


Abstract
The wealth of information that has emerged in recent years detailing the substrate specificity of hepatic transporters necessitates an investigation into their potential role in drug elimination. Therefore, an assay in which the loss of parent compound from the incubation medium into hepatocytes (“media loss” assay) was developed to assess the impact of hepatic uptake on unbound drug intrinsic clearance in vivo (CLint ub in vivo). Studies using conventional hepatocyte incubations for a subset of 36 AstraZeneca new chemical entities (NCEs) resulted in a poor projection of CLint ub in vivo (r2 = 0.25, p = 0.002, average fold error = 57). This significant underestimation of CLint ub in vivo suggested that metabolism was not the dominant clearance mechanism for the majority of compounds examined. However, CLint ub in vivo was described well for this dataset using an initial compound “disappearance” CLint obtained from media loss assays (r2 = 0.72, p = 6.3 × 10-11, average fold error = 3). Subsequent studies, using this method for the same 36 NCEs, suggested that the active uptake into human hepatocytes was generally slower (3-fold on average) than that observed with rat hepatocytes. The accurate prediction of human CLint ub in vivo (within 4-fold) for the marketed drug transporter substrates montelukast, bosentan, atorvastatin, and pravastatin confirmed further the utility of this assay. This work has described a simple method, amenable for use within a drug discovery setting, for predicting the in vivo clearance of drugs with significant hepatic uptake.
Prentis et al. (1988) highlighted the importance of drug metabolism and pharmacokinetics (DMPK) in reducing the attrition of candidate drugs in early clinical trials. This has subsequently led to a realignment of DMPK within the drug discovery process and increased use of a plethora of high-throughput screens early in lead optimization (Riley and Grime, 2004).
Arguably one of the most critical tasks within DMPK is the accurate prediction of in vivo clearance from in vitro data (Riley, 2001). Although the theory behind this process was published almost 30 years ago (Rane et al., 1977), the potential impact on drug discovery was not fully appreciated until the review by Houston (1994). Subsequently, hepatic microsomes and hepatocytes prepared from both preclinical species and humans have been used to predict in vivo clearance successfully (Obach, 1999; Soars et al., 2002; Ito and Houston, 2004; McGinnity et al., 2004; Ito and Houston, 2005; Riley et al., 2005). However, recent studies with hepatocytes have shown a significant underprediction of in vivo clearance for a distinct set of drugs, which has been attributed in some cases to hepatic uptake (Riley et al., 2005; Soars et al., 2007).
During the last decade a rapid increase has been seen in the number of publications in which researchers have investigated the role of hepatic uptake in drug clearance (Mizuno et al., 2003; Shitara et al., 2006). Perhaps the most important superfamily of enzymes for the hepatic uptake of anionic drugs is the organic anion transporting polypeptides (OATPs) (Hagenbuch and Meier, 2003, 2004). The molecular cloning of the major hepatic OATP isoforms and their expression in mammalian cell lines has generated a wealth of knowledge concerning the substrate specificity of OATPs (Hagenbuch and Meier, 2003, 2004; Mizuno et al., 2003; Shitara et al., 2006). However, the lack of suitable means to quantify data generated from recombinant cell lines directly to obtain hepatic clearance in vivo has proved more problematic than that for the cytochrome P450 enzyme superfamily, for example (Iwatsubo et al., 1997; McGinnity et al., 2000). Hepatocytes, whether plated or in suspension, have therefore become the system of choice for obtaining quantitative information regarding hepatic drug uptake (Olinga et al., 1998; Shitara et al., 2003; Hirano et al., 2004). Interestingly, most studies to date have obtained a measure of hepatic uptake by investigating the rate of appearance of radiolabeled substrate into cells, determined after a centrifugation step through oil (Olinga et al., 1998; Shitara et al., 2003; Hirano et al., 2004). Indeed, in some instances attempts have been made to use such data to predict the in vivo clearance of drugs (Olinga et al., 1998; Nezasa et al., 2003). Although this method provides robust, mechanistic data on individual compounds, it is clearly not amenable for use within an early discovery environment in which many NCEs are evaluated in parallel, and radiolabeled compounds are not routinely available.
To this end, the aims of this study were 3-fold: to develop a nonradiolabeled method to assess the impact (on clearance) of hepatic uptake in the rat; to determine whether hepatic uptake is responsible for the underprediction of in vivo clearance observed for a number of NCEs in previous studies (Riley et al., 2005; Soars et al., 2007); and to compare hepatocyte uptake rates in vitro between rats and humans for a number of NCEs and key drugs.
Materials and Methods
Chemicals and Human Hepatocytes. All chemicals and reagents used were of the highest available grade. Montelukast, bosentan, pravastatin, and atorvastatin were obtained from Sequoia Research Products Ltd. (Oxford, UK). [3H]Estrone-3-sulfate (specific activity 2120 GBq/mmol) was obtained from PerkinElmer Life Sciences (Boston, MA). All other chemicals were purchased from Sigma-Aldrich (Poole, Dorset, UK). AZ compounds were synthesized at AstraZeneca R&D Charnwood (Loughborough, UK).
Freshly isolated human hepatocytes were obtained from the UK Human Tissue Bank after appropriate consent and ethical approval (Leicester, UK). Hepatocyte viability was >80%.
Measurement of logD7.4. Partitioning of compounds (40-400 μM) between 1-octanol and 0.02 M phosphate buffer, pH 7.4, at 20°C was determined using a standard shake flask method (Leo et al., 1971). Both layers of the partition mixture were analyzed using high-performance liquid chromatography with tandem mass spectrometry detection as described below.
Preparation of Rat Hepatocytes. Isolation of rat hepatocytes was performed essentially using the two-step in situ collagenase perfusion method of Seglen (1976). Briefly, the hepatic portal vein of an anesthetized male Sprague-Dawley rat (weight 200-300 g) was cannulated just above the junction of the splenic and pyloric veins. Liver perfusion medium (Invitrogen, Paisley, UK) was perfused via the hepatic portal vein until the liver cleared to an even tan color (usually 7-8 min at a perfusion rate of 30 ml/min). Liver digestion medium (Invitrogen) was then perfused until the liver displayed evidence of extensive dissociation (usually a further 6-8 min at a perfusion rate of 30 ml/min). The liver was dissected from the rat, and cells were gently teased out of the liver capsule into a beaker containing ice-cold hepatocyte suspension buffer [2.34 g of sodium HEPES, 0.4 g of d-fructose, 2.0 g of bovine serum albumin (BSA), 1-liter powder equivalent of Dulbecco's modified Eagle's medium (Sigma, Gillingham, UK] diluted in 1 liter of water and adjusted to pH 7.4 with 1 M HCl). The cell suspension was passed through a 250-μm mesh into a precooled tube and centrifuged at 50g for 2 min at 4°C. The supernatant was decanted, the cell pellet was resuspended in suspension buffer (without BSA), and the centrifugation step was repeated. The resulting pellet of cells was resuspended in 10 ml of suspension buffer (without BSA), and an estimation of hepatocyte yield and viability was obtained using the trypan blue exclusion method. Only cells with a viability of >80% were used.
Determination of Metabolic CLint Using Rat and Human Hepatocytes. NCE stocks were prepared in dimethyl sulfoxide at 100-fold incubation concentration (100 μM), and 10 μl of this 100 μM stock was added to a vial containing 490 μl of hepatocyte suspension buffer (without serum). A vial containing either rat or human hepatocytes at a concentration of 2 million viable cells/ml was preincubated for 5 min in a shaking (80 oscillations/min) waterbath at 37°C along with the vial containing the drug-buffer mix. Reactions were initiated by adding 500 μl of hepatocyte suspension to the 500 μl of drug/buffer mix [giving a final substrate concentration of 1 μM at 1% (v/v) dimethyl sulfoxide]. Aliquots (40 μl) were removed at 0, 2, 6, 15, 30, 45, 60, and 90 min, and reactions were quenched in 120 μl of ice-cold methanol. Samples were subsequently frozen for 1 h at -20°C and then centrifuged at 2000g for 20 min at 4°C. The supernatants were removed and analyzed as described below.
Determination of Loss from Media CLint Using Rat and Human Hepatocytes. Loss from media CLint values were determined essentially as described above except that 1-ml incubations were prepared in duplicate. Aliquots (80 μl) were removed at 0, 0.5, 1, 2, 4, and 6 min from the first incubation and at 15, 30, 45, 60, 75, and 90 min from the second incubation and placed into centrifuge tubes. These aliquots were immediately centrifuged at 7000g for 30 s using a MSE MicroCentaur centrifuge (Fisher Scientific, Loughborough, UK), and 40 μl of the supernatant was pipetted into 120 μl of ice-cold methanol. Samples were then frozen for 1 h at -20°C and centrifuged at 2000g for 20 min at 4°C. The supernatants were removed and analyzed as described below.
Determination of CLint for the Appearance of [3H]Estrone-3-sulfate into Human Hepatocytes. CLint values for drug appearance into hepatocytes were determined using a method adapted from the centrifugal filtration technique of Petzinger and Fuckel (1992). A vial containing human hepatocytes at a concentration of 2 million viable cells/ml was preincubated for 5 min in a waterbath at 37°C along with a vial containing 500 μl of tritiated and unlabeled estrone-3-sulfate in suspension buffer (final concentration 3 μM, specific activity 2120 mBq/mmol). Reactions were initiated with the addition of 500 μl of hepatocyte suspension to the estrone-3-sulfate/buffer mix. Aliquots (100 μl) were removed at 10, 20, 30, and 40 s and immediately centrifuged at 7000g for 30 s through 150 μl of oil (density of 1.015 g/ml, containing 1M potassium hydroxide) using a MiniSpin centrifuge (Eppendorf, Cambridge, UK). During this process the hepatocytes pass through the oil into the alkaline solution. After an overnight incubation in the alkaline solution to dissolve the hepatocytes, each centrifuge tube was frozen in liquid nitrogen and cut, with collection of the cell pellet in a scintillation vial. After the addition of scintillation cocktail, the amount of radioactivity in the cells was determined using a Packard 2200CA Tri-Carb liquid scintillation counter (PerkinElmer Life and Analytical Sciences, High Wycombe, UK).
Analysis of Hepatocyte and logD7.4 Samples. Mass spectrometry was performed on a Micromass Quattro Ultima Platinum triple quadrupole mass spectrometer (Waters, Manchester, UK) using a Hewlett Packard 1100 high-performance liquid chromatography system (Hewlett Packard, Palo Alto, CA) for separation. Analysis was by multiple reaction monitoring using either the positive or negative ion mode. Cone voltage and collision energy were optimized for each compound.
In these analyses, chromatographic separation was achieved using a Hypersil Gold C18 (4.6 × 50 mm, 3 μm) column obtained from Thermo Electron Corporation (Basingstoke, UK) using 10 μl of each sample. The mobile phase consisted of water with 0.1% (v/v) formic acid with the organic phase being methanol containing 0.1% (v/v) formic acid. All chromatography was performed using a generic gradient (t = 0 min % organic = 5, t = 0.5 min % organic = 5, t = 2 min % organic = 100, t = 3 min % organic = 100, t = 3.1 min % organic 5, total runtime = 4 min). The flow rate was set at 1.5 ml/min, which was introduced into the mass spectrometer source at 0.4 ml/min.
Data Analysis. CLint was estimated using 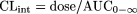
and 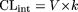
where V is the incubation volume (corrected for nonspecific binding; see below) and k is the elimination rate constant. For compounds exhibiting a monoexponential loss, these two equations give equivalent values for CLint since under these conditions, AUC0-∞ is equal to the initial drug concentration (C0) divided by the elimination rate constant (and dose/C0 = V). Nonspecific binding was determined as the difference in drug concentration between the 0 and 0.5 min time point. Therefore, the elimination rate concentration was calculated from the initial linear phase from log concentration-time plots starting from the 0.5 min time point. This method was also used for compounds exhibiting a biphasic profile. Although this represents a potential composite of uptake and metabolism, curve stripping produced similar results for a representative set of compounds (data not shown).
A schematic diagram highlighting the processes involved in a loss of parent compound from the incubation medium into hepatocytes (“media loss”) CLint determination are shown in Fig. 1. The overall CLint as viewed from the media is effectively the composite of uptake and metabolism minus any potential efflux out of the cell. Because only free drug is available for transport/metabolism, binding will modify each of these processes.
For the appearance of [3H]estrone-3-sulfate into hepatocytes, CLint was calculated from: 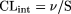
where v is the initial rate of appearance of drug into the hepatocytes, and S is the initial substrate concentration (since the reaction was performed at a low substrate concentration i.e., S ≪ Km). This equation is the differential of the equation, CLint = dose/AUC.

Schematic diagram detailing the processes involved in a “media loss” CLint determination
Determination of CLint ub in vitro. CLint ub in vitro values were calculated from CLint divided by the unbound fraction of drug in the hepatocyte incubation (fuinc). fuinc was predicted using the method of Austin et al. (2005) from a consideration of chemical class and either logD7.4 or logP. No fuinc correction was required for media loss analysis. By using physiological scaling factors to account for hepatocellularity and liver weight in the rat (Ito and Houston, 2004) and human (Riley et al., 2005), predicted CLint ub in vivo values were calculated from the values for CLint ub in vitro, derived as described above.
Determination of CLint ub in vivo. The unbound drug intrinsic clearance in vivo (CLint ub in vivo) was calculated from hepatic blood clearance using the parallel tube model (Pang and Rowland, 1977), as shown below: 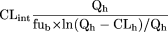
where CLh is hepatic blood clearance, fub is the fraction of drug unbound in blood, and Qh is blood flow (70 ml/min/kg in the rat and 20 ml/min/kg in human).
Rat plasma clearance was determined after the administration of an intravenous dose (1 mg/kg) to male Sprague-Dawley rats as reported previously (Weaver and Riley, 2006) and converted to CLh by dividing by the blood/plasma ratio (estimated to be 0.7 for acidic and zwitterionic compounds and 1 for the base and neutral). The fraction of drug unbound in plasma was measured by equilibrium dialysis as detailed previously (Soars et al., 2002) and converted to fub by dividing by the blood/plasma ratio. Human values for CLh, and fub were obtained from the literature (see Table 2).
Prediction of CLint ub in vivo using human hepatocytes for five marketed drugs
CLint values were determined using a media loss incubation via an initial disappearance approach and represent the mean ± S.D. from three individual human hepatocyte preparations. CLint values for estrone-3-sulfate were determined via an appearance into cells approach.
Accuracy of Predictions. Regression analyses were performed on log-transformed data for predicted and observed CLint ub in vivo for each of the rat hepatocyte assays described. The regression equation and correlation coefficient (r2) were derived, and significance was assessed using the p value (where p < 0.05 was considered significant). The accuracy of each prediction method was assessed using the average fold error (afe) with the geometric mean of prediction error providing an equal value for both under- and overpredictions.
Results
Prediction of CLint ub in vivo Using a Conventional Rat Hepatocyte Assay. A subset of 36 AZ NCEs was selected to investigate the potential of a media loss rat hepatocyte assay to predict CLint ub in vivo. The compound set comprised 6 acids, 18 zwitterions, and 1 neutral and 1 basic compound with logD7.4 values ranging from -0.2 to 3.5 (Table 1). An initial screen with a conventional rat hepatocyte assay produced a variety of predicted CLint ub in vivo values ranging from a mean value of 9 ml/min/kg for AZ19 to 315 ml/min/kg for AZ7 (Table 1). Interpreparation variability in predicted CLint ub in vivo values was acceptable (≤3-fold) for the majority of compounds investigated. Figure 2, A-C, provides examples of log concentration-time profiles for AZ10, AZ14, and AZ20. However, Fig. 3A shows the poor prediction obtained when log predicted CLint ub in vivo values calculated using the conventional rat hepatocyte assay were plotted against log CLint ub in vivo (observed) values. Only four compounds (AZ7-AZ10) were predicted within 5-fold and the afe of the dataset as a whole was 57.
Prediction of CLint ub in vivo using rat hepatocytes for 36 AZ compounds

Representative concentration-time data for AZ10 (A), AZ14 (B), and AZ20 (C). Concentration-time data were generated for AZ10 (A), AZ14 (B), and AZ20 (C) using either a conventional rat hepatocyte assay (closed symbols) or via a media loss incubation (open symbols). Concentration data have been used for illustration purposes; however, ln concentration data were used in the calculation of CLint estimates (see Materials and Methods for details).
Prediction of CLint ub in vivo Using a Media Loss Rat Hepatocyte Assay.Figure 3B shows the relationship between predicted CLint ub in vivo obtained using a media loss AUC0-∞ approach and observed CLint ub in vivo (r2 = 0.49, p = 1.9 × 10-6). Although this method reduced the underprediction of CLint ub in vivo compared with data generated using the conventional rat hepatocyte assay, the afe was still large (16-fold). However, CLint ub in vivo values calculated using an initial disappearance rate from a media loss assay produced an excellent correlation with observed CLint ub in vivo data (r2 = 0.72, p = 6.3 × 10-11). Figure 3C also shows that in general this approach produced the most accurate prediction of CLint ub in vivo (afe = 3). Preliminary experiments using hepatocytes at 4°C and inhibitor studies for several compounds confirmed that the uptake observed was active (data not shown).
Determination of fmedium Using Rat Hepatocyte Incubations.Table 1 shows the fuinc values calculated for 36 AZ compounds as described previously by Austin et al. (2005). A ratio of compound concentrations observed during the terminal phase of a media loss assay with those observed at a corresponding time point from the conventional rat hepatocyte incubation also provides the determination of fmedium values (exemplified by Fig. 2, A-C). This is effectively the fraction of drug in the incubation medium. Table 1 shows that with compounds for which CLint ub in vivo was predicted well using the conventional hepatocyte assay (AZ7-10), there was no discernible difference between fuinc and fmedium. This result is highlighted by the minimal differences in log concentration for AZ10 observed from conventional and media loss assays (Fig. 2A) However, for compounds that were actively taken up into the cell, the difference in these two values was significant (for example, AZ1 fuinc = 0.93, fmedium = 0.04) (Fig. 2, B and C).
Prediction of CLint ub in vivo Using a Media Loss Human Hepatocyte Assay. The uptake rate of [3H]estrone-3-sulfate was determined with each batch of hepatocytes to assess their suitability for uptake studies (Table 2). CLint ub in vivo values were predicted for montelukast, prazosin, pravastatin, atorvastatin, and bosentan using an initial disappearance rate from a media loss assay with three separate human hepatocyte donors. Table 2 shows that for all five drugs studied the calculated CLint ub in vivo values were within 4-fold of the observed CLint ub in vivo estimates obtained from the literature. The dataset with human hepatocytes was extended further to include the 36 AZ compounds investigated previously in the rat (Table 1). Figure 4 shows the relationship between rat and human uptake CLint for these 36 AZ NCEs determined using an initial disappearance rate constant. This analysis suggests that, in general, hepatic uptake in the rat is more rapid (up to 12-fold and 3-fold on average) than in humans for this compound set.

Prediction of CLint ub in vivo for 36 AZ compounds using a conventional rat hepatocyte assay (A), a media loss AUC0-∞ (B), or an initial disappearance approach (C). CLint estimates were determined using either a conventional rat hepatocyte assay (A) or via a media loss incubation using either an AUC0-∞ (B) or initial disappearance (C) approach (see Materials and Methods for details). Each value represents the mean of two or three determinations in rat hepatocytes (Table 1) The solid lines represent a regression analysis (A, line of best fit is given by y = 0.61x + 2.21, r2 = 0.25, p = 0.002, afe = 57; B, line of best fit is given by y = 0.83x + 1.42, r2 = 0.49, p = 1.9 × 10-6, afe = 16; C, line of best fit is given by y = 0.84x + 0.79, r2 = 0.72, p = 6.3 × 10-11, afe = 3). The dotted lines represent the line of unity.

Comparison of rat and human CLint estimates for 36 AZ compounds determined using an initial disappearance approach. CLint estimates were determined in rat and human hepatocytes using an initial disappearance approach (see Materials and Methods for details). Each value represents the mean of two or three determinations in rat hepatocytes (Table 1) and the mean values from three separate donors of human hepatocytes. The solid line represents a regression analysis (line of best fit is given by y = 0.66x + 09.29, r2 = 0.52, p = 0.25). The dotted line represents the line of unity.
Discussion
An understanding and accurate prediction of in vivo clearance for preclinical species in conjunction with robust human in vitro data provides confidence when one is extrapolating to in vivo clearance in humans for a NCE (Grime and Riley, 2006). Rat hepatocytes have been shown previously to be the in vitro system of choice for predicting in vivo clearance for compounds primarily undergoing metabolic clearance in the rat (Houston, 1994; Soars et al., 2002; Ito and Houston 2004). Recent studies have indicated that a comprehensive analysis of the interplay between metabolism and transport in hepatocytes is warranted (Lam et al., 2006). For the majority of the subset of AZ compounds investigated in this study, CLint estimates determined using conventional rat hepatocyte incubations underpredicted rat CLint ub in vivo significantly (Fig. 3A). Studies in bile duct-cannulated rats had shown biliary and renal excretion of parent compounds to be minimal for compounds in this dataset (data not shown). It has been noted that hepatic uptake can influence drug disposition, i.e., modulate the apparent volume of distribution of some drugs and enhance their clearance (Reinoso et al., 2001; Shitara et al., 2006). It was therefore postulated that hepatic uptake was the rate-determining process in the elimination of these compounds as proposed by other research groups (Yamazaki et al., 1996; Shitara et al., 2006).
The majority of studies in the literature have investigated the hepatic uptake of compounds into cells using the centrifugation of radiolabeled parent through an oil layer (Olinga et al., 1998; Shitara et al., 2003; Hirano et al., 2004). Indeed, in vitro uptake data produced with radiolabeled substrates have been used to predict the in vivo clearance of drugs such as rocuronium, digoxin, rosuvastatin, and pravastatin (Olinga et al., 1998; Nezasa et al., 2003). However, this method is not amenable for work within early drug discovery for which radiolabeled compounds are not routinely available. To this end, a simple method of incorporating the effects of hepatic uptake on clearance was sought, which obviated the need for either radiolabeled compound or the use of oil. The media loss assay used in this work focused on the disappearance of the parent drug from the incubation medium, which is effectively the inverse of monitoring for the appearance of parent drug within the cells. However by quantifying parent drug loss from the incubation medium, neither a radiolabeled drug nor an oil centrifugation step was required. An approach for investigating the appearance of nonradiolabeled drug within hepatocytes has been described in the literature recently for a limited dataset (Lam et al., 2006; Lu et al., 2006), but this method still requires the use of an oil centrifugation step.
When the clearance mechanism is hepatic and metabolic, data from this laboratory have shown that there can be a systematic underprediction of CLint ub in vivo, with an afe of 5-fold (Grime and Riley, 2006). If the hepatocyte uptake processes in vitro are similarly offset against those functioning in vivo, the dose/AUC0-∞ approach might be expected to underpredict CLint ub in vivo by a similar factor. However, the significant underprediction of CLint ub in vivo obtained for the majority of the 36 AZ compounds investigated in the rat using this method (Fig. 3B; Table 1) suggests that this is often not the case. A potential explanation for this phenomenon is that passive permeability in the isolated hepatocyte in vitro is substantially greater than that observed in vivo (Reinoso et al., 2001), artificially increasing AUC0-∞ and hence lowering CLint estimations (Fig. 2B). Up-regulation or enhanced activity of efflux transporters in such incubations would also produce a similar result and indeed some compounds in this dataset have been shown to be substrates for P-glycoprotein (PgP). Furthermore, compounds that are actively transported into the bile in vivo may be pumped back into the medium in these static hepatocyte experiments, which would also artificially increase AUC0-∞. Interestingly, a similar underprediction of CLint ub in vivo using the AUC0-∞ method was also observed using human hepatocytes (data not shown) for the known PgP substrate atorvastatin (Hochman et al., 2004) and montelukast. This is particularly pertinent because Zhao et al. (2005) have demonstrated an efficient PgP-mediated efflux using human hepatocytes.
For ∼20% of the compounds in the rat dataset, CLint ub in vivo was predicted equally well from the initial disappearance rate constant and the AUC0-∞ approach (Fig. 2C). Clearly for these compounds, drug clearance from the media is dominated by hepatocyte uptake because the initial phase contributes the majority of the total AUC. Although potential artifacts of the in vitro system may contribute to the underprediction of in vivo clearance using an AUC0-∞ method (Fig. 2B), data obtained from the terminal time points of these profiles allow the calculation of fmedium (see Results). For drugs exhibiting a significant difference between fuinc and fmedium (Table 1; Fig. 2, B and C), an assessment of hepatic uptake may be required for an accurate prediction of in vivo clearance. Hence, the calculation of fmedium provides a relatively easy initial screen to prompt further uptake studies.
A more simplistic approach for obtaining a CLint estimate from media loss data is to calculate a CLint from the “initial drug disappearance” (see Materials and Methods), synonymous with the appearance rates documented previously (Olinga et al., 1998; Shitara et al., 2003; Hirano et al., 2004). Figure 3C highlights the excellent prediction of CLint ub in vivo obtained for the compounds in Table 1 using this method. As anticipated, compounds for which conventional rat hepatocyte incubations accurately predicted in vivo clearance (AZ7-AZ10) were also predicted well using CLint estimates determined using a disappearance rate constant (Table 1). These data suggest that the use of a CLint estimate from a media loss assay provides a method suitable to predict in vivo clearance accurately, whether mediated by metabolism and/or hepatic uptake. Interestingly, although hepatic uptake has been thought to contribute most appreciably for poorly permeable compounds (Shitara et al., 2006), significant levels of uptake were observed in this study for several lipophilic (and highly permeable) compounds (e.g., acid AZ5) (Table 1).
The use of in vitro-in vivo scaling factors derived from studies in preclinical species to predict in vivo clearance in humans has been proposed by Naritomi et al. (2003). However this approach relies on a (quantitative) similarity in clearance route across the preclinical species and humans and has been contested recently (Ito and Houston, 2005). Previous studies from our laboratory (Riley et al., 2005) have demonstrated an excellent correlation between predicted CLint ub in-vivo determined for 57 drugs from conventional human hepatocyte incubations and observed CLint ub in vivo (50% drugs within 3-fold and 74% within 5-fold). However, there were several drugs for which CLint ub in vivo was significantly underpredicted, including prazosin and montelukast (30-fold). Therefore, the media loss assay was used to predict CLint ub in vivo for these drugs plus the known OATP substrates bosentan (Treiber et al., 2004), atorvastatin, and pravastatin (Shitara et al., 2006). The excellent prediction of CLint ub in vivo for each of the five drugs studied (within 4-fold) validates the use of this approach in humans. Although the CLint ub in vivo for prazosin was predicted within 2-fold using the media loss assay, CLint ub in vivo values determined via conventional human hepatocyte incubations (data not shown) also produced similar predictions. This finding suggests that the original underprediction in prazosin CLint ub in vivo was due to an underestimation in metabolic CLint rather than hepatic uptake.
Potential differences in hepatic uptake between rat and human were then investigated using the 36 AZ compounds in Table 1. Figure 4 suggests that for this dataset hepatic uptake in the rat was on average 3-fold more rapid (up to 12-fold) than in humans, confirming and expanding earlier reports (Sandker et al., 1994; Shitara et al., 2006). The uptake rate of estrone-3-sulfate, a known OATP1B1 substrate (Hirano et al., 2004), obtained in this study agreed with values determined with previous batches of human hepatocytes (Yamashiro et al., 2006), suggesting that this interspecies difference in uptake was not due to human tissue quality.
In this article, we described a media loss method for determining the hepatic uptake of drugs into rat and human hepatocytes. The utility of this method within an early discovery setting has been highlighted with reference to the successful prediction of in vivo clearance for >30 compounds in the rat. Future studies will focus on a more mechanistic approach to enable a thorough understanding into the active uptake process within hepatocytes in addition to the potential for active efflux within hepatocytes to confound clearance predictions. The aim of further work will be to build on preliminary investigations into the effect of cryopreservation on active uptake into human hepatocytes by Shitara et al. (2003).
Footnotes
-
doi:10.1124/dmd.106.014464.
-
ABBREVIATIONS: DMPK, drug metabolism and pharmacokinetics; OATP, organic anion transporting polypeptide; NCE, new chemical entity; AZ, AstraZeneca; BSA, bovine serum albumin; CLint, intrinsic clearance; AUC0-∞, area under the drug concentration-time curve from time zero to a point where the drug concentration is zero (extrapolated from the final two time points); CLint ub in vitro, unbound drug intrinsic clearance in vitro; CLint ub in vivo, unbound drug intrinsic clearance in vivo; fuinc, fraction of drug unbound in the hepatocyte incubation; fmedium, fraction of drug in the incubation medium; PgP, P-glycoprotein.
- Received December 20, 2006.
- Accepted March 1, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}