


Abstract
HepaRG is a highly differentiated cell line that displays several hepatocyte-like functions, including drug-metabolizing enzymes. In this study, the HepaRG cells were characterized and evaluated as an in vitro model to predict cytochrome P450 (P450) enzyme induction of drugs in humans. Exposure of HepaRG cells to prototypical inducers resulted in induction of CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4 mRNA, as well as phenacetin O-dealkylase, bupropion hydroxylase, diclofenac 4′-hydroxylase, and midazolam 1′-hydroxylase activities. The observed induction is consistent with the previously reported expression of the nuclear receptors pregnane X receptor, constitutive androstane receptor, and aryl hydrocarbon receptor, which are necessary for a P450 induction response. To avoid problems with toxicity and solubility, the induction potency of test compounds was evaluated by calculating the concentrations leading to a 2-fold increase of baseline mRNA or enzyme activity levels (F2 values) instead of EC50 values from full dose-response curves. For CYP3A4 mRNA, the obtained F2 values were related to the in vivo exposure [area under the plasma concentration versus time curve (AUC)] of the inducer (AUC/F2). This score was then correlated with the decrease in AUC for a CYP3A probe drug, administered before and after treatment with the inducing agent. By using this method an excellent correlation (R2 = 0.863) was obtained, which implies that the degree of CYP3A induction in vivo can be predicted from CYP3A4 mRNA induction in HepaRG cells. The present study shows that HepaRG cells are a valuable model to be used for prediction of induction of drug-metabolizing P450 enzymes in vivo in humans.
The cytochrome P450 (P450) enzymes have been shown to be responsible for the metabolism of the majority of therapeutics and are therefore important for first-pass elimination (bioavailability), clearance, and drug-drug interactions. Drug-drug interactions may be the result of both inhibition and induction of P450 enzymes. P450 inhibition studies using human liver microsomes or recombinant enzymes and model substrates for specific P450s are well established in vitro methods for quantitative predictions of inhibition potency in vivo (Ito et al., 1998; Bjornsson et al., 2003). On the contrary, quantitative predictions of induction in vivo from in vitro results are less well established. Because induction is subject to important species differences, prediction of possible induction in humans cannot rely on animal studies. Human hepatocytes have been suggested by several reports as a preferred model for induction studies (Li et al., 1997; Madan et al., 2003). Nevertheless, primary human hepatocytes are flawed with several drawbacks, such as experimentally demanding methods, phenotypic changes during culture, scarce availability, interindividual variation, and the need to evaluate multiple donors, which makes the model arduous. The demanding technique is probably the reason why no quantitative predictions of P450 induction in vivo using human hepatocytes have been published.
Induction of P450s generally occurs at the transcriptional level and is mediated by receptors such as the pregnane X receptor (PXR), constitutive androstane receptor (CAR), and the aryl hydrocarbon receptor (AhR). PXR is a transcription factor that is widely accepted as the major determinant of CYP3A4 gene regulation by xenobiotics (Lehmann et al., 1998; Gibson et al., 2002) and has also been established to be important for CYP2C9 induction (Chen et al., 2004), whereas CAR has been proposed to regulate the expression of CYP2B6 (Sueyoshi et al., 1999; Wang et al., 2004). However, during recent years there has been increasing evidence of an extensive cross-talk between nuclear receptors, indicating that CAR and PXR affect the regulation of all three P450 enzymes (CYP2B6, CYP2C9, and CYP3A4) (Lin, 2006). The CYP1A enzymes are regulated by AhR, and prototypical AhR ligands are planar, hydrophobic, and halogenated hydrocarbons such as 2,3,7,8-tetrachloro-dibenzo-p-dioxin (Denison and Nagy, 2003).
Reporter gene assays for PXR and AhR have been developed and used as convenient high-throughput tools for the determination of a compound's ability to induce CYP3A and CYP1A. Results from PXR reporter gene assays were recently successfully used to classify clinically used drugs as CYP3A inducers or noninducers (Persson et al., 2006; Sinz et al., 2006). However, because induction of P450 enzymes by drug molecules does not only depend on a single transcription factor but is mediated by a combination of several transcription factors in the cell, a human hepatocyte-like cell line would be a superior in vitro model for evaluation of P450 induction. A promising cell line is the HepaRG cells, which display several hepatocyte-like functions including drug-metabolizing enzymes, nuclear receptors, and hepatic drug transporters and can be differentiated into a hepatocyte-like morphology (Aninat et al., 2006; Le Vee et al., 2006). The HepaRG cells have also been shown to respond to prototypical P450 inducers such as 3-methylcholantrene, rifampicin, and isoniazid (Aninat et al., 2006).
The aim of the present study was to further characterize the P450 induction response in HepaRG cells and to develop a method that can be used to predict the extent of P450 induction in vivo.
Materials and Methods
Chemicals. Carbamazepine, dexamethasone, diclofenac, dimethyl sulfoxide (DMSO), nifedipine, phenobarbital, rifampicin, sulfinpyrazone, and Williams' E medium without phenol red were purchased from Sigma Chemical Co. (St. Louis, MO). Paracetamol, phenacetin, and primaquine were purchased from Aldrich Chemical Co. (St. Louis, MO). Hyperforin was purchased from Apin Chemicals Ltd. (Oxon, UK). Williams' medium E with phenol red and fetal bovine serum were obtained from Invitrogen (Carlsbad, CA). Omeprazole and troglitazone were provided by AstraZeneca R&D Mölndal (Mölndal, Sweden). 4′-Hydroxydiclofenac was obtained from BD Gentest (Woburn, MA), and 1′-hydroxymidazolam was purchased from Ultrafine (Manchester, UK). SuperScript III First-Strand Synthesis System for reverse transcription-polymerase chain reaction (PCR) and TRIzol reagent were acquired from Invitrogen (Carlsbad, CA). Midazolam was purchased from Larodan Fine Chemicals AB (Malmö, Sweden). Bupropion was purchased from Kemprotec Ltd. (Middlesbrough, UK), and hydroxybupropion was provided by Toronto Research Chemicals Inc. (North York, ON, Canada). LDH opt, reference LD401, was purchased from Randox Laboratories Ltd. (Crumlin, UK), and Triton X was purchased from Acros Organics (Geel, Belgium). The primers and probe used in this study were provided by Applied Biosystems (Cheshire, UK). Taqman Assay on Demand and Taqman Universal Master Mix were purchased from Applied Biosystems (Foster City, CA). All the other chemicals were of analytical grade and highest quality available.
Cell Culturing and Induction. The differentiated HepaRG cells (passages 14–17) were purchased from Biopredic International (Rennes, France). Twenty-four-well plates, seeded with 0.05 million cells, were used for all the experiments. The cells were initially grown in Williams' medium E with Glutamax I, supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, 5 μg/ml bovine insulin, and 50 μM hydrocortisone hemisuccinate. At confluence, 2% DMSO was added to the medium to differentiate the cells into a hepatocyte-like morphology. The cells were cultured in differentiation medium for 3 weeks before shipment to AstraZeneca. At arrival, the medium was renewed, and the cells were given 24 h to recover before the medium was changed to basal HepaRG medium (Williams' medium E with Glutamax I, supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 4 μg/ml bovine insulin, and 50 μM hydrocortisone hemisuccinate). The cells were cultured in the basal medium for 5 days before the induction started, and the medium was renewed every 24 to 48 h.
The cells were induced with rifampicin (0.004–4.0 μM), omeprazole (0.04–40 μM), troglitazone (0.024–25 μM), primaquine (0.04–40 μM), phenytoin (0.04–40 μM), phenobarbital (0.20–200 μM), carbamazepine (0.24–250 μM), dexamethasone (0.24–250 μM), nifedipine (0.24–250 μM), sulfinpyrazone (0.24–250 μM), and hyperforin (0.24–250 μM). Test compounds were dissolved in DMSO and added to the plates in basal HepaRG medium with a final DMSO concentration of 0.1% in all the incubations. Control incubations contained basal HepaRG medium and 0.1% DMSO. For mRNA measurements, the cells were harvested in TRIzol reagent (0.5 ml/well) after 24-h exposure to the study compounds, and experiments were repeated at three different occasions on separate batches of differentiated HepaRG cells. P450 activities were measured directly in the plates after 48-h exposure to the study compounds, and experiments were performed in duplicate and repeated at two different occasions on separate batches of differentiated HepaRG cells.
Viability. The viability of the cells in the dose-response experiments was tested by measuring the lactate dehydrogenase (LDH) activity before and after 24-h exposure to the study compounds. LDH was measured by using a commercial reagent kit (LDH opt) and was performed with a Cobas Bio centrifugal analyzer (Hoffman-La Roche, Basel, Switzerland).
RNA Extraction. Total RNA from HepaRG cells was prepared using TRIzol reagent according to manufacturers' instructions. Quantity and purity of the RNA were determined spectrophotometrically using a GenQuant pro RNA/DNA calculator (Biochrom, Cambridge, UK). Electrophoretic separation of 0.5 μg of total RNA on a 1% agarose gel run in Tris borate-EDTA buffer (0.09 M Tris-borate, 2 mM EDTA, pH 7.8) at 80 mV for 1 h allowed integrity assessment of the isolated RNA. Two sharp ribosomal RNA bands and absence of RNA debris were set as quality criteria to proceed to cDNA synthesis.
cDNA was prepared from 1 μg of total RNA using the SuperScript III First-Strand Synthesis System for reverse transcription-PCR with random hexamer primers according to the manufacturer's protocol.
Real-Time PCR. Real-time PCR for human P450 mRNA levels was performed using a 7500 Sequence Detector (Applied Biosystems, Foster City, CA) and manufacturer-designed Assay on Demand for CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A4, and glyceraldehyde-3-phosphate dehydrogenase. For human acidic ribosomal phosphoprotein (huPO), gene-specific double fluorescent labeled probes were used as reported previously (Persson et al., 2006). The reaction mixture (25 μl/well) contained 30 ng of cDNA, 1×Taqman Universal Master Mix, optimized concentrations of primers and probes (huPO), or 1.25 μl of Assay on Demand and RNase free water. The thermal cycle conditions were identical for all the genes analyzed and had initial steps of 50°C for 2 min and a 10-min step at 95°C, followed by 40 PCR cycles of 95°C for 15 s and 60°C for 1 min. Each sample was analyzed in triplicate, and data were analyzed using the 7500 Sequence detector software version 1.3.1 (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase and huPO were used as endogenous controls, and the amount of mRNA was determined relative to that from control samples.
Activity Measurements. For cells used for activity measurements, induction medium was renewed after 24 h. After 48 h, induction medium was removed, and the cells were washed twice with Williams' medium E without phenol red; thereafter, 500 μl of activity medium was added to each well. The activity medium consisted of Williams' medium E without phenol red, pH 7.4, and bupropion (100 μM) or a mixture of phenacetin (26 μM), diclofenac (9 μM), and midazolam (3 μM). The compounds were dissolved in methanol and added to a Falcon tube. The methanol was evaporated under nitrogen gas, and the compounds were dissolved in Williams' medium E without phenol red so that the activity medium did not include any organic solvent. After 15, 30, 45, and 60 min (for bupropion) or 0.5, 1, 2, and 3 h (for the mixture), 100 μl of sample was taken out. Fifteen microliters of cold acetonitrile with 0.8% formic acid was added, and the samples were analyzed for the CYP2B6 metabolite hydroxybupropion or the CYP1A2 metabolite paracetamol, the CYP2C9 metabolite 4′-hydroxydiclofenac, and the CYP3A4 metabolite 1′-hydroxymidazolam.
After the last sample was taken, the final medium was removed, and the cells were lysed with 200 μl of 1% Triton X. The wells were scraped, and the lysate was used for protein determination by a modified Lowry procedure (Markwell et al., 1978).
The samples were analyzed at separate occasions by means of liquid chromatography (LC)/mass spectrometry. The LC system consisted of an HP 1100 series LC pump and column oven (Agilent Technologies, Santa Clara, CA) combined with an HTS PAL injector (CTC Analytics, Zwingen, Switzerland). For hydroxybupropion, 4′-hydroxydiclofenac and 1′-hydroxymidazolam LC separations were performed on a reversed-phase HyPurity C18 column (2.1 × 50 mm, 5 μm, ThermoQuest, Runcorn, UK) with a HyPurity C18 precolumn at 40°C and with a flow rate at 750 μl/min. The mobile phase consisted of 0.1% (v/v) formic acid (A) and 0.1% (v/v) formic acid in acetonitrile (B). The organic modifier content B was increased linearly from 5 to 80% B over 3 min and then back to 5% B in 0.2 min.
For paracetamol, chromatography was performed on a Zorbax Eclipse XDB-C8 column (4.6 × 150 mm, 5 μm; Agilent Technologies) with a HyPurity C18 precolumn using the same system and mobile phase. The organic modifier content B was increased linearly from 2 to 30% B over 5 min, then from 30 to 80% over 2 min, and then back to 2% B in 0.1 min. The retention times of hydroxybupropion, 4′-hydroxydiclofenac, 1′-hydroxymidazolam, and paracetamol were 2.0, 2.9, 2.4, and 6.4 min, respectively. Detection was performed with a triple quadrupole mass spectrometer, API4000, equipped with electrospray interface (Applied Biosystems/MDS Sciex, Concord, ON, Canada). The mass spectrometry parameters were optimized using each analyte. The compound dependent parameters were as follows: the collision energy was set at 20, -15, 39, and 21 V for hydroxybupropion, 4′-hydroxydiclofenac, 1′-hydroxymidazolam, and paracetamol, respectively. Collision-activated dissociation gas was at 5, 5, 7, and 5, respectively. The multiple reaction monitoring transitions chosen were 256.1 > 237.8 for hydroxybupropion, 309.9 > 265.9 for 4′-hydroxydiclofenac, 342.0 > 202.7 for 1′-hydroxymidazolam, and 152.3 > 110.0 for paracetamol. A dwell time of 200 ms was used. Instrument control, data acquisition, and data evaluation were performed using Applied Biosystems/MDS Sciex Analyst 1.4 software.
Metabolite formation was calculated in the linear range of the time curve, and for hydroxybupropion the formation rate at 30 min was used. For paracetamol, 4′-hydroxydiclofenac, and 1′-hydroxymidazolam, the formation rate at 2, 1, and 1 h was used, respectively.
Curve-Fitting and Calculation of Induction Response. All the curve-fittings were carried out with XLfit 4.1.1 (ID Business Solutions, Emeryville, CA). Dose-response data were fitted to a Hill equation for one-site dose-response as follows: 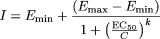
where Emin is the background, Emax the maximum effect, EC50 is the concentration giving 50% of Emax, k is the slope of the curve, and C is the drug concentration. A fit to the data points is created even if a plateau in the effect is not reached, but Emax values are only reported if a plateau is obtained. From this fit the F2 values also were calculated. The F2 value is the concentration leading to a 2-fold increase of the baseline levels. This approach was used by Weiss and Haefeli (2006) and found useful for evaluation of inhibition of P-glycoprotein.
The F2 values were related to the in vivo exposure of the test drug, represented by the area under the plasma concentration versus time curve (AUC). This score, AUC/F2, was then ranked from lowest to highest value.
In vivo induction is measured by administration of a drug metabolized by the enzyme of interest, called a probe drug. Examples of probe drugs are caffeine for CYP1A2, tolbutamide for CYP2B6, and midazolam for CYP3A4. The probe drug is administered before and after administration of the potential inducer, and the induction is measured as the decrease in AUC of the probe drug. The correlation between in vitro and in vivo induction in the present study was assessed by plotting AUC/F2 versus the percentage decrease in AUC of a coadministered CYP3A probe drug. The data were fitted to an equation as follows: 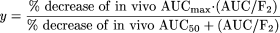
Results
Viability. The viability of the cells was analyzed by measuring LDH activity in the medium before and after 24-h exposure. No effect on the cells, as evidenced by no increased LDH activity in the medium, was detected after incubations with carbamazepine, dexamethasone, omeprazole, phenobarbital, primaquine, rifampicin, and troglitazone. Hyperforin, nifedipine, phenytoin, and sulfinpyrazone increased LDH activity at all the tested concentrations, although not in a dose-related fashion, and dose-response curves were still achieved. Hyperforin, nifedipine, and troglitazone displayed the highest induction at 62.5, 0.4, and 6.25 μM, respectively, whereas the induction response was weaker at the highest concentration (250, 2, and 25 μM, respectively). The weaker induction response by troglitazone at the highest concentration was not reflected by LDH leakage, suggesting that the induction response by troglitazone in this study was not affected by acute cell toxicity.
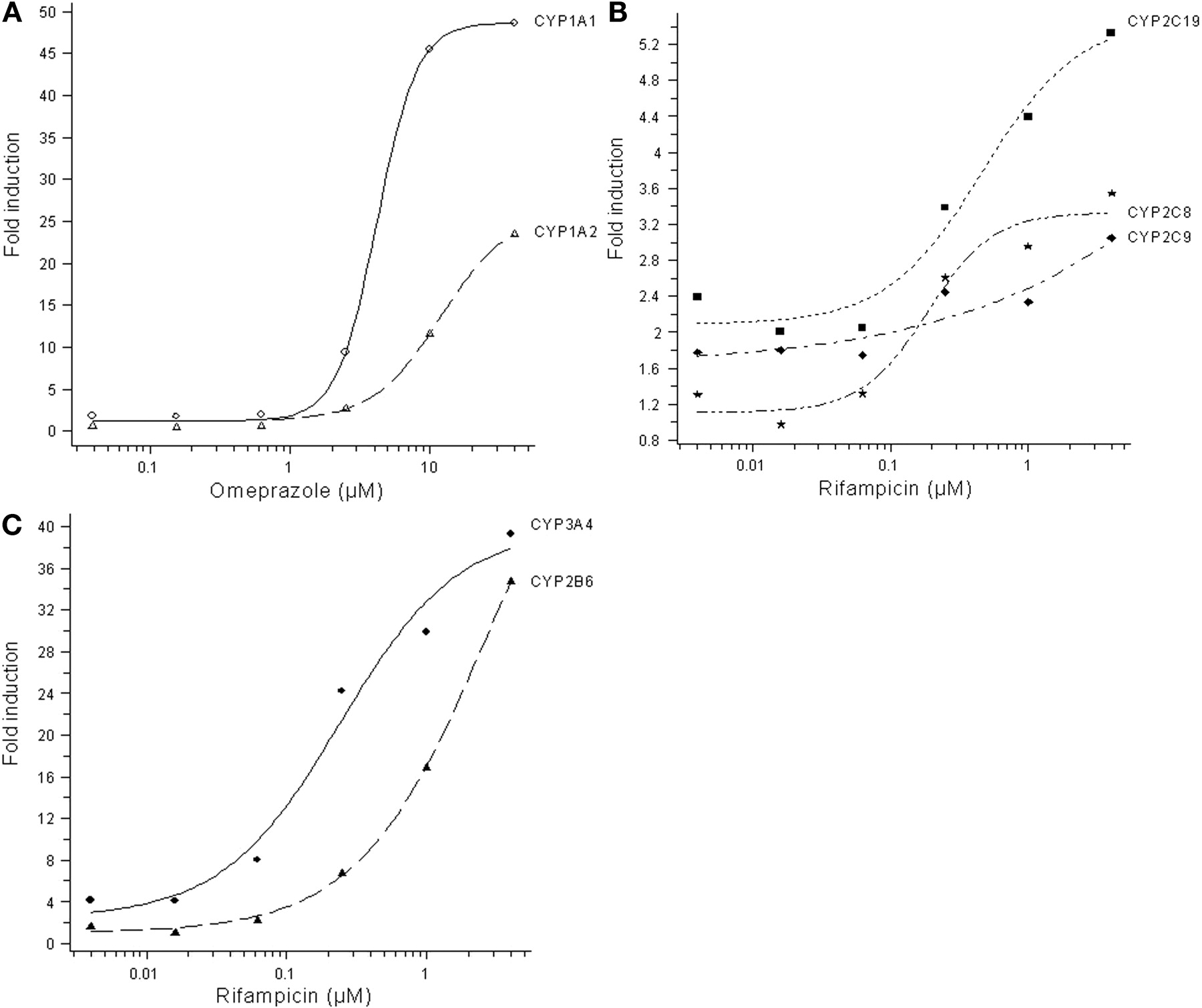
Induction of P450 mRNA. In initial experiments, HepaRG cells were treated with omeprazole, 2,3,7,8-tetrachloro-dibenzo-p-dioxin, phenobarbital, and rifampicin. Results indicated that the induction of CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4 mRNA peaked after 24 h. No induction was seen for CYP2D6 mRNA, and the mRNA for CYP2D6 was not analyzed in subsequent experiments. Therefore, the HepaRG cells were exposed to the study compounds rifampicin, omeprazole, troglitazone, primaquine, phenytoin, and phenobarbital for 24 h for mRNA experiments. Representative dose-response curves for CYP1A1 and CYP1A2 mRNA induction by omeprazole and for CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4 mRNA induction by rifampicin are shown in Fig. 1. In Tables 1 and 2, the F2 values and the -fold induction at the highest response measured for CYP1A1, CYP1A2, and CYP2B6 mRNA by the test compounds are presented. When a full dose-response curve was obtained and no sign of toxicity or solubility problems was observed, the EC50 value also was calculated. CYP1A1 and CYP1A2 mRNA was induced by omeprazole and primaquine, whereas no effect was seen by the other tested compounds (Table 1). CYP2B6 mRNA was strongly induced by rifampicin, omeprazole, troglitazone, phenytoin, and phenobarbital, whereas primaquine did not affect CYP2B6 transcription (Table 2). Induction of CYP2C8, CYP2C9, and CYP2C19 mRNA was seen after exposure to rifampicin (Table 3), whereas omeprazole, troglitazone, phenytoin, and primaquine did not significantly affect the CYP2C mRNA levels. Phenobarbital gave a weak induction of CYP2C mRNA, but the average -fold induction at the highest concentration was low (2.6-, 1.7-, and 2.5-fold for CYP2C8, CYP2C9, and CYP2C19, respectively), and no average F2 values could be calculated. A larger set of compounds was tested for CYP3A4 mRNA induction. Rifampicin, omeprazole, troglitazone, phenytoin, phenobarbital, carbamazepine, dexamethasone, hyperforin, nifedipine, and sulfinpyrazone all induced CYP3A4 mRNA, whereas primaquine did not affect CYP3A4 mRNA levels (Table 4).
F2 values and -fold induction at highest concentration tested (given in parenthesis) for induction of CYP1A1 and CYP1A2 mRNA in HepaRG cells treated with rifampicin, omeprazole, troglitazone, phenytoin, primaquine, or phenobarbital F2 is the concentration resulting in a 2-fold increase of baseline mRNA levels. EC50 values are presented when achieved. Values are mean ± S.D. of three separate batches of differentiated HepaRG cells.
F2 values and -fold induction at highest concentration tested (given in parenthesis) for induction of CYP2B6 mRNA in HepaRG cells treated with rifampicin, omeprazole, troglitazone, phenytoin, primaquine, or phenobarbital F2 is the concentration resulting in a 2-fold increase of baseline mRNA levels. EC50 values are presented when achieved. Values are mean ± S.D. of three separate batches of differentiated HepaRG cells.
F2 values and -fold induction at highest concentration tested (given in parenthesis) for induction of CYP2C8, CYP2C9, and CYP2C19 mRNA in HepaRG cells treated with rifampicin F2 is the concentration resulting in a 2-fold increase of baseline mRNA levels. Values are mean ± S.D. of three separate batches of differentiated HepaRG cells.
F2 values, -fold induction at highest concentration tested (given in parenthesis), and AUC/F2 for induction of CYP3A4 mRNA in HepaRG cells treated with rifampicin, omeprazole, troglitazone, phenytoin, primaquine, phenobarbital, carbamazepine, dexamethasone, hyperforin, nifedipine, or sulfinpyrazone F2 is the concentration resulting in a 2-fold increase of baseline mRNA levels. EC50 values are presented when achieved. Values are mean ± S.D. of three separate batches of differentiated HepaRG cells. Published values of in vivo exposure (AUC) for the same drugs are listed.
Induction of P450-Dependent Activities. The induction of P450-specific enzyme activities was more pronounced after 48-h exposure than after 24-h exposure to the test compounds; therefore, 48-h incubation was used for enzyme activity experiments. Induction of phenacetin O-dealkylase activity by omeprazole and bupropion hydroxylase, diclofenac 4′-hydroxylase, and midazolam 1′-hydroxylase activities by rifampicin gave clear dose-response curves over the concentration ranges tested (Fig. 2). In Table 5, the F2 values, the -fold induction at the highest response measured, and, when obtained, EC50 values for phenacetin O-dealkylase, bupropion hydroxylase, and midazolam 1′-hydroxylase activities after treatment with rifampicin, omeprazole, troglitazone, phenytoin, primaquine, and phenobarbital are presented.
F2 values and -fold induction at highest concentration tested (given in parenthesis) for induction of phenacetin O-dealkylase, bupropion hydroxylase, and midazolam 1′-hydroxylase activities in HepaRG cells treated with rifampicin, omeprazole, troglitazone, phenytoin, primaquine, or phenobarbital F2 is the concentration resulting in a 2-fold increase of baseline activities levels. EC50 values are presented when achieved. Values are mean ± S.D. of duplicates in two separate batches of differentiated HepaRG cells.
Induction of the CYP1A2-dependent phenacetin O-dealkylase activity was only detected by omeprazole. Bupropion hydroxylase activity, which is catalyzed by CYP2B6, was induced by rifampicin, phenytoin, and phenobarbital. Omeprazole and troglitazone did not result in measurable effects on bupropion hydroxylase activity, although they induced CYP2B6 mRNA. CYP2C9-catalyzed diclofenac 4′-hydroxylase activity was induced by rifampicin, and the average -fold induction at the highest concentration was 1.7-fold. CYP3A4-dependent midazolam 1′-hydroxylase activity was strongly induced by rifampicin and phenobarbital and weakly by troglitazone.
In Vitro-in Vivo Correlation. To investigate the relationship between P450 induction in HepaRG cells and induction in vivo, the same approach as for the previously reported PXR assay was applied (Persson et al., 2006), in which the in vivo exposure of the test compound was related to the EC50 from the PXR reporter gene assay (AUC/EC50). This method correctly identified compounds known to induce CYP3A in vivo. We recalculated the previously published PXR results to F2 values and found the same ranking and correct classification of the tested compounds when using F2 values instead of the EC50 values, which indicates that the F2 values could be a useful indicator for assessing induction potential.

Representative results of mRNA induction in HepaRG cells. Induction of CYP1A1 and CYP1A2 by omeprazole (A), CYP2C8, CYP2C9, and CYP2C19 by rifampicin (B), and CYP2B6 and CYP3A4 by rifampicin (C).
In the present study, the in vivo AUC for the investigated drugs was related to F2 values (AUC/F2) obtained for the induction of CYP3A4 mRNA in the HepaRG cells (AUC, F2, and AUC/F2 values are listed in Table 4). By using the results from HepaRG cells, the same ranking of compounds was obtained as with the AUC/F2 values from the PXR reporter gene assay (Persson et al., 2006). In Fig. 3, the AUC/EC50 and the AUC/F2 from activation of PXR in the reporter gene assay and the AUC/F2 from CYP3A4 mRNA induction in HepaRG cells are shown normalized to rifampicin, which was set to 100%. The same ranking of compounds was obtained for all three ratios.
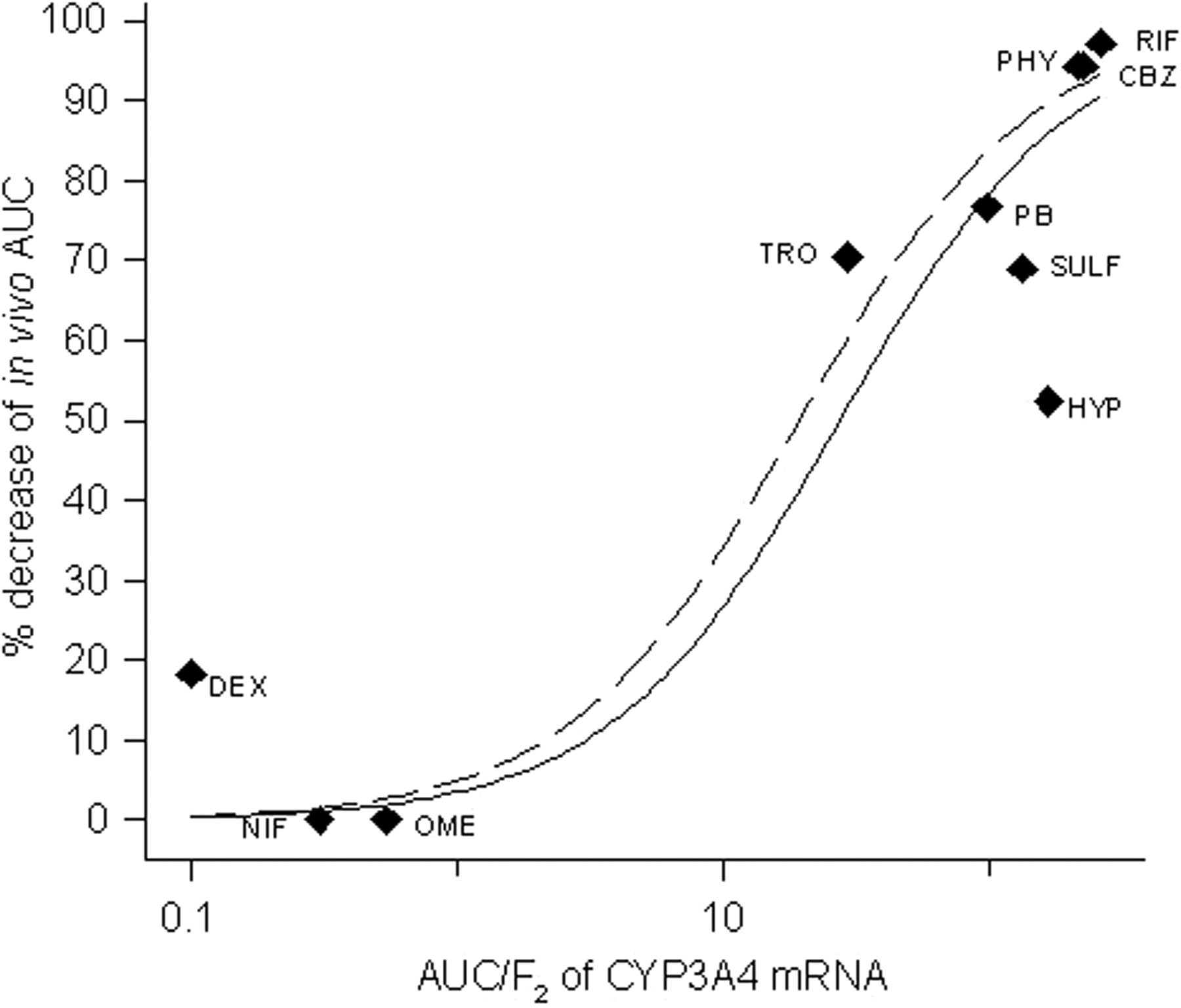
Induction of CYP3A4 mRNA in HepaRG cells by drugs was investigated as a quantitative measure of induction potency of the same drugs in vivo. Ten compounds (listed in Table 4) were selected on the basis that acceptable information on their induction potential in vivo is published. The in vivo induction is represented by the decrease in AUC for a CYP3A probe drug, administered before and after treatment with the inducing agent. The in vivo probes used for the correlation were midazolam for rifampicin (Backman et al., 1996a, 1998), phenytoin (Backman et al., 1996b), carbamazepine (Backman et al., 1996b), and hyperforin (St. John's wort) (Wang et al., 2001); verapamil for phenobarbital (Rutledge et al., 1988) and sulfinpyrazone (Wing et al., 1985); terfenadine for troglitazone (Loi et al., 1998); and triazolam for dexamethasone (Villikka et al., 1998). These probe drugs are moderate or high clearance drugs (Greenblatt et al., 1998; Wong et al., 1998; Lin, 2006), and the clearance and bioavailability are dependent on CYP3A4 metabolism. Omeprazole and nifedipine have not been found to induce CYP3A in vivo (Soons et al., 1992; Bowles et al., 1993). The correlation of AUC/F2 values from CYP3A4 mRNA induction and the in vivo induction (Fig. 4) show an excellent correlation (R2 = 0.863). When excluding hyperforin, a constituent of St. John's wort, the correlation factor increased to 0.943 (Fig. 4).
Using unbound AUC values gave a slightly lower in vitro-in vivo correlation (R2 = 0.859, data not shown). When the maximum plasma concentration in vivo (Cmax and Cmax,u) was used instead of AUC and AUCu, the R2 values were 0.741 and 0.812, respectively.
The corresponding evaluation of the predictive value for the induction of the other investigated P450s in the HepaRG cells is hampered by the lack of in vivo data.
Discussion
HepaRG is a new human hepatoma cell line developed from a liver carcinoma. The aim of the present study was to further evaluate the induction properties of the HepaRG cells and to develop a method to predict the magnitude of induction in vivo. In the present study, we show that the drug-metabolizing enzymes CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4 are inducible in the HepaRG cells. The compounds tested are known to mediate the induction response via the AhR, CAR, and/or PXR. Thus, the results are consistent with the reported expression of relevant transcription factors in the HepaRG cells (Aninat et al., 2006).
The results are from three separate batches of differentiated HepaRG cells. The small variations in -fold induction and F2 values obtained after exposure to test compounds indicate that the HepaRG cells will give reproducible and consistent results.
CYP1A1 and CYP1A2 mRNA were highly induced in HepaRG cells by the tested compounds previously known to induce CYP1A. The EC50 value for induction of CYP1A1 mRNA by omeprazole (5.9 μM) was slightly lower than the EC50 value (18 μM) for the activation of AhR by omeprazole previously reported in a reporter gene assay (Persson et al., 2006). The F2 values for the induction of phenacetin O-dealkylase activity was in the same order of magnitude as the F2 values for induction of CYP1A1 and CYP1A2 mRNA. Although the -fold induction of CYP1A1 and CYP1A2 mRNA was extensive after exposure to 40 μM omeprazole, the corresponding -fold induction of phenacetin O-dealkylase activity at 48-h exposure in the present study was only 3.5-fold. Omeprazole has in many in vitro studies been used as a positive control for activation of AhR and induction of CYP1A. However, omeprazole, or any other drug on the market that induces CYP1A or activate AhR in vitro, has not been found to induce CYP1A in vivo at relevant concentrations. Thus, any translation of the in vitro results for CYP1A induction to the in vivo situation is uncertain.
CYP2B6 mRNA and bupropion hydroxylase activity was highly inducible in HepaRG cells by rifampicin, phenobarbital, and phenytoin. Rifampicin and phenytoin are considered to be selective activators of PXR and CAR, respectively, whereas phenobarbital activates both receptors (Moore et al., 2000; Wang et al., 2004). The present results, together with previously reported mRNA expression of PXR and CAR (Aninat et al., 2006), indicate that HepaRG cells express both a functional PXR and a functional CAR.
In in vivo studies using P450-selective probes, the induction of CYP2C9 results in lower induction than induction of CYP3A when treating subjects with the same activators, such as rifampicin. This implies that CYP2C9 will be induced to a lesser degree than CYP3A by the same compounds. In accordance with the in vivo findings, the F2 values were higher and the -fold induction lower for CYP2C9 mRNA induction in HepaRG cells by rifampicin compared with CYP3A4 mRNA induction. The same phenomenon has also been shown in induction studies with primary human hepatocytes (Gerbal-Chaloin et al., 2001).
Induction of CYP2C8 and CYP2C19 mRNA in HepaRG cells by rifampicin is in the same range as induction of CYP2C9 mRNA, which corresponds with results from human hepatocytes (Gerbal-Chaloin et al., 2001). Both CYP2C8 and CYP2C19 are also induced in vivo by rifampicin (Zhou et al., 1990; Niemi et al., 2004; Park et al., 2004).
CYP3A4 was highly induced by all the compounds known to activate PXR and/or CAR. The EC50 for rifampicin induction of CYP3A4 mRNA (0.42 μM) and midazolam 1′-hydroxylase activity (0.12 μM) in HepaRG cells is consistent with the EC50 value obtained in the PXR reporter gene assay (0.20 μM) by the same drug (Persson et al., 2006), which suggests that PXR is the major receptor regulating the rifampicin induction of this gene product.
One important application of in vitro P450 induction studies in drug discovery is to predict the risk for induction of drug metabolism in vivo. In a previous study it was shown that the EC50 values from a PXR reporter gene assay could be used to distinguish between CYP3A inducers and noninducers when related to the in vivo exposure of the test compound (Persson et al., 2006). In the present study, F2 values from CYP3A4 mRNA induction related to in vivo AUC of the test compounds were found to give the same correct ranking of inducers and noninducers. The results suggest that instead of EC50 values from full dose-response curves, F2 values could be used to evaluate the induction response in the cell system. This is an advantage because an Emax many times cannot be achieved or may be confounded as a result of toxicity or solubility problems. For example, troglitazone did not affect viability but showed a decrease in CYP3A4 induction response at the highest concentration, which may affect the Emax value and as a consequence the EC50.
The AUC/F2 values for CYP3A4 mRNA induction were further evaluated as possible predictors of the magnitude of induction in vivo. In the present study we used results from published in vivo induction studies utilizing different probe drugs for measuring CYP3A4-dependent clearance. The probe drugs used in the current in vivo studies are all intermediate or high clearance drugs, metabolized and cleared mainly by CYP3A4. Therefore, they should be affected to a similar extent by CYP3A4 induction (Lin, 2006). Such an assumption is supported by the fact that the effect of rifampicin on AUC for the high/medium clearance drugs midazolam, verapamil, and triazolam after p.o. dosing is comparable (Backman et al., 1996a, 1998; Fromm et al., 1996; Villikka et al., 1997). An excellent correlation was found between the decrease of in vivo AUC for CYP3A probe drugs and AUC/F2 for the tested drugs. Thus, the results indicate that the AUC/F2 values for CYP3A4 mRNA response in HepaRG cells could be used as a reliable in vitro measure for evaluating CYP3A induction potency of compounds in vivo.
In the in vitro-in vivo correlation analysis, a component of St. John's wort, hyperforin, was included. The manufacture of herbal remedies is not as rigorously controlled as production of drugs, which results in varying concentration of the components in different extracts. This means that in vivo data for hyperforin could be confounded by varying concentrations of hyperforin in the extracts. If hyperforin is excluded from the correlation, a higher correlation factor is obtained, which could indicate that this may be the case. Besides hyperforin, the compound deviating the most from the correlation is dexamethasone, a more potent inducer in vivo than what was predicted from AUC/F2 values. The reason for this is obscure and proposes further investigations of this compound.
The Fa2N-4 cells were investigated by Ripp et al. (2006) as a model for predicting CYP3A induction in vivo. The in vitro Emax and EC50 values for a number of potential CYP3A drug inducers were related to in vivo Cmax for the same drugs. The induction score obtained showed an excellent correlation with the degree of induction in vivo. In the present study on HepaRG cells, using Cmax instead of AUC values for calculation of induction scores gave a poorer correlation with the degree of induction in vivo for the tested drugs.
According to the Food and Drug Administration guideline (http://www.fda.gov/cder/guidance/6695dft.pdf), a test compound should reach an induction level equal to or greater than 40% of a positive control to be considered as relevant for the in vivo situation. In the present study all the compounds known to induce CYP3A4 in vivo reached at least 40% of rifampicin induction, except the well known CYP3A inducer phenytoin. Phenytoin has previously been shown to be a weak inducer in PXR reporter gene assays and human hepatocytes, exhibiting an induction response close to or less than 40% of rifampicin response (Luo et al., 2002; Persson et al., 2006). Therefore, substances like phenytoin could be overlooked if using a conservative cut-off value for in vivo relevance. In the present study, 40 μM phenytoin gave an induction response that was only 14% of maximum rifampicin induction. However, using the AUC/F2 value for the CYP3A4 mRNA induction response in HepaRG cells correctly predicted the degree of induction in vivo by phenytoin. Furthermore, strong in vitro CYP3A4 inducers like omeprazole and nifedipine (50 and 92% of maximum rifampicin induction, respectively) were correctly classified as noninducers when using AUC/F2 values. These results emphasize the importance to relate the in vitro results to in vivo exposure for both seemingly “strong” and “weak” inducers in the in vitro system.
In conclusion, we have shown that HepaRG cells respond to PXR, CAR, and AhR activators, resulting in induction of CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4. Further, it was shown that the F2 values for CYP3A4 mRNA induction in HepaRG cells could be used not only to classify compounds as inducers or noninducers of CYP3A but also to predict the extent of induction in vivo in humans. Thus, the HepaRG cell line could be used as an important in vitro model for investigations of enzyme induction in drug discovery.
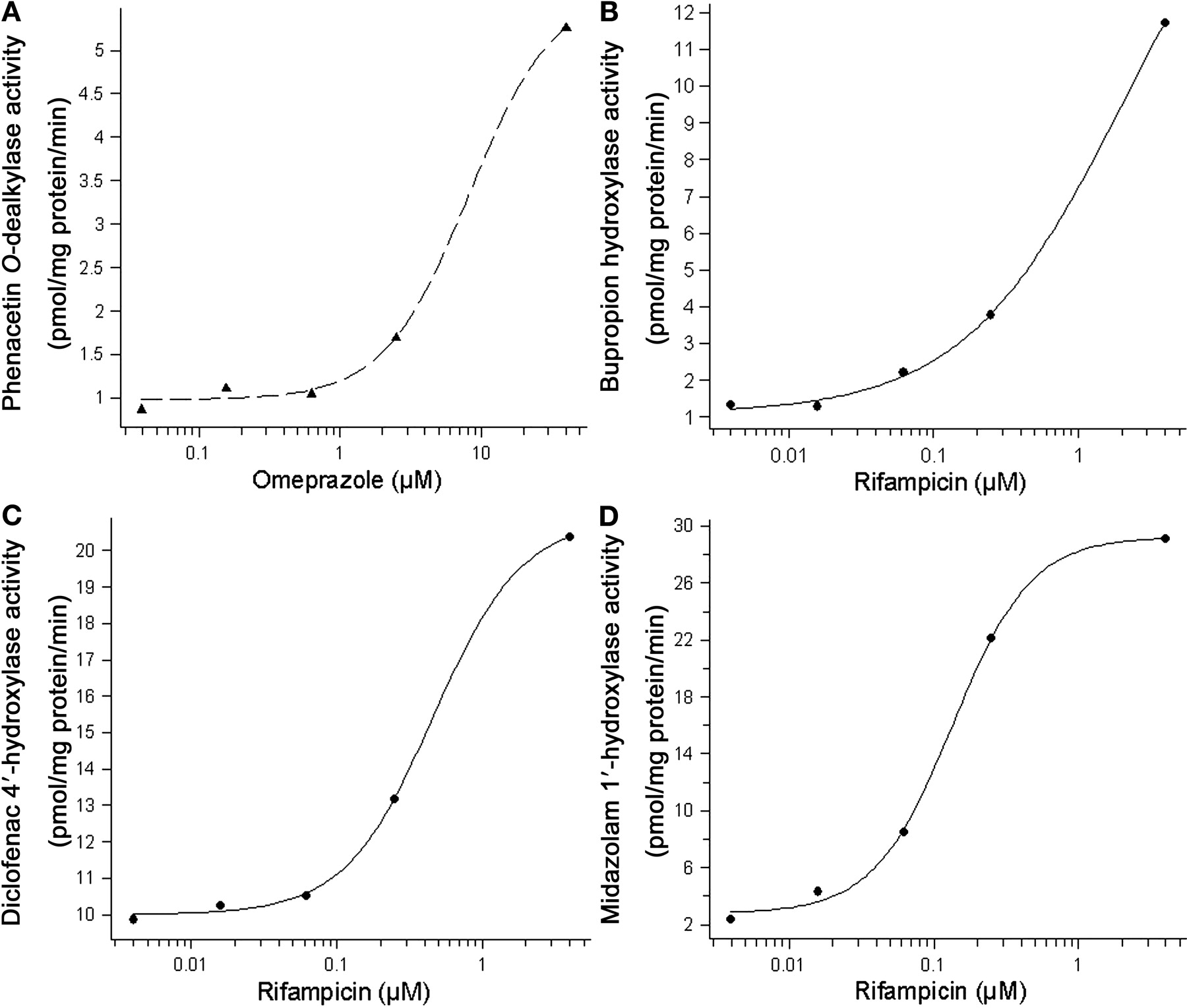
Representative results of induction of enzyme activities in HepaRG cells. Induction of phenacetin O-dealkylase (CYP1A2) activity by omeprazole (A), bupropion hydroxylase (CYP2B6) activity by rifampicin (B), diclofenac 4′-hydroxylase (CYP2C9) activity by rifampicin (C), and midazolam 1′-hydroxylase (CYP3A4) activity by rifampicin (D).

Ranking of in vitro results. The AUC/EC50 from PXR assay (white bars), AUC/F2 from PXR assay (gray bars) [data from (Persson et al., 2006)], and AUC/F2 CYP3A4 mRNA in HepaRG cells (black bars). The study compounds were normalized to the rifampicin response, which were set to 100%.
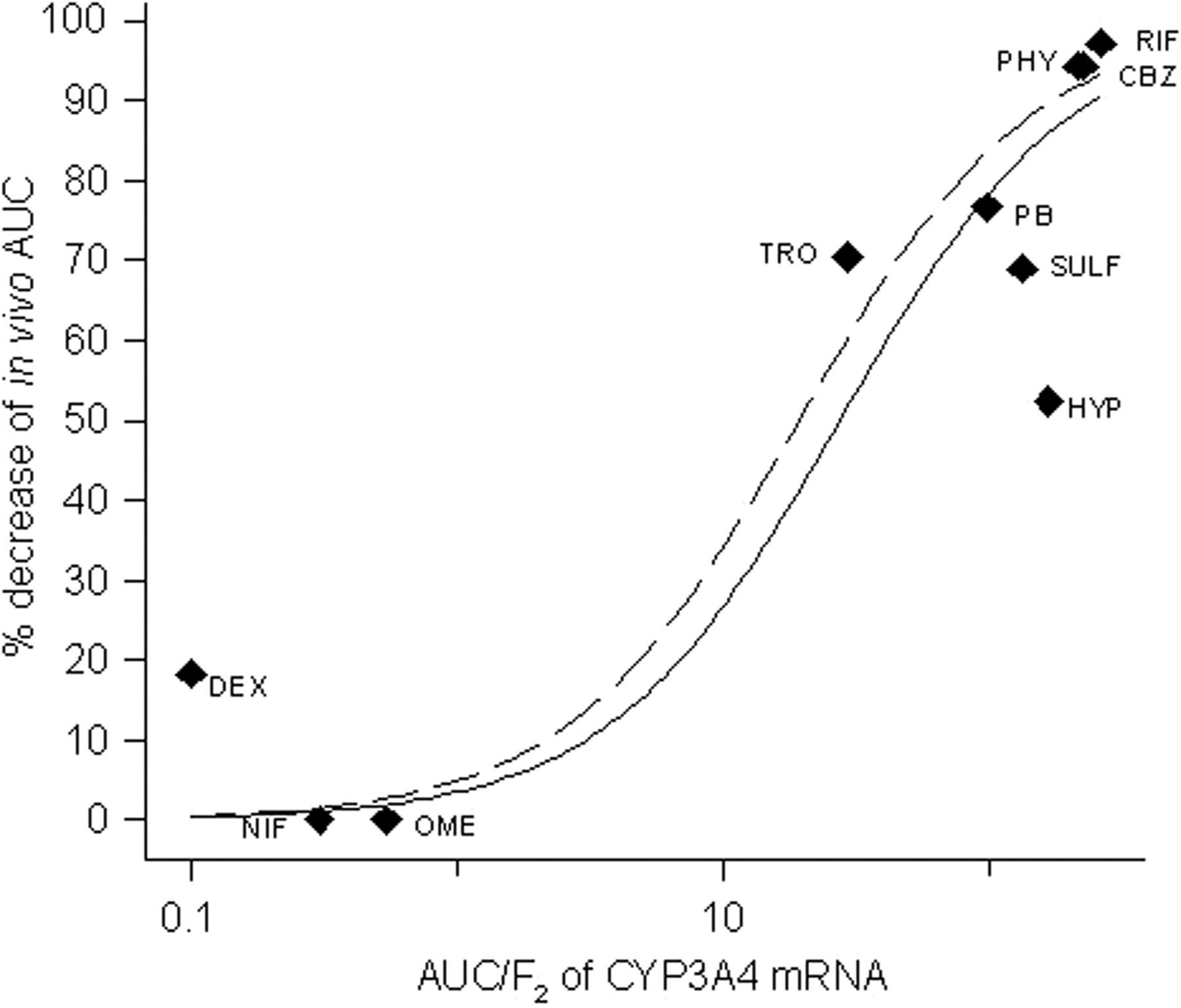
Correlation of AUC/F2 of CYP3A4 mRNA in HepaRG cells with percentage decrease of in vivo AUC for CYP3A probe drugs (whole line), R2 = 0.863. The equation used is described under Materials and Methods. Correlation when hyperforin is excluded (dashed line), R2 = 0.943. Compound abbreviations: CBZ, carbamazepine; DEX, dexamethasone; HYP, hyperforin; NIF, nifedipine; OME, omeprazole; PB, phenobarbital; PHY, phenytoin; RIF, rifampicin; SULF, sulfinpyrazone; TRO, troglitazone.
Acknowledgments
We thank Gisela Häggblad, Anne-Cristine Carlsson, and Dr. Lennart Nilsson for measurement of LDH activity.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.017418.
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; CAR, constitutive androstane receptor; AhR, aryl hydrocarbon receptor; DMSO, dimethyl sulfoxide; PCR, polymerase chain reaction; LDH, lactate dehydrogenase; huPO, human acidic ribosomal phosphoprotein; LC, liquid chromatography; AUC, area under the plasma concentration versus time curve.
- Received June 26, 2007.
- Accepted October 17, 2007.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}