namely Tris, phosphate, sulfate, carbonate, acetate, human plasma, deproteinized human liver cytosol, and Williams E medium. The data showed that carbonate and Williams E medium, more physiologically relevant buffers, yielded the highest rates of AZT glucuronidation. Km observed in HLM/carbonate was 240 μM, closer to that found in hepatocytes, suggesting that matrix differences might cause the kinetic differences observed between liver preparations. Caution should be exercised when extrapolating metabolic lability via glucuronidation or inhibition of UGT enzymes from human liver microsomes, since this system appears to underpredict the degree of lability or inhibition, respectively, due in part to an apparent decrease in substrate affinity.
In vitro experiments using human liver microsomes (HLMs) are typically used to predict in vivo drug clearance by phase I metabolizing enzymes (McGinnity and Riley, 2001). Human liver microsomal preparations contain the major drug-metabolizing cytochromes P450 (P450s), flavin-containing monooxygenases, and UDP-glucuronosyltransferases (UGTs). However, experiments are typically performed with only NADPH to reflect P450 and flavin-containing monooxygenases activity. Clearance values generated in these experiments can be successfully scaled to predict the hepatic metabolic clearance observed in vivo (Obach et al., 1997). Because of this concordance, a human liver microsomal lability screen can be applied in drug discovery to identify drug candidates with acceptable P450 metabolic lability (Eddershaw et al., 2000). Moreover, as chemical space is explored to minimize P450-mediated metabolism, other clearance pathways, such as phase 2 metabolism, have become relatively more prevalent (Fisher et al., 2000; Obach, 2001). However, the ability of our in vitro methods to quantitatively predict these alternative clearance pathways has not been as thoroughly characterized as for P450s (Fisher et al., 2001). The most common phase 2 clearance pathway is glucuronidation by UGTs (Evans and Relling, 1999; Williams et al., 2004).
The UGTs are a superfamily of enzymes that catalyze the addition of glucuronic acid to substrates. Glucuronidation can convert a hydrophobic xenobiotic to a more hydrophilic compound, which assists in detoxification and excretion of a possible toxin (Radominska-Pandya et al., 2001). UGTs are also responsible for glucuronidation of endogenous compounds such as steroids, thyroid hormones, and bilirubin. In contrast to the P450s, UGTs are localized to the luminal side of the endoplasmic reticulum; thus, membrane-disrupting agents are commonly used in microsomal incubations to enhance observable enzyme activity (Fisher et al., 2000). More than a dozen UGT isoforms have been identified in humans, with the majority being involved in drug metabolism to some extent (Tukey and Strassburg, 2001). Although some intestine-specific isoforms exist, such as UGT1A8 (Tukey and Strassburg, 2000), the liver is considered to be the major site of drug glucuronidation. UGT2B7 is largely a hepatic isoform and is involved in the metabolism of most glucuronidated drugs (Williams et al., 2004), such as morphine, codeine, 3′-azido-3′-deoxythymidine (AZT), and naproxen (Guillemette, 2003). Morphine had traditionally been used as a probe substrate for UGT2B7 (Fisher et al., 2000; Coffman et al., 2001). However, recently, AZT glucuronidation has been shown to be most selective for UGT2B7 (Barbier et al., 2000), suggesting that AZT glucuronide (AZTG) formation could be used as an in vitro and in vivo probe for this isoform (Court et al., 2003).
Although methods exist to observe glucuronide formation by incubating liver microsomes with the cofactor UDP-glucuronic acid (UDPGA) and a latency-removing agent such as alamethicin or a detergent (Fisher et al., 2000), the quantitative relevance of this approach has not been validated. A few reports exist for glucuronidated drugs demonstrating a large difference between clearance values predicted from human liver microsomes with UDPGA added and observed in vivo clearance. Mistry and Houston (1987) first demonstrated this for primarily glucuronidated compounds in rat. More recently, Soars et al. (2002) have shown human liver microsomes to underpredict for many drugs, the majority of which were metabolized by UGT2B7, while also demonstrating that human hepatocytes predict in vivo hepatic clearance more accurately for the same compounds.
A few recent reports have investigated the effect of incubation conditions on UGT activity (Boase and Miners, 2002; Soars et al., 2003). Research by Boase and Miners specifically focused on investigating incubation conditions for glucuronidation of the UGT2B7 probe AZT. Results presented suggest that incubation conditions, such as buffer type and concentration, affect AZT glucuronidation rates. Also, it was found that clearance predicted from the human liver microsome incubations underestimates in vivo hepatic clearance, with optimal conditions still yielding a significant underprediction of in vivo clearance (Boase and Miners, 2002). Since this work demonstrated that the in vitro conditions could have a large effect on the quality of the clearance prediction, we sought to more completely understand this phenomenon. Specifically, since the intrinsic clearance (Vmax/Km) from HLMs appears to be significantly less than both hepatocyte and in vivo values, we wanted to determine whether this was due to an alteration in Km, Vmax, or both. The goals of this work are to compare the enzyme kinetics for metabolism and inhibition of UGT2B7 using AZT as its substrate in human liver microsomes, recombinant UGT2B7 enzyme, and human hepatocytes, and to understand the effects different buffer systems have on the activity and kinetics of UGT2B7 as a possible explanation for the different kinetics observed in liver microsomes versus intact cells.
Materials and Methods
Materials. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless specified otherwise. UDPGA triammonium salt was obtained from Fluka (packaged in Buchs, Switzerland). 3′-Azido-3′-deoxythymidine β-d-glucuronide was purchased from Sigma-Aldrich, Cerilliant (Round Rock, TX), and biosynthesized in-house. AZT was obtained from Toronto Research Chemicals Inc. (North York, ON, Canada). Williams E medium (WEM) was obtained from Invitrogen (Carlsbad, CA). HPLC-grade acetonitrile and water were purchased from J. T. Baker (Phillipsburg, NJ).
Cryopreserved human hepatocyte lots 84, 88, 117, YAA, 98, and NQT were purchased from In Vitro Technologies (Baltimore, MD), and human liver microsomes and cytosol were obtained from an in-house supply prepared from a mixture of 56 different human livers (Foti and Fisher, 2004). Recombinant UGT2B7 enzymes were obtained from PanVera Corp. (Madison, WI). Human plasma was freshly isolated from heparinized blood from one donor by centrifugation. UGT2B7 and CYP3A4 immunoblotting kits were purchased from BD Biosciences (San Jose, CA) and a Western blotting detection kit was obtained from Amersham Biosciences Inc. (Piscataway, NJ).
Human Liver Microsome Incubations. Microsomal incubations contained 0.78 mg/ml HLMs, 0.025 mg of alamethicin, 1 mM MgCl2, 250 mM Tris-HCl (pH 7.4), and 2.5 mM UDPGA to give a total volume of 700 μl. Alamethicin was prepared by dissolution in an 80:1 ratio of water to methanol solution. Before the addition of UDPGA, the mixture was preincubated for 5 min at 37°C. The substrate, AZT, was between 0 and 5 mM, and incubations were performed at least in duplicate. To initiate the reaction, UDPGA was added to a final concentration of 2.5 mM, and 100-μl aliquots were removed at 0, 5, 10, 15, 20, and 30 min. Chlorzoxazone (500 ng/ml) internal standard in acetonitrile was used to stop the reaction at each time point. Recombinant UGT2B7 enzyme incubations were performed the same way as HLMs except that 0.5 mg/ml protein was used.
Microsomal inhibition experiments followed the same protocol as the HLM incubations except that AZT was run at its Km concentration found in HLMs or in recombinant UGT2B7 enzymes. The inhibitors used were naloxone, valproic acid, codeine, ibuprofen, morphine, and diclofenac. Inhibitor concentrations ranged from 0.025 to 5.0 mM and were performed in triplicate. Human liver microsomal incubations with different buffers were performed as stated above. The buffers tested were 250 mM Tris HCl, 100 mM sodium phosphate, 100 mM sodium acetate, 100 mM sodium carbonate, 100 mM sodium citrate, human liver cytosol [deproteinized by ultrafiltration in a Centricon YM-10 (10,000 mol. wt. cutoff; Millipore Corporation, Billerica, MA)], fresh human plasma, and Williams E medium, and were performed in triplicate. All buffers were freshly prepared at pH 7.4.
Hepatocyte Incubations. Prior to experimentation, glassware was treated with Sigmacote. Three different lots of hepatocytes were used per incubation and thawed in a water bath set at 37°C. Cells were washed with WEM (pH 7.4) and centrifuged at 50g for 3 min to gently pellet cells. The supernatant was removed, cells were resuspended in 10 ml of media, and cell viability was assessed by trypan blue exclusion (viability ≥0.76) and diluted to 500,000 viable cells/ml using WEM. Concentrations of 0 to 150 μM AZT were incubated in duplicate, since preliminary experiments found that the concentration range used for liver microsome kinetic experiments was too high for hepatocytes. Morphine (10 μM) was used as a control, since the liver preparations used in this study had previously demonstrated morphine glucuronidation activity. A 300-μl aliquot was taken at 0, 15, 30, 60, 120, and 240 min and flash-frozen in liquid nitrogen. A 100-μl aliquot was taken from the thawed time point and added to internal standard. Drug disappearance and glucuronide formation were monitored. The hepatocyte incubation protocol above was also used for inhibition studies. AZT was run at the Km concentration obtained experimentally. Inhibitor concentration ranged from 0 to 400 μM in triplicate incubations, and stock solutions were made in DMSO. Inhibitor was added to hepatocyte matrix prior to addition of substrate.
Immunoblotting. An immunoblot analysis was conducted to compare the relative amount of UGT2B7 present in human liver microsomes, human hepatocytes, and recombinant 2B7 enzymes. Dilutions were performed to equalize the amount of liver tissue loaded between the enzyme sources, based on microsomal protein. Using the conversion factor of 1.35 × 108 cells per 45 mg of liver microsomes (Houston, 1994), it was determined that 20 μg of HLMs is equivalent to about 60,000 hepatocytes. Therefore, hepatocytes were added to a volume of water so that 60,000 hepatocytes would be loaded onto the gel. Nupage LDS sample buffer (60 mM Tris, pH 6.8, 2% sodium dodecyl sulfate, 2% 2-mercaptoethanol, 0.001% bromphenol blue), Nupage reducing agent, and deionized water were added to samples. Samples were heated at 70°C for 5 min to denature the protein present and ensure cell lysis. An aliquot containing 20 μg of microsomal protein equivalents for each sample was added onto the gel. BD Gentest (Woburn, MA) conditions and procedures were followed. The rabbit α-UGT2B7 polyclonal primary antibody and horseradish peroxidase-conjugated secondary antibody were utilized. Detection solutions 1 and 2 were used for band development, and chemiluminescence was used to develop the membrane. Visualization of the polyvinylidene difluoride membrane was performed using a Lumi-Imager (Roche Diagnostics, Mannheim, Germany). To test the method of cell lysis, a separate immunoblot was performed to test for CYP3A4 in HLMs, hepatocytes, and recombinant 3A4 enzymes using rabbit α-CYP3A4 polyclonal antibody from BD Gentest.
Liquid Chromatography-Mass Spectrometry. Incubation samples were placed in 96-well format and analyzed using a Sciex API3000 MS/MS triple quadrupole mass spectrometer (PerkinElmerSciex Instruments, Boston MA). A total of 20 μl of each sample was injected onto a Phenomenex Synergi 4-μm Polar-RP 80A, 50 × 2 mm (Phenomenex, Torrance, CA). The column was equilibrated with solvent A (A, 95% water/5% acetonitrile/0.1% formic acid). After the sample injection, the flow rate increased from 1 ml/min to 1.5 ml/min over a period of 2 min. Initial flow rate begins at 100% solvent A and immediately changes to 100% solvent B (B, 95% acetonitrile/5% water/0.1% formic acid). AZT and AZTG eluted at 0.8 and 0.6 min and were detected by monitoring m/z 273→273 and 442→273 in negative ionization mode at collision energies of –5.0 and –30.0, respectively. Chlorzoxazone internal standard was detected using the transition 168→132. Morphine, morphine-3-glucuronide, and the internal standard codeine were detected using multiple reaction monitoring (MRM) in positive ionization mode, using the transitions 286→286 (Slawson et al., 1999), 462→286, and 300→165 at collision energies of 5, 36, and 36 kV, respectively. Codeine was analyzed in a manner similar to morphine using estazolam as internal standard. Codeine-6-glucuronide was detected in positive ionization mode using the MRM transition 476→300. The estazolam MRM transition was 295→267. Collision gas was nitrogen. For quantitation, peak area ratio of analyte to internal standard was compared with ratios obtained from a standard curve containing known amounts of analyte.
Data Analysis.Km and Vmax values were determined by fitting the data to the Sigmoid Emax model using WinNonlin v3.2 (Pharsight, Mountain View, CA). The sigmoid model routinely gave better fits of the data as defined by visual inspection of the fit and the error in the parameter estimates. Hill coefficients for enzyme kinetics were always 1 to 1.1. IC50 values and Hill coefficients were determined using the inhibitory effect Sigmoid Emax model. Large deviations from Michaelis-Menten behavior were observed for inhibition, with Hill coefficients both greater than and less than 1.0, but the reasons for this are unknown. Kinetic parameters were reported as parameter and the error in the curve fitting. Vmax is reported in nanomoles per minute per milligram of human liver microsomes or recombinant UGT. The conversion factor of 1.35 × 108 cells per 45 mg of liver microsomes (see above) was used to convert hepatocyte rates to liver microsome rates for comparison purposes. Statistical analyses, including mean, standard deviation, and analysis of variance, were performed by Microsoft Excel (Microsoft Corporation, Redmond, WA). Scaling of in vitro data to predicted clearance values was performed as previously described (Fisher et al., 2002). Log P and log D were estimated using ACD Labs v6.0 (Advanced Chemistry Development, Inc., Toronto, ON Canada). Correction for free fraction was not performed since AZT shows negligible binding to HLMs (J.A. Williams, Pfizer, Ann Arbor, MI, personal communication), the physical properties of the other compounds (morphine, codeine) were of sufficiently low log P/D to suggest minimal incubation binding (Austin et al., 2002, 2005), and they are known not to appreciably bind to plasma (Hardman et al., 2001). Ibuprofen has been shown not to bind to HLMs (Andersson et al., 2004).
Results
Preliminary experiments to predict clearance of AZT, morphine, and codeine using the disappearance half-life at 1 μM substrate concentration in human liver microsomes, alamethicin-pretreated and supplemented with UDPGA, resulted in no measurable disappearance (half-life >120 min). For drugs with high in vivo clearance values due to extensive glucuronidation (>15 ml/min/kg) like AZT and morphine, disappearance half-life should have been <14 min under the experimental conditions used, assuming in vitro-in vivo scalability of the reactions. Interestingly, morphine, AZT, and codeine clearances scaled from analogous experiments performed in cryopreserved human hepatocytes (10.1, 18.2, and 5.8 ml/min/kg, respectively) yielded values similar to in vivo clearances (19, 19, and 5.5 ml/min/kg, respectively). Since some in vitro conditions had been shown to significantly affect AZT glucuronidation rates (Boase and Miners, 2002), the initial goals of this work were to evaluate the enzyme kinetics of UGT2B7 using AZT as its substrate to assess the differences in enzyme activity in human hepatocytes, human liver microsomes, and recombinant UGT2B7.
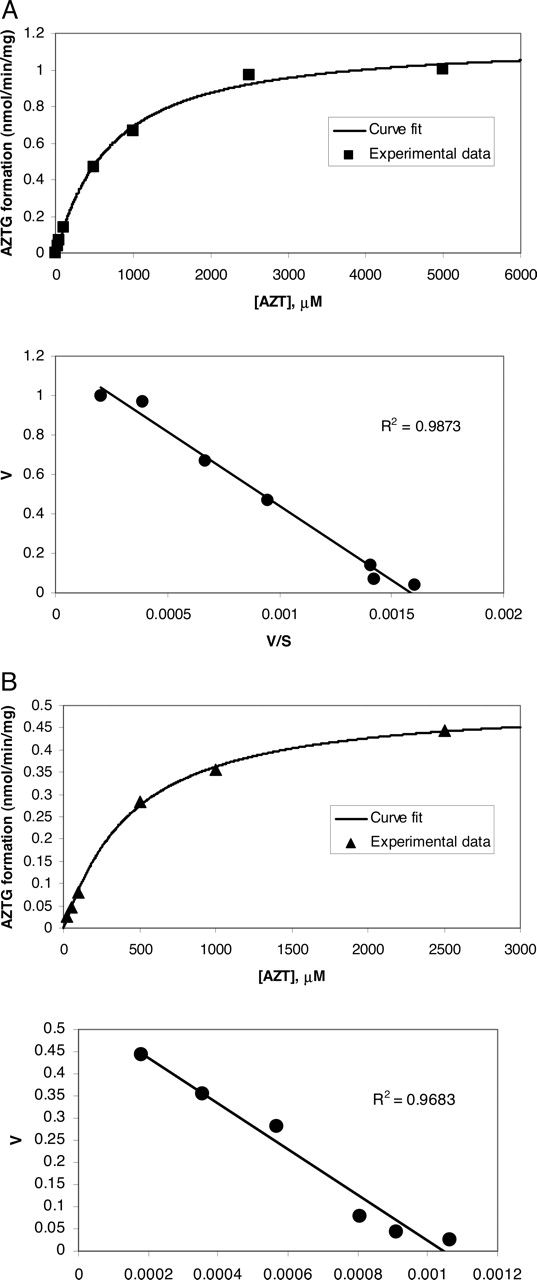
Figure 1, A and B, shows the substrate concentration versus velocity curve for AZT glucuronidation in human liver microsomes and in recombinant UGT2B7 enzymes. Kinetic analysis results in an apparent Vmax value of 1.19 ± 0.04 nmol/min/mg and an apparent Km value of 0.76 ± 0.07 mM for AZT in human liver microsomes, and an apparent Vmax value of 0.53 ± 0.01 nmol/min/mg and an apparent Km value of 0.49 ± 0.03 mM for AZT in recombinant UGT2B7. To assess inhibitor potency for UGT2B7, IC50 values against AZTG formation at its observed Km were obtained for several 2B7 inhibitors in human liver microsomes (Table 1). IC50 values ranged from 0.06 to 3.0 mM, with diclofenac having the lowest IC50 value of all the inhibitors tested. For compounds previously reported to inhibit UGT2B7, the IC50 values were similar to reported values (Kiang et al., 2004). Figure 2 shows a graphical representation of diclofenac inhibition of AZTG formation.
IC50 summary of UGT2B7 in the presence of different inhibitors

Enzyme kinetics of AZTG formation. Incubations were performed and samples were analyzed by HPLC-MS/MS as described under Materials and Methods. AZT concentration versus AZTG formation rate is shown, along with an Eadie-Hofstee plot of the same data. In HLMs (A), results showed a Vmax of 1.19 ± 0.04 nmol/min/mg and an apparent Km value of 0.76 ± 0.07 mM. In recombinant UGT2B7 (B), results showed a Vmax of 0.53 ± 0.01 nmol/min/mg and an apparent Km value of 0.49 ± 0.03 mM.
Kinetic characterization of AZT in human hepatocytes was also performed, and the resulting substrate versus rate of formation of AZTG in hepatocytes can be found in Fig. 3. The apparent Km for AZT in hepatocytes was found to be 0.087 ± 0.01 mM, which is about 9-fold lower than the Km value observed in human liver microsomes (p < 0.05). The apparent Vmax value obtained from the hepatocyte incubation was 5.74 ± 0.33 nmol/min/mg, which is about 5-fold higher than the Vmax observed in human liver microsomes (p < 0.05).
To further characterize the kinetics of UGT2B7 in hepatocytes using AZT as its substrate at its observed Km, an inhibition experiment was performed using ibuprofen as the inhibitor. Ibuprofen was selected since it was a moderate potency inhibitor among the compounds tested in liver microsomes, and so significant increases or decreases in inhibition potency would be easily detected over a similar concentration range. Analysis shows an IC50 value of 0.17 ± 0.03 mM, which is about 6-fold lower (p < 0.05) than the IC50 value for ibuprofen obtained in human liver microsomes, 0.98 ± 0.19 mM.
An immunoblot was performed to determine the relative amount of enzyme present in HLMs and human hepatocytes, with recombinant UGT2B7 as a control. Figure 4 is a depiction of an immunoblot that shows immunodetectable UGT2B7 present in the three different matrices. The blot demonstrates that human hepatocytes contain less immunodetectable UGT2B7 compared with HLM when equivalent amounts of liver tissue are loaded (see Materials and Methods). The recombinant UGT2B7 at all concentrations (2, 1, and 0.5 μg/ml) was overloaded (data not shown). Although human hepatocytes apparently contained less UGT2B7 compared with HLMs, the apparent Vmax in hepatocytes was 5-fold greater than in HLMs. Thus, when corrected for enzyme content (Vmax = kcat · [E]), the kcat is likely to be much greater in hepatocytes. As a positive control for cell lysis and loading, an immunoblot from a separate gel demonstrated similar amounts of immunodetectable CYP3A4 among liver microsomes, cryopreserved human hepatocytes, and recombinant CYP3A4 (data not shown), suggesting that the method of cell lysis was adequate.
In an attempt to understand the effects that buffer incubation conditions have on UGT2B7 activity with AZT in HLMs as a possible explanation for altered enzyme kinetics in hepatocytes, AZTG formation rate in different buffer systems was determined. All buffers were adjusted to pH 7.4 immediately before incubation. Figure 5 graphically compares the amounts of AZTG formed over the incubation to the amount of AZTG formed under our standard incubation conditions (Tris buffer, 250 mM, pH 7.4). Both 100 mM sodium carbonate buffer and WEM produced significantly higher amounts of AZTG (p < 0.05) than did Tris buffer and the other six incubation buffers examined. Sodium carbonate buffer ultimately produced the largest amount of AZTG and was therefore further characterized. Kinetic analysis resulted in an apparent Km value of 0.24 ± 0.09 mM, which is 3-fold lower (p < 0.05) than the Km value observed in the HLM incubation in Tris buffer (0.76 mM), and is about 3-fold higher than the apparent Km value obtained from the hepatocyte incubation. Conversely, the apparent Vmax in carbonate buffer (1.25 ± 0.10 nmol/min/mg) was not significantly different from that obtained in Tris.

Inhibition of AZTG formation by diclofenac in HLMs. Incubations were performed and samples were analyzed by HPLC-MS/MS as described under Materials and Methods. AZT was incubated at its Km concentration determined in Fig. 1.

Enzyme kinetics of AZTG formation in cryopreserved human hepatocytes. Incubations were performed and samples were analyzed by HPLC-MS/MS as described under Materials and Methods. AZT concentration versus AZTG formation rate is shown, along with an Eadie-Hofstee plot of the same data. Results showed a Vmax of 5.74 ± 0.33 nmol/min/mg and an apparent Km value of 0.087 ± 0.01 mM. Vmax units of cell number were converted to milligrams of microsomal protein equivalents using scaling factors as described under Materials and Methods.

The effect of various buffers and matrices on AZTG formation rate in HLMs. Incubations were performed and samples were analyzed by HPLC-MS/MS as described under Materials and Methods. All buffers were adjusted to pH 7.4 immediately before incubation. Cytosol was deproteinized by ultracentrifugation through a dialysis membrane (mol. wt. cutoff 10,000). Asterisk denotes statistically significant differences from Tris buffer.
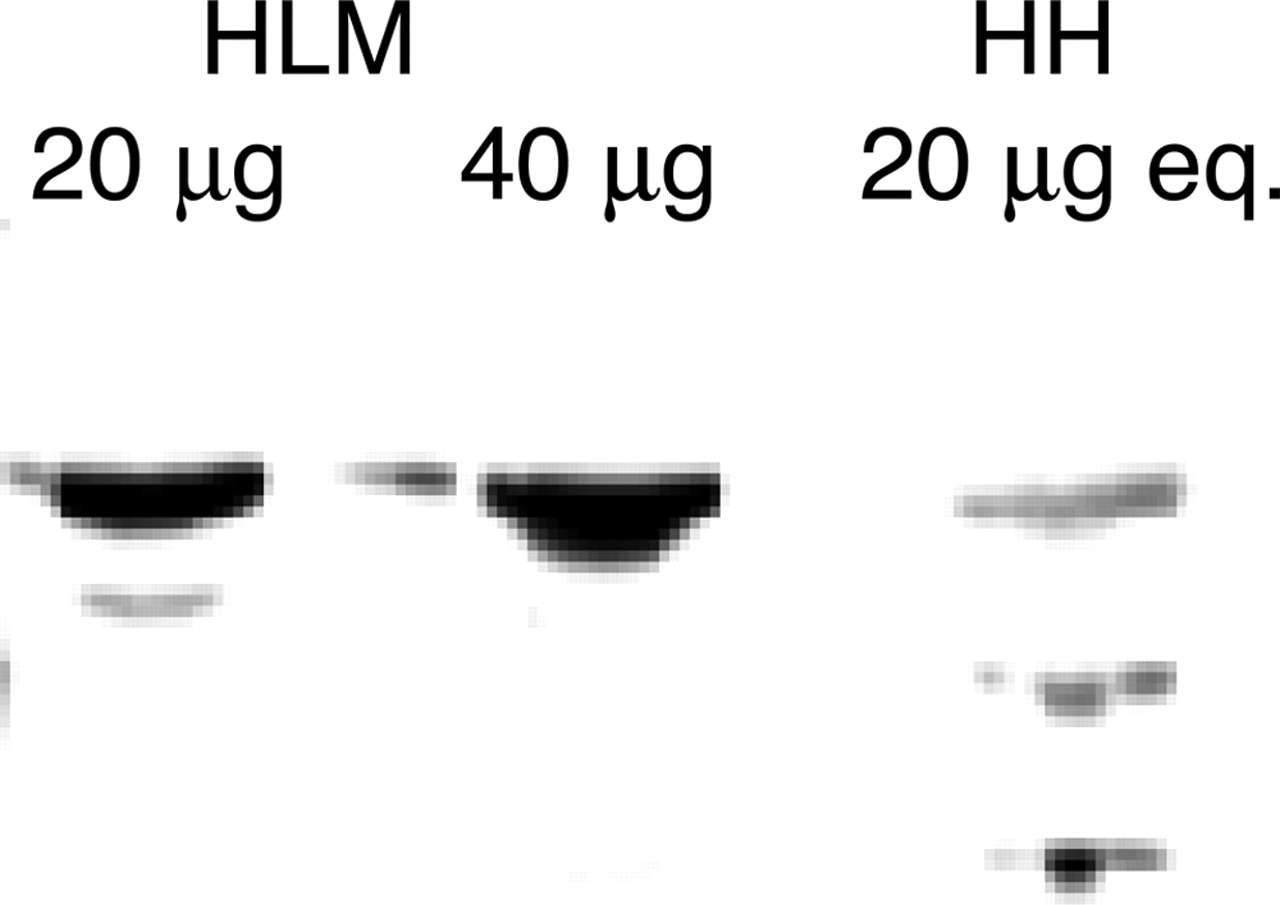
UGT2B7 relative expression in HLMs and human hepatocytes. Samples were prepared and immunoblotted with α-UGT2B7 antisera as described under Materials and Methods. Using commonly applied scaling factors (see Materials and Methods), lysed hepatocyte loading was normalized to an equivalent amount (20 μg) of microsomal protein.
Predicted clearance calculations were performed using kinetic parameters from HLM and hepatocyte incubations. Hepatocytes predict an in vivo clearance of 15.1 ml/min/kg, whereas in vivo clearance of AZT has been reported to be 19 ml/min/kg. Predicted in vivo clearance from HLMs is 1.7 ml/min/kg, which is 9 times lower then the predicted clearance obtained from hepatocytes and similar to previous reports (Boase and Miners, 2002). Using previously utilized acceptance criteria for scaling in vitro to in vivo (±2-fold error) (Obach, 1999), the in vivo clearance is accurately predicted from hepatocytes but is significantly underpredicted using HLMs.
Discussion
Human liver microsome lability data have been used for decades to make hepatic clearance predictions for cytochrome P450 enzymes (Houston, 1994; Obach, 2001). The degree of success has improved with the understanding of factors that can affect the in vitro result, such as microsomal protein binding (Obach, 1997). This utilization of liver microsomes has increased to a point where medium to high-throughput microsomal lability screens are in place to assay up to hundreds of compounds per week in drug discovery, thus providing medicinal chemists with the ability to move analog efforts toward chemical space with reduced phase 1 metabolism. Thus, other metabolism pathways are becoming relatively more common in new drug candidates, and less is known about the ability to accurately scale these pathways from in vitro data.
To our knowledge, only three publications in the last 20 years have reported scaling of microsomal rates of glucuronidation to predict in vivo clearance. Mistry and Houston (1987) reported a 10- to 30-fold underprediction of clearance due to glucuronidation in rats for compounds now known to be UGT2B7 substrates in human. Recently published data have also shown a significant underprediction of in vivo hepatic clearance due to glucuronidation in humans from HLMs (Soars et al., 2001). Although the majority of compounds in that report were UGT2B7 substrates, other forms (UGT1A1 with ethynylestradiol and UGT1A4 for imipramine) were represented and demonstrated a similar underprediction.
Boase and Miners (2002) examined the effects of incubation conditions on AZTG formation rates and kinetic constants from human liver microsomes. In that work, they focused mostly on different activators to remove latency, with examination of only the nonphysiological Tris and phosphate buffers. Only Vmax was found to be significantly affected by buffer type and concentration. The work presented in this article attempts to confirm and extend these data with a more complete examination of liver preparations and buffer conditions to identify the factors affecting enzyme behavior in HLMs that cause underprediction of in vivo clearance. AZT is an ideal substrate to work with because it is cleared almost completely via hepatic metabolism (PDR, 2002), it is not appreciably bound in plasma or liver tissue (Hardman et al., 2001; Quevedo et al., 2001), and its clearance is mediated by just one enzyme in humans (Court et al., 2003). Large differences exist in kinetic parameters for AZT between HLMs and hepatocytes that affect the predicted clearance. The apparent Km value from human hepatocytes was ∼9-fold lower than that from human liver microsomes, whereas the apparent Vmax value from hepatocytes was 5-fold higher. Despite this higher apparent Vmax, immunoblot analysis demonstrated that less UGT2B7 enzyme was present in human hepatocytes from a similar amount of liver compared with liver microsomes, suggesting an even larger difference in turnover (Vmax = kcat · [E]). Although lower molecular weight immunoreactive bands were most apparent in the hepatocyte lane, suggesting that proteolysis may have occurred, it is unclear whether this actually is degraded UGT2B7, given the robust enzyme activity observed in this system. Given these factors, the enzyme appears to be in a more active state in a whole cell system compared with a microsomal preparation, with Vmax/Km differing by ∼30-fold, and kcat/Km >30-fold higher in hepatocytes. Interestingly, the kinetics in recombinant UGT2B7 also appeared to be different from that of liver microsomes. However, this may have been due to the recent observation of functional alterations in UGT activity due to heterodimerization (Ishii et al., 2001; Kurkela et al., 2004), and was not pursued since we focused on the large difference between HLMs and hepatocytes.
We observed a lower Km in a whole cell system, which could be explained by UGT2B7 possessing altered biochemical properties due to altered buffer matrix. However, we recognize that whole cell systems can possess active transport processes that can affect the equilibrium between media and intracellular space. It has been reported that AZT is a substrate for uptake (Hong et al., 2001) and efflux (Wang et al., 2004) transporters. A lower apparent Km could possibly be explained by uptake transporters concentrating AZT intracellularly and lowering the observed Km. However, these incubations were performed in suspension, whereas cell-cell contacts are often required for expression of cell membrane transporters and cell polarity to be maintained (Talamini et al., 1997; Liu et al., 1998, 1999). Even complex culturing systems may not allow for recovery of expression (Liu et al., 1998, 1999). Also, hepatocytes in suspension are thought to be advantageous due to a “rapid distribution of drug to metabolizing enzymes” (Griffin and Houston, 2005). Moreover, our hypothesis was that the difference in intrinsic activity between HLMs and hepatocytes arose from the actual buffer matrix to which the enzyme was exposed. To explore the potential effect of different matrices on AZTG from HLMs, several buffers and conditions were assessed, including human plasma and deproteinized cytosol. As shown in Fig. 5, sodium carbonate yielded the highest rate, followed by WEM, which is carbonate-based. Interestingly, apparent Km values from HLMs in carbonate were closer to the Km observed in hepatocytes, suggesting that this, and not active uptake, was at least part of the difference. However, apparent Vmax values were relatively unchanged compared with HLMs in Tris. The effects of different buffers on enzyme kinetics have been examined more commonly with P450s. CYP2D6 was shown to be significantly affected by carbonate relative to phosphate for some reactions, and the effect was mostly on Vmax (Hutzler et al., 2003). CYP3A4 has been shown to be highly sensitive to matrix components such as lipid composition, cytochrome b5, divalent metals, glutathione, and others (Imaoka et al., 1992; Shaw et al., 1997; Kim and Kim, 1998; Schrag and Wienkers, 2000). Maenpaa et al. (1998) showed a significant effect of buffer (Tris versus phosphate) on the Km for midazolam 1- and 4-hydroxylation, although it is unclear which value more closely reflects the in vivo enzyme behavior. In the case of AZTG formation, kinetic parameters in hepatocytes and in HLMs in carbonate buffers are consistent with enzyme behavior that is more physiologically relevant than Tris or phosphate buffers, based on predicted clearance extrapolated from in vitro kinetic data. More work is required to see if this effect is substrate/enzyme-independent, and what factors will increase apparent Vmax in HLMs to reflect that observed in hepatocytes.
A surprising, yet important, finding was that the apparent IC50 value was significantly lower in hepatocytes relative to HLMs. Although AZT in these two tissues gave different Km values, inhibition experiments were performed at the observed Km for that tissue. Although lower apparent IC50 is consistent with more efficient binding of ligand to enzyme, as was apparently observed for Km, it has important repercussions. Inhibition potencies for UGTs have been reported numerous times in the literature (Ammon et al., 2000; Kiang et al., 2004), and some calculations have attempted to predict or explain in vivo findings for UGT drug interactions (Trapnell et al., 1998; Williams et al., 2004). However, these inhibition potencies have been performed in liver microsomes and/or recombinant UGTs. It is unclear which IC50 is closer to the “true” value that would reflect the in vivo environment. Although our studies in human hepatocytes were performed with only one substrate-inhibitor pair and we determined IC50 instead of Ki, partially to minimize the expense of hepatocytes, we feel that more data needs to be gathered to see if other inhibitors demonstrate more potent inhibition in hepatocytes.
This alteration of enzyme and substrate interaction can be seen by the hepatic clearance predictions produced from HLMs and hepatocytes for AZT. HLMs predict an in vivo clearance value of 1.7 ml/min/kg, whereas hepatocytes predict 15.1 ml/min/kg. This is about a 9-fold difference. The actual in vivo clearance of AZT is 19 ml/min/kg, which is comparable to the clearance predicted from hepatocytes in this study (Boase and Miners, 2002). HLMs significantly underpredict hepatic clearance. Therefore, based on these and other data, human liver microsomes should not be used for predicting clearance due to glucuronidation, or any kinetic parameters for metabolism or inhibition involving UGT enzymes, at least until this phenomenon can be more completely understood.
Acknowledgments
We acknowledge Drs. Michael L. Schrag (Amgen Biologicals, Thousand Oaks, CA) and Cliff Fisher (Barker Institute, Groton, CT) for many helpful discussions.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.005058.
-
ABBREVIATIONS: HLM, human liver microsome; P450, cytochrome P450; UGT, UDP-glucuronosyltransferase; AZT, 3′-azido-3′-deoxythymidine; AZTG, AZT-glucuronide; UDPGA, UDP-glucuronic acid; WEM, Williams E medium; MRM, multiple reaction monitoring; HPLC-MS/MS, high-performance liquid chromatography-tandem mass spectrometry.
- Received April 7, 2005.
- Accepted July 22, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}