


Abstract
To verify the availability of pharmacokinetic parameters in cynomolgus monkeys, hepatic availability (Fh) and the fraction absorbed multiplied by intestinal availability (FaFg) were evaluated to determine their contributions to absolute bioavailability (F) after intravenous and oral administrations. These results were compared with those for humans using 13 commercial drugs for which human pharmacokinetic parameters have been reported. In addition, in vitro studies of these drugs, including membrane permeability, intrinsic clearance, and p-glycoprotein affinity, were performed to classify the drugs on the basis of their pharmacokinetic properties. In the present study, monkeys had a markedly lower F than humans for 8 of 13 drugs. Although there were no obvious differences in Fh between humans and monkeys, a remarkable species difference in FaFg was observed. Subsequently, we compared the FaFg values for monkeys with the in vitro pharmacokinetic properties of each drug. No obvious FaFg differences were observed between humans and monkeys for drugs that undergo almost no in vivo metabolism. In contrast, low FaFg were observed in monkeys for drugs that undergo relatively high metabolism in monkeys. These results suggest that first-pass intestinal metabolism is greater in cynomolgus monkeys than in humans, and that bioavailability in cynomolgus monkeys after oral administration is unsuitable for predicting pharmacokinetics in humans. In addition, a rough correlation was also observed between in vitro metabolic stability and Fg in humans, possibly indicating the potential for Fg prediction in humans using only in vitro parameters after slight modification of the evaluation system for in vitro intestinal metabolism.
Because the development of new drugs is a cost- and labor-intensive task, the selection of candidates with good pharmacokinetic profiles is becoming increasingly common. This practice minimizes the number of drug candidates dropped due to pharmacokinetic problems during the clinical phase (Wishart, 2007).
When predicting human pharmacokinetics, the fraction absorbed (Fa), intestinal availability (Fg), and hepatic availability (Fh) are the main factors to consider. Fh prediction has become considerably accurate since several mathematical prediction models have been established, including the physiological model, well stirred model, parallel tube model, and dispersion model (Iwatsubo et al., 1996; Naritomi et al., 2001; De Buck et al., 2007). For FaFg, however, no quantitative prediction method has ever been established, although several qualitative prediction methods using human intestinal microsomes have been reported (Chiba et al., 1997; Shen et al., 1997; Fagerholm, 2007; Fisher and Labissiere, 2007; Yang et al., 2007). For these reasons, we have mainly used animal pharmacokinetic parameters to predict human FaFg in the drug discovery stage.
It has been regarded as natural that monkey metabolism is the most similar to that of humans, and cynomolgus monkeys have been widely used in pharmacokinetic or drug-safety studies for that reason. In the last decade, however, cynomolgus monkeys have often been found to have a poorer bioavailability (F) than other animal species for many compounds (Tabata et al., 2009).
More recently, several reports have stated that the intestinal transit process, namely Fa or Fg, is a major contributor to the low F in cynomolgus monkeys (Sakuda et al., 2006; Takahashi et al., 2008). However, unlike Fh, which can be easily calculated via conventional pharmacokinetic analysis, Fa and Fg are difficult to evaluate separately, particularly in the intestine. Consequently, few systemic studies have explored the usefulness of using monkey FaFg parameters to predict human pharmacokinetics.
Chiou and Buehler (2002) reported that the Fa and total clearance, corrected by hepatic blood flow rate, correlated well between humans and monkeys. This finding suggests that the species difference may be caused by Fg. In addition, our laboratory reported that midazolam (MDZ) had a markedly lower F (2.0%) in cynomolgus monkeys than in humans (24–46%), which was caused by high first-pass intestinal metabolism (Sakuda et al., 2006). Similar results reported by Nishimura et al. (2007) showed that extensive metabolism in the intestine is the cause of MDZ's low F in cynomolgus monkeys.
In the present study, the following studies were performed to further investigate the species differences between humans and cynomolgus monkeys. Thirteen commercially available drugs for which the human pharmacokinetic parameters are known were selected and classified into five categories according to cytochrome P450 (P450) isozyme selectivity and p-glycoprotein (P-gp) affinity.
The 13 drugs were intravenously and orally administered to cynomolgus monkeys to obtain the in vivo pharmacokinetic parameters (F, Fh, and FaFg) for each drug, which were then compared with those in humans. In addition, we also obtained the in vitro parameters for all 13 drugs, including protein binding, blood-to-plasma concentration ratio (Rb), membrane permeability, in vitro intrinsic clearance (CLint) in liver microsomes (CLintliver), CLint in intestine microsomes (CLintintestine), and P-gp affinity.
In this report, we discuss the main factor affecting the species difference between humans and cynomolgus monkeys indicated by these results. We also discuss the adequacy of cynomolgus monkeys as an animal model for predicting human pharmacokinetics.
Materials and Methods
Chemicals.
MDZ (Dormicam, 5 mg/ml solution for intravenous injection) was obtained from Astellas Pharma Inc. (Tokyo, Japan). Tacrolimus (TAC), which was synthesized at our laboratory, was used. Lithium carbonate (Li) was purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). Hydrochlorothiazide (HT), verapamil (VER), propranolol (PRO), and amitriptyline (AMI) were purchased from Wako Pure Chemicals (Osaka, Japan). Dexamethasone (DEX), nifedipine (NIF), quinidine (QID), timolol (TIM), and ibuprofen (IBU) were purchased from Sigma-Aldrich (St. Louis, MO). Liver and intestine microsomes from humans and cynomolgus monkeys were purchased from XenoTech, LLC (Lenexa, KS). All other reagents and solvents were commercial products of analytical grade.
Animals.
Male cynomolgus monkeys (Shin Nippon Biomedical Laboratories, Ltd., Kagoshima, Japan, and Astellas Research Technology, Osaka, Japan) weighing approximately 5 kg were used. The animal experiment was conducted according to the ethical rules of each company.
Selected Drugs and Categorization.
We allocated the 13 drugs into five categories (Type A–E), according to their pharmacokinetic properties in humans, as follows: membrane permeability, P450 isozyme selectivity, and P-gp affinity (Yu, 1999; Kivist et al., 2004; Yang et al., 2006) (Table 1).
Classification of each drug based on P450 isozyme selectivity and P-gp affinity
Type A.
The drugs categorized as Type A are indicator drugs that undergo no metabolism in humans and are not P-gp substrates. For each of these, almost all of the absorbed drug is excreted into urine as the unchanged form. Li, which has a high F in humans (94.5%) (Arancibia et al., 1986), and HT, which has a moderate F in humans (60.2%) (Patel et al., 1984), were assigned to this category.
Type B.
The drugs categorized as Type B are CYP3A4 substrates, and they have very weak, if any, affinity for P-gp. DEX, which has a high F in humans (81.4%) (Duggan et al., 1975), NIF, and MDZ, which have a moderate F in humans [41.2% (Holtbecker et al., 1996) and 30.0% (Thummel et al., 1996), respectively] were assigned to this category.
Type C.
The drugs categorized as Type C are substrates of both CYP3A4 and P-gp. QID, which has a high F in humans (79.5%) (Greenblatt et al., 1977), as well as TAC and VER, which have a moderate F in humans [23.3% (Moller et al., 1999) and 18.0% (McAllister and Kirsten, 1982), respectively], were assigned to this category.
Type D.
Digoxin (DIG), which is substrate of P-gp but not CYP3A4, was categorized as Type D. DIG has a high F in humans (65.3%) (Hinderling and Hartmann, 1991) and undergoes almost no metabolism in the human body, i.e., it undergoes only P-gp efflux during the absorption process in the intestine.
Type E.
The drugs categorized as Type E are mainly metabolized by the P450 isozyme (except CYP3A4) and have very weak, if any, affinity for P-gp. IBU and TIM, which have a high F in humans [100% (Martin et al., 1990) and 61.0% (Wilson et al., 1982), respectively], as well as AMI and PRO, which have a moderate F in humans [47.7% (Schulz et al., 1983) and 29.0% (Borgstrom et al., 1981), respectively], were assigned to this category. See Table 1 for P450 isozymes that contribute to each drug metabolism.
Pharmacokinetic Study in Cynomolgus Monkeys.
Intravenous and oral administrations were performed with a washout period of at least 7 days between each type of administration. Animals were fasted for approximately 17 h before dosing. Blood samples were collected from the antebrachial vein, kept in an ice-water bath, and then centrifuged at 10,000 rpm for 1 min at 4°C. The plasma samples were kept in a deep freezer (approximately −20°C) until analysis. The experimental conditions for the pharmacokinetic studies, including doses, dosing solution, dosing volume, and sampling time for each drug, are shown in Table 2. Values obtained from the literature were used as the pharmacokinetic parameter values for all selected drugs in humans as well as those for MDZ in cynomolgus monkeys.
Experimental conditions of cynomolgus monkey pharmacokinetic studies
The intravenous and oral administrations conditions for MDZ were taken from Sakuda et al., 2006.
Measurement of Model Compounds Plasma Concentration in Cynomolgus Monkeys.
The concentrations of model drugs in cynomolgus monkey plasma were determined by using atomic absorption, enzyme immunoassay analysis, or high-performance liquid chromatography (LC) coupled with tandem mass spectrometry (MS/MS) with sample pretreatment.
Atomic absorption method: Lithium.
The lithium level in the plasma was determined by using atomic absorption in accordance with the method of Pybus and Bowers (1970).
Enzyme immunoassay analysis: Dexamethasone and tacrolimus.
The DEX level in the plasma and the TAC level in the blood were determined by using enzyme immunoassay. After extraction (see below), an aliquot was used as the sample for analysis by enzyme immunoassay (Tamura et al., 1987).
A 50-μl aliquot of plasma was buffered with 1% skim milk/phosphate-buffered saline. After the addition of 1 ml of distilled water, the mixture was extracted with 5 ml of diethyl ether, and the solvent was removed under a stream of nitrogen gas. The residue was then dissolved in 250 μl of skim milk (1%)/phosphate-buffered saline.
LC-MS/MS analysis.
The plasma concentrations of all other drugs were determined by using LC-MS/MS. The LC-system comprised a LC-VP/LC-10A series (Shimadzu, Kyoto, Japan) or HP-1100 series HPLC (Agilent Technologies, Santa Clara, CA). The MS/MS experiments were conducted by using API-2000 or API-3000 LC/MS/MS systems (Applied Biosystems, Foster, CA). The details of the LC-MS/MS conditions, including the machines and columns used for each drug, are shown in Table 3.
Apparatus and LC-MS/MS analytical conditions for each drug during the determination of concentration in cynomolgus monkey plasma
Hydrochlorothiazide.
A 200-μl aliquot of plasma was buffered with 500 μl of phosphate buffer (10 mM) adjusted to pH 3.0. After adding 100 μl of acetonitrile and 20 μl of internal standard solution (1 μg/ml diclofenac in 50% acetonitrile), the mixture was extracted with 4 ml of ethyl acetate, and the solvent was removed under a stream of nitrogen gas. Then, the residue was dissolved in 100 μl of mobile phase, and a 40-μl aliquot was injected into the LC-MS/MS (molecular>product: m/z = 296 > 269 [M+H]−).
Nifedipine.
A 50-μl aliquot of plasma, 50 μl of acetonitrile (50%), and 100 μl of internal standard solution (1 μg/ml of in-house compound A in acetonitrile) were mixed well and then centrifuged to remove precipitated protein. The supernatant (100 μl) was then decanted, and 30 μl was injected into the LC-MS/MS (molecular>product: m/z = 347 > 315 [M+H]+).
Quinidine, Verapamil, Propranolol, Amitriptyline, and Timolol.
A 200-μl aliquot of plasma was buffered with 500 μl of saturated sodium bicarbonate solution. After the addition of 50 μl of acetonitrile and 50 μl of internal standard solution (1 μg/ml of in-house compound B in 50% acetonitrile), the mixture was extracted with 3 ml of tert-butyl methyl ether, after which the solvent was removed under a stream of nitrogen gas. The residue was then dissolved in 200 μl of mobile phase, and a 20-μl aliquot was injected into the LC-MS/MS (molecular>product: QID m/z = 325 > 307 [M+H]+, VER m/z = 455 > 165 [M+H]+, TIM m/z = 317 > 261 [M+H]+, AMI m/z = 278 > 117 [M+H]+, PRO m/z = 260 > 116 [M+H]+).
Digoxin.
A 200-μl aliquot of plasma was buffered with 500 μl of phosphate buffer (10 mM) adjusted to pH 3.0. After the addition of 100 μl of acetonitrile and 50 μl of internal standard solution (1 μg/ml digitoxin in 50% acetonitrile), the mixture was extracted with 3 ml of ethyl acetate, and the solvent was removed under a stream of nitrogen gas. The residue was then dissolved in 100 μl of mobile phase, after which a 20-μl aliquot was injected into the LC-MS/MS (molecular>product: m/z = 798 > 391 [M+NH4]+).
Ibuprofen.
A 200-μl aliquot of plasma was buffered with 500 μl of phosphoric acid (5 mM). After the addition of 50 μl of acetonitrile and 50 μl of internal standard solution (1 μg/ml of diclofenac in 50% acetonitrile), the mixture was extracted with 3 ml of tert-butyl methyl ether, and the solvent was removed under a stream of nitrogen gas. The residue was then dissolved in 200 μl of mobile phase, and a 20-μl aliquot was injected into the LC-MS/MS (molecular>product: m/z = 205 > 161, [M+H]−).
Blood-to-Plasma Concentration Ratio.
One milliliter of human and cynomolgus monkey blood was spiked with 10 μl of standard solution (100 μg/ml; 1000 ng/ml final) and preincubated in a shaking water bath at 37°C for 10 min. A 200-μl aliquot was then analyzed to determine the drug concentration in the blood. The remaining samples were centrifuged at 1800g for 10 min at 4°C, after which the drug concentration in 200-μl aliquots of plasma was determined. The Rb was then calculated from the concentrations of drug per milliliter of blood and plasma. All data regarding TAC level in humans and cynomolgus monkeys were determined by blood level base because the Rb value of TAC has been reported to be nonlinear, with values between 10 and 40 depending on the drug concentration in humans (Wallemacq et al., 1993).
Parallel Artificial Membrane Permeability Assay.
The parallel artificial membrane permeability assay (PAMPA) method was carried out by using a PAMPA Evolution instrument from pION INC. (Woburn, MA) (Avdeef et al., 2005). The lipid solution consisted of a 20% (w/v) dodecane solution and lecithin mixture. The donor solutions consisted of test compounds dissolved in 10 mM dimethylsulfoxide diluted in pH 6.5 buffer (final concentration of 50 μM). The acceptor plate was filled with 1% (w/v) SDS in water, and the pH was adjusted to 7.4 with 1N hydrochloric acid. The test plate was incubated for 120 min at 30°C. The concentration of each test compound in the reference, donor, and acceptor plates was measured with a UV plate reader. The permeability coefficient was calculated by using Evolution Library Manager software version 2.2 (pION INC.).
Plasma Protein Binding.
The plasma protein binding (unbound drug fraction in plasma) was determined by using the equilibrium dialysis method or ultracentrifugation method and the following equations: protein binding (%) = (1 − fp) × 100, and fp = concentration in filtrate or supernatant/concentration in serum, where fp is the unbound drug fraction in plasma. The unbound drug fraction in blood (fb) was calculated by dividing fp by Rb.
Equilibrium dialysis method.
A DIANORM dialysis device (Diachema, Zürich, Switzerland), which is impermeable to substances with molecular weights greater than 10,000, was used. Aliquots (3.5-ml) of human and cynomolgus monkey plasma were spiked with 35 μl of standard solution (100 μg/ml; 1000 ng/ml final) and preincubated in a 37°C shaking water bath for 10 min.
One milliliter of mixture and isotonic phosphate buffer solution (pH 7.4) was put into the dialyzing cell and receptor cell, respectively. After 4-h incubation at 37°C, the plasma mixture and buffer sample were stored in 100-μl aliquots at −20°C until analysis.
Ultracentrifugation method.
Ten microliters of standard solution (100 μg/ml) was added to 1000 μl of human or cynomolgus monkey plasma. The calibration samples were prepared by adding 17 μl of acetonitrile (50%) to 1700 μl of human or cynomolgus monkey plasma. These samples were then centrifuged at 436,000g for 140 min at 37°C by using a Beckman Optimal TL ultracentrifuge (Beckman Coulter, Fullerton, CA). After ultracentrifugation, the unbound fp was calculated by dividing the concentration of drugs in the supernatant by that in the plasma.
In Vitro Metabolism in Liver and Intestine Microsomes.
Metabolism study conditions.
The time courses of the unchanged drugs were obtained. Each drug was incubated at 37°C with a reaction mixture (1 ml) containing 500 μl of potassium-phosphate buffer (200 mM; pH 7.4), 100 μl of 1 mM EDTA-NaOH (pH 7.4), 100 μl of liver or intestine microsomes solution (the final concentration of microsomal protein was 0.05 mg/ml for TAC, 0.5 mg/ml for HT and DIG, and 0.2 mg/ml for all other drugs), 190 μl of distilled water, and 10 μl of each compound solution in 50% acetonitrile (final concentration: 0.2 μM).
After a 5-min preincubation, the reaction was initiated by the addition of 100 μl of an NADPH-generating system. The reaction was terminated by adding 100 μl of reaction mixture to 200 μl of acetonitrile including the internal standard at various time periods. After stopping the enzyme reaction, the reaction mixture of TAC and DIG was extracted with 3 ml of tertiary butyl methyl ether, and the solvent was removed under a stream of nitrogen gas. The residue was then dissolved in 150 μl of mobile phase, and a 10-μl aliquot was injected into the LC-MS/MS. The reaction mixture of DEX and NIF was centrifuged at 10,000g for 5 min. The supernatant (100 μl) was then decanted, and 30-μl aliquots were injected into the LC-MS/MS.
The reaction mixtures of all other drugs were centrifuged at 10,000g for 5 min. The supernatants (100 μl) were decanted, and 10-μl aliquots were injected into the LC-MS/MS.
In this experiment, the unchanged concentrations of all drugs were determined by using LC-MS/MS analysis. Mass number of molecular ion and product ion for each compounds were identified as follows (polarity, molecular>product): HT m/z = 296 > 269 [M+H]−; DEX m/z = 393 > 91 [M+H]+; NIF m/z = 347 > 315 [M+H]+; MDZ m/z = 326 > 291 [M+H]+; QID m/z = 325 > 307 [M+H]+; TAC m/z = 821 > 769 [M+NH4]+; VER m/z = 455 > 165 [M+H]+; DIG m/z = 780 > 85 [M+H]−; IBU m/z = 205 > 161 [M+H]−; TIM m/z = 317 > 261 [M+H]+; AMI m/z = 278 > 117 [M+H]+; PRO m/z = 260 > 116 [M+H]+.
The Prominence 2000 series (Shimadzu) was used as the LC-system. The MS/MS analyses were conducted on an API-3200 LC-MS/MS system (Applied Biosystems). For TAC, an Alliance HT Waters 2790 separations module and Micromass Quattro Ultima (Waters Corporation, Milford, MA) were used for the LC-MS/MS analysis.
The Supelco RP-Amide (3 μm, 3.0 × 31 mm; Supelco, Inc., Bellefonte, PA) was used as the analysis column for HT and DIG. The Capcell PAK MG (3 μm, 2.0 × 35 mm; Shiseido Corporation, Kyoto, Japan) HPLC column was used for all other drugs.
The flow rate was 0.3 ml/min. The column temperature was 50°C. The gradient system was used, starting with an ammonium acetate concentration of 20 mM (pH 4.8)/acetonitrile (9:1) for 0.5 min, and increasing the ratio of acetonitrile to 20 mM ammonium acetate (pH 4.8)/acetonitrile (1:9) over 0.5 min, which was then held for 2.5 min. The initial conditions were restored over 0.1 min, after which the column was re-equilibrated for 1 min.
Calculation of CLintliver.
CLintliver was calculated by using the following equation based on the time course of the residual ratio of the unchanged drugs as determined using least-squares linear regression (Naritomi et al., 2001): CLintliver (ml/min/mg protein) = ke/microsomal protein concentration, where ke is the disappearance rate constant.
In the case of liver microsomes study, the units of CLintliver values were converted to per kilogram of body weight by using the following equation: CLintliver (ml/min/kg) = CLintliver (ml/min/mg protein) × SF1 (mg protein/g liver) × SF2 (g liver/kg body weight), where SF1 is the microsomal protein content per gram of liver [48.8 was used for both species (Naritomi et al., 2001), assuming that the SF1 in cynomolgus monkeys is the same as in humans] and SF2 is the liver weight per kilogram of body weight (25.7 and 30.0 were used for humans and cynomolgus monkeys, respectively) (Davies and Morris, 1993).
Calculation of in vitro intrinsic clearance in intestine.
CLintintestine was calculated by using the following equation based on the time course of the residual ratio of the unchanged drugs as determined using least-squares linear regression (Naritomi et al., 2001): CLintintestine (μl/min/mg protein) = ke/microsomal protein concentration.
P-gp ATPase Assay.
Each drug was dissolved in dimethylsulfoxide (0.1–100 μM final) and preincubated for 5 min with 2 μg/ml human P-gp membrane (BD Gentest, Woburn, MA) in 50 mM MES buffer (pH 6.8 adjusted with Tris) containing 2 mM EGTA, 2 mM dithiothreitol, 50 mM potassium chloride, and 5 mM sodium azide. Then, the ATPase reaction was started by the addition of 50 mM Mg-ATP solution. After 20-min incubation at 37°C, the reaction was stopped by adding 20 μl of sodium dodecyl sulfate (10%) containing Antifoam A (Sigma-Aldrich). Subsequently, 200 μl of ammonium molybdate/zinc acetate was added for color development, and the mixture was incubated for another 20 min at 37°C. After incubation, the amount of liberated phosphate was measured by using the UV absorption method (630 nm). Baseline activity was determined by reading incubated sodium orthovanadate (100 μM). Finally, ATPase activity was determined as the amount of liberated phosphate per milligram protein per minute. VER was evaluated in all ATPase assays, and the ATPase activity of each drug was normalized by dividing by the VER ATPase activity for each experiment.
Calculation of in Vivo Pharmacokinetic Parameters.
Plasma concentration data were analyzed individually at each point in time, and pharmacokinetic parameters were calculated by using a model-independent method. F, FaFg, and Fh were then calculated from these pharmacokinetic parameters and Rb (see Blood-to-Plasma Concentration Ratio under Materials and Methods) by using the formulas shown below. For Li and HT, we assumed that these drugs underwent almost no in vivo metabolism and that their FaFg values (meaning Fa in this case) were equal to F. The F values for the drugs in cynomolgus monkeys were determined by using the following equation: F(%) = {AUCinf (p.o.)/AUCinf (i.v.)} × (Dose i.v./Dose p.o.) × 100, where AUCinf (i.v.) and AUCinf (p.o.) are the area under the plasma concentration-time curve calculated using the trapezoidal rule with extrapolation from the last measured plasma concentration to infinity after intravenous and oral administrations, respectively.
The Fh of drugs was determined by using the following equation and assuming that the elimination of drugs from the body after intravenous administration consisted of liver metabolism and renal excretion: Fh = 1 − {(CLh/Rb)/Qh}, CLh = CLt × (1 − fe); fe = urinary excretion of unchanged of unchanged drug after intravenous administration, where Qh is the blood flow rate in the liver (the human and cynomolgus monkey Qh values were 20.7 and 43.6 ml/min/kg, respectively) (Davies and Morris, 1993), CLh is hepatic clearance, CLt is total clearance, and fe is the urinary excretion ratio of the unchanged drug after intravenous administration. In cases where the fe value was not available, the CLh was assumed to be equal to the CLt.
The drug FaFg values were determined by using the following equations, assuming that the F was expressed as the product of FaFg and Fh: F(%) = Fa × Fg × Fh × 100, FaFg = {F(%)/100}/Fh. The F, FaFg, and Fh values of each drug in humans were also calculated in a similar manner by using the reported pharmacokinetic parameters.
Results
Comparison of Pharmacokinetic Parameters between Humans and Cynomolgus Monkeys.
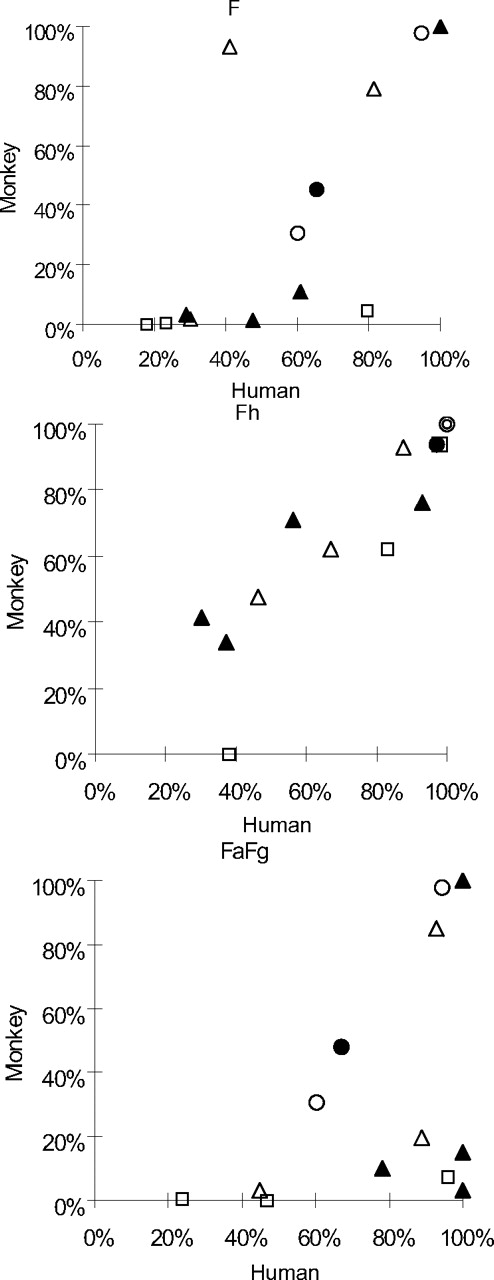
The in vivo pharmacokinetic parameters, F, FaFg, and Fh, for all 13 drugs are summarized in Table 4. Each drug's cynomolgus monkey F, FaFg, and Fh values are plotted against those in humans in Fig. 1.
Summary of in vivo pharmacokinetic parameter values in humans and cynomolgus monkeys
Mean values were used for CLh, F, and FaFg. For Li and HT, it was assumed that fe after oral administration was equal to F. MDZ in cynomolgus monkey was taken from Sakuda et. al., 2006.

Correlation of F, FaFg, and Fh in humans and cynomolgus monkeys. Open circle, open triangle, open square, closed circle, and closed triangle represent category Types A–E, respectively.
Correlation of the F between Humans and Cynomolgus Monkeys.
The F values of all drugs observed in cynomolgus monkeys were compared with those in humans. The results showed that the F value for Li, DEX, and IBU in humans and cynomolgus monkeys were similar, and that the F value for HT and DIG were almost similar (<2-fold). In contrast, with the exception of DEX and IBU, many of the P450 substrate drugs had a markedly lower F in cynomolgus monkeys than in humans.
Type A.
The F values for Li in humans and cynomolgus monkeys were similar (94.5%/97.9%), and HT showed slightly lower F values in cynomolgus monkeys (30.7%) than in humans (60.2%).
Type B.
For DEX, the F values in humans and cynomolgus monkeys were similar (81.4 and 78.9%, respectively). However, the F values for NIF and MDZ in cynomolgus monkeys were markedly lower [9.3 and 2.0% (Sakuda et al., 2006), respectively] than those in humans (41.2 and 30.0%, respectively).
Type C.
The Type C drugs, QID, TAC and VER, which are known to be substrates for both CYP3A4 and P-gp in humans, had markedly lower F values (4.5, 0.5, and 0%, respectively) in cynomolgus monkeys than in humans (79.5, 23.3, and 18.0%, respectively).
Type D.
The DIG, which is a typical substrate of P-gp, had a slightly lower F value in cynomolgus monkeys (45.0%) than in humans (65.3%). This finding was similar to that for HT.
Type E.
Whereas the F value of IBU was almost the same in both species, that for TIM, AMI, and PRO was lower in cynomolgus monkeys (10.8, 1.3, and 3.3%) than in humans (61.0, 47.7, and 29.0%). These findings were similar to those for Type B drugs. No significant correlation between the P450 isozyme selectivity of drugs and their F values in cynomolgus monkeys was observed.
Correlation of the Fh between Humans and Cynomolgus Monkeys.
The correlations between the human and cynomolgus monkey Fh values for the 13 drugs are shown in Fig. 1, Fh. The Fh values in cynomolgus monkeys were similar to those in humans for all drugs except VER (Fh was calculated as 0 in cynomolgus monkeys), because the plots for the drugs were the same or nearly the same (Fig. 1, Fh; Table 4). Li and HT underwent almost no in vivo metabolism; therefore, the Fh values were considered to be 1.
Correlation of the FaFg between Humans and Cynomolgus Monkeys.
As shown in Fig. 1, FaFg, the FaFg values for Li, DEX, and IBU were similar in both humans and cynomolgus monkeys (0.95/0.98, 0.93/0.85, and 1/1, respectively). For HT and DIG, the FaFg values in cynomolgus monkeys were slightly lower than those in humans (0.60/0.31 and 0.67/0.48 in humans and cynomolgus monkeys, respectively).
For the other 7 drugs (except VER), the F in cynomolgus monkeys was low, and a markedly low FaFg was observed. These tendencies correlated well with those of the F values (assuming Fh = 1 for Li and HT, which means F = FaFg).
In Vitro Parameters.
In this study, some additional in vitro assays were performed to evaluate the drugs' (except Li) in vitro pharmacokinetic properties. These assays included determination of the blood-to-plasma concentration ratio, membrane permeability, in vitro metabolic stability assay using human and cynomolgus monkey liver microsomes, plasma protein binding, and P-gp affinity. The results are summarized in Table 5.
Summary of in vitro pharmacokinetic parameters of all tested drugs in humans and cynomolgus monkeys
Lithium was excluded from all in vitro studies.
Membrane permeability.
As shown in Table 5, almost all drugs except HT and DIG showed good membrane permeability (>apparent permeability coefficient of more than 10). Taking the F values into consideration, the HT and DIG were speculated to be absorbed moderately in cynomolgus monkeys. These results suggest that all tested drugs were well absorbed or relatively well absorbed in cynomolgus monkeys, even though many drugs had a low F.
Metabolic stability in liver microsomes.
For HT and DIG, no depletion was observed, and the intrinsic clearance for DEX MDZ, and IBU in both humans and cynomolgus monkeys were almost the same (66/24 ml/min/kg, 877/1422 ml/min/kg, and 38/25 ml/min/kg, respectively). Intrinsic clearance values for the other seven drugs were higher in cynomolgus monkeys than in humans (Table 5). Although Fh correlated well between humans and cynomolgus monkeys for all tested drugs except VER, these drugs were metabolized more rapidly in cynomolgus monkey microsomes than in human microsomes. Furthermore, the fb × CLintliver/Qh for NIF, VER, PRO, and AMI were found to be higher (>4) after taking fb and blood flow rate in the liver into consideration, indicating that these drugs might undergo rapid metabolism in the livers of cynomolgus monkeys.
Metabolic stability in intestine microsomes.
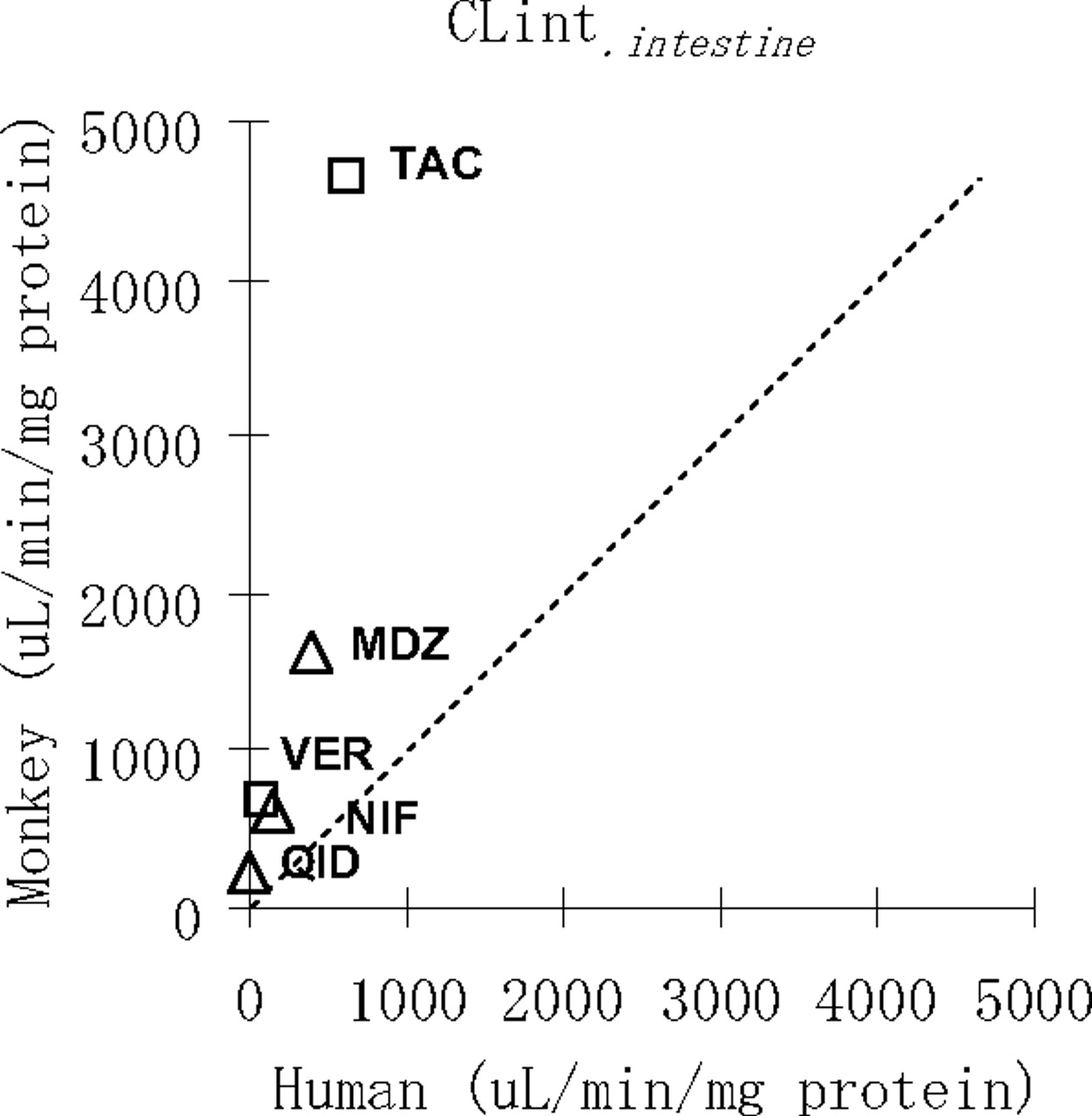
The CLintintestine was expressed by μl/min/mg protein because there is no widely used physiological conversion model from μl/min/mg protein to μl/min/kg in intestine. The CLintintestine values for NIF, MDZ, QID, TAC, and VER in cynomolgus monkey were 612, 1635, 212, 4663, and 696 μl/min/mg protein, respectively. As well as in human, the values were 138, 385, no depletion, 625, and 69 μl/min/mg protein for each (Fig. 2; Table 5). In contrast, no significant decreases in other drugs were observed in both human and cynomolgus monkey intestine microsomes.

Correlation of CLintintestine in humans and cynomolgus monkeys. NIF, MDZ, QID, TAC, and VER represent nifedipine, midazolam, quinidine, tacrolimus, and verapamil, respectively.
ATPase assay.
The ATPase activity of all drugs was normalized by dividing them by the VER value. As shown in Table 5, the ATPase activity of QID, DIG, and TAC was higher than that of VER. For PRO, AMI, TIM, and IBU, the ATPase activity values were similar to the VER value, whereas the HT, DEX, NIF, and MDZ were lower. There was no significant correlation between P-gp affinity and F values in cynomolgus monkeys observed.
Discussion
Although cynomolgus monkeys are often used for pharmacokinetic studies for drug discovery, it remains unclear whether this is a useful animal species for predicting human pharmacokinetics. In this study, we investigated the pharmacokinetic profile of 13 commercially available drugs in cynomolgus monkeys and compared their pharmacokinetic parameters with those in humans. The results showed that the majority of the drugs tested (8 of 13) had a markedly lower F in cynomolgus monkeys (<15%). We explored the reasons for these species differences and suggest some possibilities as listed below.
Species Differences in Hepatic Metabolism.
The Fh values in humans and cynomolgus monkeys were almost the same for the 12 drugs (except VER). No obvious species differences were revealed for hepatic metabolism, regardless of P450 isozyme selectivity. These results suggested that the values obtained from cynomolgus monkeys after intravenous administration were useful for predicting human pharmacokinetic parameters, such as CLt or Fh. These findings agreed with the consistency seen between the species with regard to P450 isozyme amino acid sequence (over 90% agreement) (Uno et al., 2007).
A species difference in Fh was apparent for VER, which was explained by the difference in the rate of hepatic metabolism. The fb × CLintliver/Qh of VER in cynomolgus monkeys was much higher than that in humans, which agreed with the in vivo observation.
Species Differences in the Intestinal Transit Process.
The fact that all drugs with a low F in cynomolgus monkeys had low FaFg values indicates that the low FaFg is attributable to the low F, in cynomolgus monkeys specifically. The FaFg values for Li, DEX, and IBU were correlated well between humans and cynomolgus monkeys. The common properties of these three drugs are as follows: 1) they have good membrane permeability (Li is absorbed via a paracellular pathway); 2) they are not P-gp substrates; and 3) they undergo little or no in vivo metabolism (see Tables 4 and 5).
Subsequently, the FaFg correlation between humans and cynomolgus monkeys was found to be weak for both HT and DIG. The FaFg values for these drugs in cynomolgus monkeys were slightly lower than those in humans. The common properties of these two drugs are as follows: 1) they have moderate membrane permeability, and 2) they undergo almost no in vivo metabolism (Tables 4 and 5). Although HT is not a P-gp substrate, DIG was found to cause high activity in the ATPase assay. These results suggest that membrane permeability and P-gp efflux are partial contributors to the low F in cynomolgus monkeys.
In contrast, the other seven drugs (except VER), which had a markedly low FaFg in cynomolgus monkeys, were metabolized by P450 enzymes and had relatively high CLintliver or CLintintestine values in cynomolgus monkeys. These drugs also showed good membrane permeability (Table 5).
These findings suggest the possibility that these drugs were extensively metabolized in the cynomolgus monkey intestine, and the low FaFg was caused by intestinal metabolism rather than poor absorption. In fact, all of five drugs, which observed good FaFg correlation in both species, undergo little or no in vivo P450 metabolism.
The Major Species Difference Factor between Humans and Cynomolgus Monkeys.
There have been several reports that focused on the species differences between humans and monkeys (Chiou et al., 2002; Sakuda et al., 2006; Takahashi et al., 2008). However, the present study showed that drugs that satisfy the following properties have similar FaFg or F values in both humans and cynomolgus monkeys: 1) good membrane permeability; 2) not a P-gp substrate; and 3) undergoes little or no in vivo metabolism.
In contrast, drugs that are P450 substrates and are relatively or rapidly metabolized in cynomolgus monkeys could have markedly low F values because of their low FaFg values, even if the drugs have a low CLt. The potential reasons for these findings are as follows: 1) the amount of P450 enzyme expressed in cynomolgus monkey intestine is higher than that in humans, even though CYP3A4 is a major intestinal enzyme in humans; and 2) the enzyme expressed in cynomolgus monkey intestine has higher activity (Vmax/Km) than that in humans. To clearly understand these speculations, additional in vitro studies using intestine microsome were conducted with the same condition as the liver microsomes study. In cynomolgus monkey, the values of CLintintestine for NIF, MDZ, QID, TAC, and VER were 612, 1635, 212, 4663, and 696 μl/min/mg protein, respectively. As well as in human, the values were 138, 385, no depletion, 625, and 69 μl/min/mg protein for each. These five compounds, which have low F in cynomolgus monkey, showed markedly larger values in cynomolgus monkey than those in human (Fig. 2). In contrast, no significant decreases in other drugs were observed in both human and cynomolgus monkey intestine microsomes.
Whereas the cynomolgus monkey P450 isozyme corresponding to human CYP3A4 is CYP3A8 (Uno et al., 2007), it is unclear whether CYP3A8 is also a major enzyme in the cynomolgus monkey intestine. In fact, a lower FaFg in cynomolgus monkeys was also observed for Type E drugs (mainly metabolized by CYP 2C9, 2C19, or 2D6).
Although it is possible that glucuronide conjugates contributed to the low F obtained for PRO (Walle et al., 1979), further studies are needed to explain this observation. Because all drugs with a low F in cynomolgus monkeys show good membrane permeability in the present study, first-pass intestinal metabolism must be the most critical factor affecting species differences between humans and cynomolgus monkeys.
We also investigated the pharmacokinetics of several drugs in rats and/or dogs, and the FaFg in rats or dogs correlates better with humans than cynomolgus monkeys (Tabata et al., 2009). Further studies are needed to clarify the species differences for FaFg, including the contribution of permeability, intestinal first-pass metabolism, and P-gp excretion.
The Usability of Cynomolgus Monkey Pharmacokinetic Parameters for Predicting Pharmacokinetic in Humans.
These results suggest that a go/no go decision does not have to be made immediately, even if a candidate has a markedly low F in cynomolgus monkeys. In such cases, the main factor causing low F in cynomolgus monkeys may be evaluated separately from Fa, Fg, and Fh. If the cause is found to be Fg, the candidate could still have an acceptable pharmacokinetic profile in humans.
Recognition of the importance of intestinal metabolism has increased over recent years. Many studies using intestinal microsomes are in progress in our laboratory in an attempt to establish a system for evaluating human Fg.
It is noteworthy that a rough correlation was observed between CLintliver and Fg in humans (Fig. 3) in this study, indicating the possibility that Fg prediction in humans using only in vitro parameters may be possible with slight but elaborated modification of the evaluation system for in vitro intestinal metabolism. In fact, when evaluation of intestinal metabolism was inadequate, we successfully predicted the human pharmacokinetics for several in-house candidate drugs with a markedly low F in cynomolgus monkeys by using human in vitro parameters for each candidate, including membrane permeability, metabolic stability in liver microsomes, and P-gp affinity (in-house data). These low values for F in cynomolgus monkeys were virtually thought to be due to low Fg.

Correlation of FaFg and CLintliver in humans. Open triangle, open square, and closed triangle represent category Types B, C, and E, respectively.
In conclusion, many drugs had a markedly low F in cynomolgus monkeys despite having relatively good F in humans. These findings are speculated to be attributable mainly to first-pass intestinal metabolism. Consequently, the pharmacokinetic parameters obtained for a candidate after oral administration to cynomolgus monkeys are not adequate for directly predicting human pharmacokinetics.
The accurate prediction of Fg in humans eventually becomes necessary to predict human pharmacokinetics with more accuracy. In addition, the slight but elaborated modification of the evaluation system for in vitro intestinal metabolism, which is under development in our laboratory (Kadono et al., 2007), may enable us to estimate the Fg in humans, and subsequently it becomes possible to predict accurate human pharmacokinetics in the near future.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.028829.
-
- Fa
- fraction absorbed
- Fg
- intestinal availability
- Fh
- hepatic availability
- F
- bioavailability
- MDZ
- midazolam
- P450
- cytochrome P450
- P-gp
- p-glycoprotein
- Rb
- blood-to-plasma concentration ratio
- CLint
- intrinsic clearance
- CLintliver
- CLint in liver microsomes
- CLintintestine
- CLint in intestine microsomes
- TAC
- tacrolimus
- Li
- lithium carbonate
- HT
- hydrochlorothiazide
- VER
- verapamil
- PRO
- propranolol
- AMI
- amitriptyline
- DEX
- dexamethasone
- NIF
- nifedipine
- QID
- quinidine
- TIM
- timolol
- IBU
- ibuprofen
- DIG
- digoxin
- LC
- liquid chromatography
- MS/MS
- mass spectrometry
- HPLC
- high-performance liquid chromatography
- PAMPA
- parallel artificial membrane permeability assay
- fp
- unbound drug fraction in plasma
- fb
- unbound drug fraction in blood
- ke
- disappearance rate constant
- AUC
- area under the plasma concentration-time curve
- fe
- urinary excretion ratio of unchanged
- CLh
- hepatic clearance
- CLt
- total clearance
- Qh
- blood flow rate in the liver
- SF
- scaling factor
- Papp
- apparent permeability.
- Received June 5, 2009.
- Accepted November 11, 2009.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}