


Abstract
The current study assesses hepatic and intestinal glucuronidation, sulfation, and cytochrome P450 (P450) metabolism of raloxifene, quercetin, salbutamol, and troglitazone using different in vitro systems. The fraction metabolized by conjugation and P450 metabolism was estimated in liver and intestine, and the importance of multiple metabolic pathways on accuracy of clearance prediction was assessed. In vitro intrinsic sulfation clearance (CLint, SULT) was determined in human intestinal and hepatic cytosol and compared with hepatic and intestinal microsomal glucuronidation (CLint, UGT) and P450 clearance (CLint, CYP) expressed per gram of tissue. Hepatic and intestinal cytosolic scaling factors of 80.7 mg/g liver and 18 mg/g intestine were estimated from published data. Scaled CLint, SULT ranged between 0.7 and 11.4 ml · min−1 · g−1 liver and 0.1 and 3.3 ml · min−1 · g−1 intestine (salbutamol and quercetin were the extremes). Salbutamol was the only compound with a high extent of sulfation (51 and 28% of total CLint for liver and intestine, respectively) and also significant renal clearance (26–57% of observed plasma clearance). In contrast, the clearance of quercetin was largely accounted for by glucuronidation. Drugs metabolized by multiple pathways (raloxifene and troglitazone) demonstrated improved prediction of intravenous clearance using data from all hepatic pathways (44–86% of observed clearance) compared with predictions based only on the primary pathway (22–36%). The assumption of no intestinal first pass resulted in underprediction of oral clearance for raloxifene, troglitazone, and quercetin (3–22% of observed, respectively). Accounting for the intestinal contribution to oral clearance via estimated intestinal availability improved prediction accuracy for raloxifene and troglitazone (within 2.5-fold of observed). Current findings emphasize the importance of both hepatic and intestinal conjugation for in vitro-in vivo extrapolation of metabolic clearance.
Introduction
The prediction of drug clearance via P450 enzymes is well established. In contrast, much less attention has been given to non-P450 reactions, in particular conjugation, or to the situation in which multiple enzymes contribute to drug clearance. Prediction of in vivo hepatic clearance from in vitro microsomal glucuronidation data results in a general underprediction trend but to a degree similar to that with P450s (Ito and Houston, 2005; Miners et al., 2006; Mohutsky et al., 2006; Cubitt et al., 2009; Kilford et al., 2009). For some drugs (substrates of UGT1A9 and UGT2B7), predictions have been improved by addition of albumin as a result of sequestration of inhibitory long-chain unsaturated fatty acids released during the incubation. These fatty acids have been reported to competitively inhibit certain, but not all, UGT enzymes (e.g., no effect on UGT1A1) (Rowland et al., 2007, 2009). In addition, clearance of raloxifene (UGT1A9 substrate) was underpredicted even when intrinsic clearance (CLint) was determined in the presence of albumin (Kilford et al., 2009). Poor prediction of in vivo clearance from in vitro hepatic microsomal glucuronidation data may be explained by the potential contribution of other conjugation pathways (e.g., sulfation).
There is increasing interest in the sulfotransferases (SULTs) and other cytosolic enzymes and their contribution to drug clearance (Zhang et al., 2007; Riches et al., 2009a,b; Pryde et al., 2010; Zientek et al., 2010). Published in vitro sulfation studies (Walle et al., 1993; Pacifici et al., 1997; Honma et al., 2002) generally focus on activity data rather than on full kinetic characterization; this is particularly evident with intestinal cytosolic studies. Reported studies show large variations in experimental conditions, including concentration of human cytosolic protein, concentration of the cofactor PAPS, buffer type, and pH range. In addition, published in vitro cytosolic studies so far have not addressed in vitro-in vivo extrapolation of generated data, either in isolation or combined with corresponding microsomal data on other contributing metabolic pathways.
A reliable analysis of in vitro clearance data requires accurate scaling factors to express clearance to per gram of organ. This has been described previously for hepatic (Barter et al., 2007) and intestinal microsomal scaling factors (Cubitt et al., 2009). To our knowledge, the current study is the first instance in which sources of available cytosolic scaling factors have been collated for both liver and intestine. Limitations in the sources of these data and implications on prediction of in vivo clearance are discussed.
In addition to liver, several conjugative enzymes (UGT1A8, UGT1A10, and SULT1A3) are expressed predominantly in the intestine (Tukey and Strassburg, 2001; Lindsay et al., 2008; Riches et al., 2009b), and the importance of intestinal glucuronidation has been demonstrated for some drugs (Dalvie et al., 2008; Cubitt et al., 2009). For oral clearance, an underprediction trend may therefore result from ignoring the contribution of extrahepatic glucuronidation or sulfation to the overall clearance.
The overall aim of the current study was to assess multiple metabolic pathways of quercetin, raloxifene, salbutamol, and troglitazone and estimate the hepatic and intestinal fraction metabolized by SULT, UGT, and P450 enzymes for these drugs. To allow this, intestinal and hepatic sulfation was assessed in vitro, using human liver and intestinal cytosol and a substrate depletion method. Scaling of the data was performed using the estimated cytosolic scaling factors for liver and intestine from published sources, and data were analyzed in conjunction with previously determined microsomal CLint, UGT and CLint, CYP values (Cubitt et al., 2009). The four drugs selected showed differential specificity for intestinal and hepatic SULT, UGT, and P450 enzymes or poor prediction success based solely on hepatic P450 and UGT clearance data. Ultimately, the prediction of intravenous and oral clearance values (in the latter case accounting for intestinal contribution) from in vitro data was explored, and the contributing role of the multiple different hepatic and extrahepatic pathways to the clearance prediction was assessed.
Materials and Methods
Chemicals.
All solvents were purchased from VWR International (Lutterworth, UK). All other compounds and reagents were purchased from Sigma-Aldrich (Dorset, UK).
Source of the Subcellular Fractions.
Pooled human liver cytosol (HLC) (n = 20 donors) and intestinal cytosol (HIC) (n = 8 donors) were purchased from XenoTech, LLC (Lenexa, KS). The HIC was prepared by enterocyte elution mainly of the jejunum section. No enzyme activity data were reported for either of the cytosolic fractions. Glucuronidation and P450 clearance values obtained in the pooled intestinal microsomes (n = 10), HLM pool A (n = 30) for quercetin and salbutamol, and HLM pools A, B, and C (n = 30, 22, and 33, respectively) for raloxifene and troglitazone were reported previously (Cubitt et al., 2009). The current study included additional CLint, UGT and CLint, CYP data for quercetin and salbutamol obtained in the same HLM pools B and C. All HLM pools were purchased from BD Gentest (Woburn, MA).
Preliminary Assessment of Cytosolic Experimental Conditions.
Experimental protocols for sulfation studies reported in the literature vary in the concentration of the cofactor PAPS (range 0.4–50 μM), buffer type (Tris or phosphate), and pH (range pH 6.2–9.5), as detailed in the supplemental data. Therefore, incubation conditions in HLC were initially optimized using troglitazone unbound CLint by sulfation in human liver cytosol as a marker. A range of conditions were assessed including different buffer types (50 mM Tris-HCl, 50 mM phosphate, and 100 mM phosphate buffer), cytosolic protein concentration (0.5–1.5 mg/ml), and concentration of PAPS (5–400 μM). A protein concentration of 1.5 mg/ml resulted in high average troglitazone CLint (45.8 μl · min−1 · mg−1), a linear depletion profile, and the lowest coefficient of variation. An increase in PAPS concentration between 5 and 50 μM resulted in a corresponding increase in the troglitazone CLint values between 12.0 and 48.9 μl · min−1 · mg−1. A further increase in PAPS concentration had no effect on troglitazone clearance or resulted in reduced and more variable estimates (up to 62%), and hence the concentration of 50 μM was selected. Cytosolic conditions of 1.5 mg/ml HLC or HIC protein concentration, 100 mM phosphate buffer, pH 7.4, and 50 μM PAPS resulted in the most reproducible troglitazone intrinsic clearance by sulfation (CLint, SULT) values and were selected for further sulfation studies.
Incubation Procedure.
All cytosols were stored at −80°C and rapidly thawed just before use. Incubations for all four compounds were performed in duplicate and on three separate occasions using an Eppendorf Thermomixer at 37°C and 1400 rpm. All substrates were preincubated for 5 min at 37°C with HLC or HIC and 0.1 M phosphate buffer, pH 7.4. Reactions were initiated by the addition of PAPS (final concentration 50 μM) to give a final incubation volume of 800 μl. Substrate concentrations in the final incubation were 1 μM, and cytosolic protein concentration was 1.5 mg/ml for both HLC and HIC. Glucuronidation and P450 incubations for quercetin and salbutamol (HLM pools B and C) were performed under the conditions reported previously (Cubitt et al., 2009). The mean intrinsic clearance by glucuronidation (CLint, UGT) value from three microsomal pools was 5011 and 9.3 μl · min−1 · mg−1 for quercetin and salbutamol, respectively. The corresponding salbutamol intrinsic clearance by cytochrome P450 (CLint, CYP) was 7.7 μl · min−1 · mg−1, whereas quercetin had negligible P450 metabolism. The final concentration of organic solvent in the incubation media was <0.1%. No cofactor incubations were performed for drugs to account for any potential cofactor-independent loss of a drug over the incubation time. For each time point, 100 μl of the incubation was removed, and the reaction was terminated by the addition of 100 μl of ice-cold acetonitrile containing the internal standard, as specified previously (Cubitt et al., 2009). The total length of the cytosolic incubations was 60 min for all compounds except for quercetin, which only required an incubation of 16 min. Samples were centrifuged at 1400g (Mistral 3000i centrifuge; MSE Ltd., London, UK) for 10 min, and the parent compound was analyzed by LC-MS/MS.
LC-MS/MS.
The LC-MS/MS system used consisted of a Waters 2790 with a Micromass Quatro Ultima triple quadruple mass spectrometer (Waters, Elstree, UK). Varying gradients of four mobile phases were used, the compositions of which were 1) 90% water and 0.05% formic acid with 10% acetonitrile, 2) 10% water and 0.05% formic acid with 90% acetonitrile, 3) 90% water and 10 mM ammonium acetate with 10% acetonitrile, and 4) 10% water and 10 mM ammonium acetate with 90% acetonitrile. A Luna C18 column (3 μm, 50 × 4.6 mm; Phenomenex, Macclesfield, UK) was used for chromatographic separation of analytes. The flow rate was 1 ml/min, and this was split to 0.25 ml/min before entry into the mass spectrometer. Further analytical parameters were as described previously (Cubitt et al., 2009). The ion chromatograms were integrated and quantified using Micromass QuanLynx software (Waters).
Correction for Nonspecific Protein Binding.
To correct CLint, SULT for nonspecific protein binding in the incubation, fraction unbound from protein in the incubation (fuinc) values for raloxifene, salbutamol, and troglitazone were experimentally determined using human liver cytosol. The fuinc values were determined in HLC at protein concentrations of 0.5, 1, and 1.5 mg/ml using the high-throughput dialysis method, as described previously (Gertz et al., 2008). With 0.1 M phosphate buffer, the drugs were at a concentration of 1 μM. Dialysis membranes had a 12 to 14 kDa molecular mass cutoff and were purchased from HTDialysis, LLC (Gales Ferry, CT). The fuinc values for quercetin were predicted using an algorithm proposed by Hallifax and Houston (2006) because of compound degradation during the duration of equilibrium dialysis (6 h). Correction of intestinal CLint values for nonspecific binding assumed the equality of fuinc between intestinal and liver cytosol/microsomes.
Data Analysis.
Sulfation intrinsic clearance was calculated by eq. 1, using GraFit 5 (Erithacus Software, Horley, UK):
 where CLint, SULT is intrinsic clearance by sulfation (microliters per minute per milligram of protein) and k is the depletion rate constant (minute−1). The clearance was corrected for experimentally determined fuinc to give an unbound value for CLint, SULT.
where CLint, SULT is intrinsic clearance by sulfation (microliters per minute per milligram of protein) and k is the depletion rate constant (minute−1). The clearance was corrected for experimentally determined fuinc to give an unbound value for CLint, SULT.
Determination of Human Cytosolic Scaling Factors.
Hepatic and intestinal cytosolic scaling factors were derived from literature sources quoting cytosolic protein abundance or activity data in both the cytosolic fraction and tissue homogenate (Wynne et al., 1992; Boogaard et al., 1996; Gibbs et al., 1998; Renwick et al., 2002; Mutch et al., 2007). Hepatic CLint, SULT values obtained in the current study were scaled using 80.7 mg/g liver, whereas intestinal CLint, SULT values were scaled using 18 mg/g intestine, a value derived from only one literature source available and based on data from 12 donors (Gibbs et al., 1998).
Comparison of Intestinal and Hepatic Extent of Sulfation.
To allow valid comparison of sulfation between the intestine and liver, clearance data were expressed per gram of tissue using collated cytosolic scaling factors. Microsomal P450 and UGT clearance data were scaled using hepatic and intestinal microsomal recovery of 40 mg/g liver and 20.6 mg/g intestine, respectively (Barter et al., 2007; Cubitt et al., 2009). Clearance values scaled to per gram of tissue were used to estimate extent of each metabolic process in vitro; fraction metabolized by sulfation (fmSULT) values were calculated from mean CLint, SULT and previously reported CLint, UGT and CLint, CYP using eq. 2. Previously reported fraction metabolized by glucuronidation (fmUGT) and fraction metabolized by P450 metabolism (fmCYP) values for the drugs investigated have been reassessed, taking into account the additional contribution of the sulfation process (eqs. 3 and 4). For the hepatic estimates of fractions metabolized, CLint, UGT and CLint, CYP values used represent the mean of three HLM pools (total n = 85), whereas for intestine, data were obtained in a single intestinal microsomal pool. Details of the microsomal pools have been published previously (Cubitt et al., 2009).

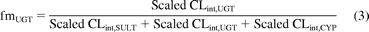

Prediction of Intravenous and Oral Clearance.
CLint values expressed per gram of liver were further scaled using liver weight of 21.4 g liver/kg (Ito and Houston, 2005) to give a predicted CLint in units of milliliters per minute per kilogram. For multiple pathways, total in vitro CLint values were calculated as a sum of unbound CLint values for all individual metabolic pathways after scaling to per gram of liver. Oral and intravenous clearance values, renal clearances, fraction unbound in plasma (fup), and blood/plasma concentration ratios (RB) were collated from the literature for all compounds investigated. When multiple clinical studies were available, mean clearance values were calculated weighted for the number of subjects in the study. References for all clinical studies considered are available in the supplemental data. Hepatic blood clearance (CLH) after intravenous administration was calculated by correction of plasma clearance for renal excretion and RB (Ito and Houston, 2005; Gertz et al., 2010). Estimated CLH was used to calculate in vivo hepatic intrinsic clearance (CLint, h) using the well stirred liver model (eq. 5) and average hepatic blood flow (QH) of 20.7 ml · min−1 · kg−1. For raloxifene, no observed intravenous clearance data were available, and, therefore, intravenous clearance of 14.7 ml · min−1 · kg−1 was estimated from oral clearance (735 ml · min−1 · kg−1) and reported bioavailability of 2% (Hochner-Celnikier, 1999; Mizuma, 2009):
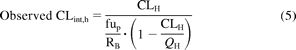 Observed CLint, h after oral administration was calculated as shown in eq. 6:
Observed CLint, h after oral administration was calculated as shown in eq. 6:
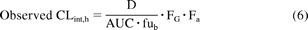 where D/AUC represents the hepatic blood clearance obtained from the mean plasma data after correction for renal clearance (when applicable) and RB. fub represents fraction unbound in blood, Fa represents fraction absorbed, and FG represents intestinal availability. The following Fa values were used: 0.54 for quercetin (Petri et al., 2003), 0.60 for raloxifene (Dalvie et al., 2008; Mizuma, 2009), and 0.80 for salbutamol (Mizuma, 2008). For troglitazone, Fa of 0.69 was estimated from animal data (Izumi et al., 1996), because of the lack of any human data. The contribution of the small intestine to oral clearance was incorporated using the FG values estimated from intravenous or oral data, as shown in eq. 7:
where D/AUC represents the hepatic blood clearance obtained from the mean plasma data after correction for renal clearance (when applicable) and RB. fub represents fraction unbound in blood, Fa represents fraction absorbed, and FG represents intestinal availability. The following Fa values were used: 0.54 for quercetin (Petri et al., 2003), 0.60 for raloxifene (Dalvie et al., 2008; Mizuma, 2009), and 0.80 for salbutamol (Mizuma, 2008). For troglitazone, Fa of 0.69 was estimated from animal data (Izumi et al., 1996), because of the lack of any human data. The contribution of the small intestine to oral clearance was incorporated using the FG values estimated from intravenous or oral data, as shown in eq. 7:
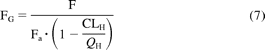
Estimation of Enterocytic Drug Concentration.
The concentration of drug present in the enterocytes was estimated using eq. 8 (Obach et al., 2006; Galetin et al., 2007), on the basis of typical in vivo doses for quercetin, raloxifene, salbutamol, and troglitazone (500, 60, 4, and 400 mg, respectively). Fa values used were as outlined above. A mean value of 0.3 l/min (Yang et al., 2007; Galetin et al., 2008) was used for enterocytic blood flow (Qent). Absorption rate constants (ka) of 0.009 min−1 were available for quercetin and raloxifene (Czock et al., 2005; Moon et al., 2008), whereas the value of this parameter for troglitazone (0.004 min−1) was estimated from animal data and Izumi et al. (1996). There were no published ka data available for salbutamol, and the generic value of 0.03 min−1 was used (Dalvie et al., 2008). All references associated with in vivo data are listed in the supplemental data.
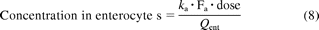 Estimated enterocytic drug concentrations were 31, 2, 1, and 9 μM for quercetin, raloxifene, salbutamol, and troglitazone, respectively.
Estimated enterocytic drug concentrations were 31, 2, 1, and 9 μM for quercetin, raloxifene, salbutamol, and troglitazone, respectively.
Results
Depletion Profiles.
Figure 1 shows sulfation depletion profiles for all four compounds in HLC and HIC using substrate concentrations less than the expected Km (1 μM). Monoexponential time profiles were observed for the intestinal and hepatic sulfation of raloxifene and the hepatic sulfation of both quercetin and troglitazone. The hepatic sulfation of salbutamol and the intestinal sulfation of both quercetin and troglitazone followed a biphasic profile. In these instances, the initial linear phase was used to estimate k and obtain CLint, SULT. Salbutamol and quercetin had the lowest and highest unbound CLint, SULT, respectively, in both organs, with a range between 8.7 and 141 μl · min−1 · mg−1 HLC and 8.0 and 186 μl · min−1 · mg−1 HIC (Table 1). Values of cytosolic CLint, SULT were based on triplicate determinations using a pool of 8 and 20 donors for HIC and HLC, respectively, and were subject to experimental variability of 11 to 44% (quercetin and troglitazone, respectively). The average coefficients of variation of CLint, SULT for the four drugs were comparable between cytosol from both tissues.

Comparison of sulfation depletion profiles in human liver and intestinal cytosol for four drugs: ▴, hepatic sulfation; ○, intestinal sulfation. A, quercetin. B, raloxifene. C, salbutamol. D, troglitazone. Data points represent the mean ± S.D. of three separate sulfation experiments in pooled liver and intestinal cytosol.
SULT specificity, nonspecific protein binding, and sulfation clearance obtained in liver and intestinal cytosol for quercetin, raloxifene, salbutamol, and troglitazone
Correction for Nonspecific Cytosolic Protein Binding.
CLint, SULT values were corrected for nonspecific cytosolic protein binding using experimentally determined fuinc values in human liver cytosol (Table 1). The extent of nonspecific protein binding in cytosol differed from that reported previously in microsomes at the same protein concentration (Cubitt et al., 2009). In consideration of the small dataset, no particular trends could be identified, because experimental fuinc was higher in the cytosol for raloxifene and troglitazone, whereas the opposite was seen for salbutamol. Therefore, direct use of microsomal fuinc is not ideal for the correction of cytosolic kinetic data. Experimental fuinc could not be determined for quercetin because of the instability of this compound during the equilibrium dialysis procedure and was therefore estimated using the Hallifax and Houston (2006) algorithm. For comparison, predicted cytosolic fuinc values for the remaining drugs are also shown in Table 1. Although discrepancies are evident, in particular for raloxifene, the use of a predicted value for quercetin was felt not to be of concern in subsequent calculations of unbound CLint. Unlike drugs such as raloxifene, quercetin is not particularly lipophilic (logP = 1.6) and minimal binding is to be expected.
Human Cytosolic Scaling Factors.
To compare sulfation clearance in the intestine and liver, CLint, SULT values were scaled from per milligram of cytosolic protein to per gram of tissue. To achieve this, intestinal and hepatic cytosolic scaling factors were collated from sources reporting measurement of a specific marker protein activity (glutathione transferase or alcohol dehydrogenase) in both tissue homogenate and subcellular fraction. Data from 52 donors in total were collated from five independent sources and are considered in the current analysis, as illustrated in Table 2. The hepatic cytosolic scaling factor ranged from 45 to 134 mg of cytosolic protein per gram of liver. Mean value weighted according to the number of livers used in each study (80.7 mg of cytosolic protein per gram of liver) was used in the current study for scaling of hepatic cytosolic data. Estimates obtained from the study by Wynne et al. (1992) indicated higher cytosolic recovery from male donors; however, overall data on the gender of donors, as well as age, were limited across studies to ascertain any impact on the estimated cytosolic recovery. For the intestine, data from only one study in 12 donors were available (Gibbs et al., 1998), resulting in a cytosolic scaling factor of 18 mg of cytosolic protein per gram of intestine. No information on the potential variation of the scaling factor along the intestine was available, as reported for intestinal microsomal recovery (ranges from 16.6 to 30.6 mg protein/g intestine for ileum and duodenum, respectively) (Cubitt et al., 2009).
Hepatic cytosolic scaling factors collated from published literature sources and corresponding information on liver donors
M and F represent male and female donors, respectively, where this information was stated.
Comparison of Intestinal and Hepatic Scaled Intrinsic Clearance by Sulfation, Glucuronidation, and P450 Metabolism.
Scaled CLint, SULT values (expressed per gram of tissue) were compared for the intestine and liver and also relative to microsomal glucuronidation and P450 clearance data. Unbound microsomal CLint, UGT and CLint, CYP values were scaled as reported previously (Cubitt et al., 2009). Comparison of scaled intestinal and hepatic CLint, SULT, CLint, UGT, and CLint, CYP values is shown Fig. 2. Scaled CLint, SULT values ranged from 0.70 to 11.4 ml · min−1 · g−1 liver and 0.14 to 3.34 ml/ · min−1 · g−1 intestine for salbutamol and quercetin, respectively. Mean scaled hepatic CLint, SULT values were 3-fold (quercetin and troglitazone) and 5-fold (raloxifene and salbutamol) higher than the intestinal values. Salbutamol was the only compound predominantly cleared by sulfation in both the liver and intestine. Of the four drugs, raloxifene and troglitazone showed higher clearances by both glucuronidation and P450 metabolism than by sulfation in both the intestine and liver (Fig. 2). Quercetin had the highest hepatic scaled CLint, UGT, negligible P450 metabolism, and sulfation clearance of 11.4 and 3.3 ml · min−1 · g−1 liver and intestine, respectively. Raloxifene had the highest intestinal CLint, UGT value (88 ml · min−1 · g−1 intestine) of the drugs investigated, a value that was >40-fold higher than its intestinal CLint, SULT and CLint, CYP.

Comparison of scaled unbound CLint by sulfation, glucuronidation, and P450 (expressed per gram of organ) in the intestine and liver for four drugs. ■, CLint, SULT; □, CLint, UGT; dark gray bar, CLint, CYP. A, liver. B, intestine. Data represent the mean ± S.D. of three separate sulfation experiments using the same pool of HLC or HIC. Glucuronidation and P450 metabolism represent either the mean ± S.D. of CLint, UGT and CLint, CYP obtained in three different HLM pools or data from a single intestinal microsomal pool.
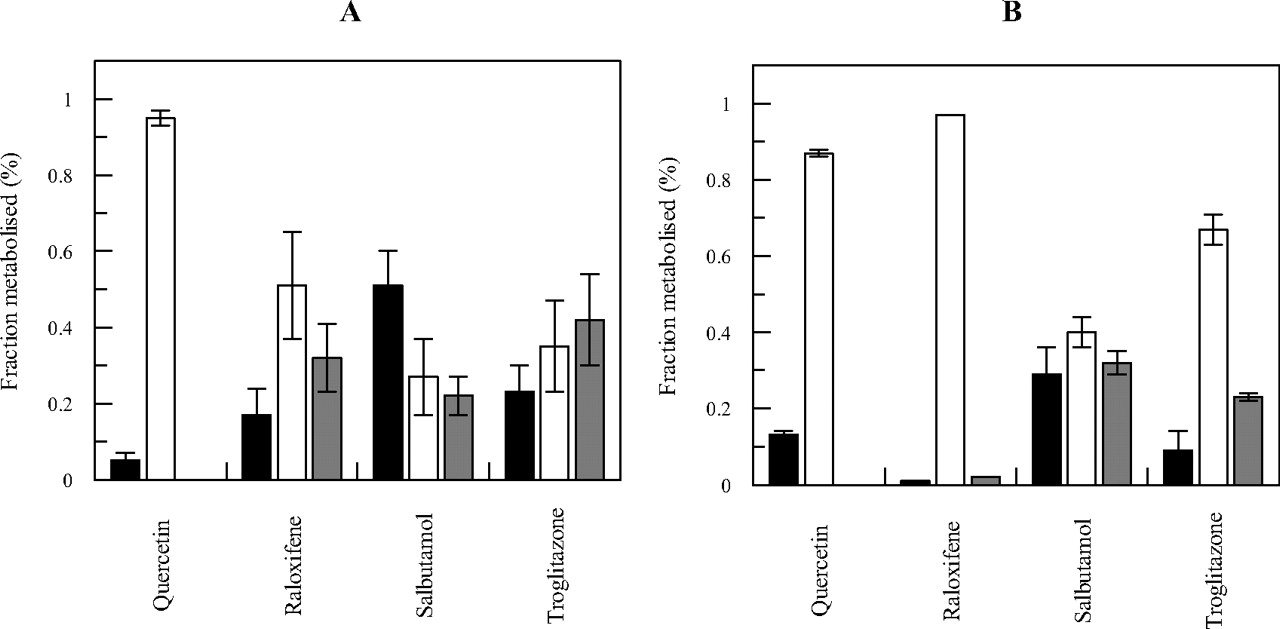
Comparison of Individual Metabolic Pathways in the Liver and Intestine.
In vitro fmSULT, fmUGT, and fmCYP estimates were calculated from mean scaled CLint, SULT, CLint, UGT, and CLint, CYP values. A comparison of these fractions for each of the drugs investigated is shown in Fig. 3, A and B, for the liver and intestine, respectively. A small contribution of sulfation to overall CLint was evident for quercetin in the liver and for raloxifene in the intestine (<5%), whereas up to 51 and 28% was seen for the respective hepatic and intestinal metabolism of salbutamol. The extent of glucuronidation ranged from 27 and 40% (hepatic and intestinal metabolism of salbutamol, respectively) up to 97% (intestinal metabolism of raloxifene). Mean fmCYP ranged from zero in either organ for quercetin, up to 42 and 32% (liver and intestine for troglitazone and salbutamol, respectively).

Comparison of estimated in vitro intestinal and hepatic fraction metabolized by sulfation, glucuronidation, and P450 metabolism for quercetin, raloxifene, salbutamol, and troglitazone. ■, fmSULT; □, fmUGT; dark gray bar, fmCYP. A, liver. B, intestine. The contribution of each metabolic process in vitro was estimated from mean clearance values scaled to per gram of tissue, as defined in eqs. 2 to 4.
Quercetin was the compound with the highest CLint, SULT in both liver and intestine (Fig. 2). However, the extent of sulfation was fairly marginal relative to glucuronidation of this drug and resulted in fmSULT <15% in both liver and intestine (Fig. 3). Quercetin was the only drug for which fmSULT was higher in intestine than in liver (13 and 5%, respectively), and this could be a consequence of the reported high affinity (Km of 2 μM) for SULT1E1 (Teubner et al., 2007; Riches et al., 2009a). Salbutamol was predominantly cleared by sulfation in comparison with glucuronidation and P450 metabolism (Fig. 3) in both the liver and the intestine. Salbutamol is reported to be a substrate for SULT1A3 (Mizuma, 2008), and, therefore, higher intestinal CLint, SULT would be expected, in contrast with current findings. However, there are no recombinant SULT data available to support specificity for this particular SULT. The intestinal fmCYP of salbutamol was comparable to the extent of sulfation, in contrast to liver, in which the contribution of P450 was lower (22% of the total clearance). The contribution of sulfation to the total in vitro metabolism of troglitazone was 23 and 9% in the liver and intestine, respectively (Fig. 3). The scaled CLint, SULT of troglitazone was half of the scaled CLint, UGT and CLint, CYP (Fig. 2) in both organs, in contrast to in vivo data, suggesting that troglitazone sulfate is the main metabolite (Loi et al., 1999). Hepatic P450 metabolism (42%) was comparable to glucuronidation (35%) for this compound, whereas in the intestine glucuronidation was the dominant process (67%); the intestinal fmCYP of troglitazone was only half of the hepatic value. Glucuronidation was the predominant intestinal pathway for raloxifene (>90%), whereas hepatic fmUGT represented only half of the intestinal value and approximately 30% of the hepatic in vitro clearance was attributed to the P450 metabolism and 17% to sulfation. Both intestinal fmSULT and fmCYP were negligible and represented <5% of the hepatic clearance (Fig. 3). This finding is surprising, considering that the recombinant SULT data suggest high affinity for SULT1A1 and SULT1A3 (<3 μM), comparable to that for recombinant UGTs (5–59 μM) (references in the supplemental data).
Prediction of In Vivo Clearance from Cytosolic and Microsomal In Vitro Data.
In vivo CLint, h values used for the assessment of prediction accuracy were calculated from a number of reported intravenous and oral studies using a well stirred liver model. Intravenous clearance data were generally less available, with the exception of data for salbutamol. Oral clearance data were available from >1000 individuals for raloxifene (single study) and from four to seven different studies for quercetin, salbutamol, and troglitazone (Table 3 and supplemental data). Salbutamol had the lowest in vivo observed CLint, h (4.7 and 8.1 ml · min−1 · kg−1 from intravenous and oral clearance data, respectively) of the four compounds and was the only drug in the dataset with a substantial contribution of renal elimination. Raloxifene and quercetin CLint after both intravenous and oral administration were >100-fold higher in comparison with salbutamol, as shown in Table 4.
Mean intravenous and oral plasma clearance data, number of subjects, blood/plasma ratios, and fraction unbound in plasma for drugs investigated
References for all the clinical data are listed in the supplemental data.
Observed and predicted hepatic intrinsic clearance for quercetin, raloxifene, salbutamol, and troglitazone
Predicted clearance was obtained using either individual or combined hepatic sulfation, glucuronidation, and P450 in vitro data.
Further analysis assessed the accuracy of predicted intravenous or oral in vivo clearance, using in vitro data from both cytosolic and microsomal sources. Multiple metabolic pathways contributing to clearance of the selected drugs (CLint, SULT, CLint, UGT, and CLint, CYP) were considered once expressed per gram of liver. For quercetin, accuracy of prediction on the basis of the combined SULT, UGT, and P450 hepatic in vitro clearances (Fig. 4) showed no improvement relative to the use of only glucuronidation data. This finding was consistent with the high estimated contribution of glucuronidation to the total hepatic clearance (hepatic fmUGT of 95%) (Fig. 3). Predicted quercetin CLint, h was 6-fold higher than the observed intravenous CLint, h. Accounting for intestinal glucuronidation (FG estimated as 0.03 from intravenous and oral data) resulted in a comparable degree of overprediction of oral CLint, h to intravenous data. In contrast, raloxifene, salbutamol, and troglitazone, with lower primary pathway involvement (<60% of total in vitro hepatic metabolism), showed a significant difference in predictions performed combining clearances from all metabolic pathways in comparison with those using data from the primary pathway in isolation. Prediction of intravenous clearance of both raloxifene and troglitazone was poor (22–36% of observed, respectively) when in vitro hepatic CLint data corresponding to their primary pathway were used. Incorporation of additional SULT, UGT, and P450 hepatic in vitro data improved prediction accuracy of intravenous clearance, in particular for troglitazone, as the predicted clearance from combined data represented 86% of the observed value. For raloxifene, predicted clearance from the combined approach was marginally outside of the 2-fold of in vivo intravenous clearance (44% of the intravenous clearance) (Fig. 4). In contrast to the success achieved with troglitazone and raloxifene, overprediction of salbutamol intravenous CLint, h was observed using data from all metabolic pathways. For this drug, in vitro CLint data corresponding to the primary sulfation pathway overpredicted intravenous clearance by 3-fold, and thus incorporation of additional pathways increased this error further (626% of observed). The oral clearance of this compound was predicted well from in vitro hepatic SULT CLint (within 2-fold), whereas inclusion of other pathways reduced the accuracy (364% of observed).

Comparison of predicted and observed CLint, h values for quercetin, raloxifene, salbutamol, and troglitazone. A, clearance prediction accuracy using intravenous data for four drugs investigated. B, comparison of observed and predicted CLint values from oral data before (light gray bars) and after (■) correction for in vivo FG. The sum of scaled CLint, SULT, CLint, UGT, and CLint, CYP values per gram of liver was used. Observed intravenous CLint values were calculated using eq. 5, whereas eq. 6 was applied for oral data, assuming either no intestinal first pass (FG = 1) or accounting for estimated FG from intravenous and oral studies.
Ignoring intestinal first pass resulted in significant underprediction of oral CLint (predicted estimates were 5 and 31% of the observed data for raloxifene and troglitazone, respectively). FG estimated from intravenous and oral data were 0.03, 0.12, 0.76, and 0.35 for quercetin, raloxifene, salbutamol, and troglitazone, respectively. Accounting for FG in the oral clearance decreased the overall degree of underprediction seen for these drugs, resulting in good comparability in prediction accuracy between oral and intravenous data, as illustrated in Fig. 4.
Discussion
Contribution of more than one metabolic elimination mechanism (hepatic and extrahepatic) is increasingly becoming a more common scenario, with implications on the in vitro-in vivo extrapolation of clearance and assessment of drug-drug interaction potential (Mohutsky et al., 2006; Rostami-Hodjegan and Tucker, 2007; Zhang et al., 2007; Hinton et al., 2008; Cubitt et al., 2009; Kilford et al., 2009; Gan et al., 2010; Zientek et al., 2010). So far, few attempts have been made to include in vitro data corresponding to cytosolic enzymes such as SULTs in prediction of in vivo clearance in combination with microsomal P450 or UGT data. The aim of the current study was to compare the extent of metabolism by sulfation in the intestine and liver in conjunction with previously published microsomal UGT and P450 data for four selected drugs. A standardized experimental design and appropriate scaling of in vitro cytosolic clearance data were used. Sulfation clearance obtained in the current study covered a 16- to 23-fold range in hepatic and intestinal cytosol, respectively. In both tissues, salbutamol and quercetin had the lowest and highest CLint, SULT, respectively. Experimental variability in cytosolic CLint, SULT estimates of the four drugs, although comparable between liver and intestinal cytosol, was more pronounced in comparison with microsomal CLint, UGT and CLint, CYP (Cubitt et al., 2009); this could be partly due to instability of the cofactor PAPS and inhibition of SULT activity by adenosine 3′,5′-bisphosphate (Nováková et al., 2004).
Appropriate scaling factors are particularly important when data on multiple metabolic pathways obtained in different subcellular fractions are accounted for to avoid propagation of error in the combined approach. Consideration of interindividual and experimental variability in scaling factors is important, and this has been addressed for hepatic and to a lesser extent for intestinal microsomal recovery (Barter et al., 2007; Cubitt et al., 2009). The human hepatic cytosolic scaling factors used here were estimated from several literature sources reporting measurement of a marker protein in both tissue homogenate and subcellular fraction (Table 2). Hepatic cytosolic scaling factors were subject to variation across five studies investigated (52 donors in total), with a coefficient of variation of 38%. No individual data were available to distinguish between the experimental and interindividual variability. No information on variability was reported in the study quoting intestinal cytosolic data in 12 donors (Gibbs et al., 1998). As for intestinal microsomes (Paine et al., 1997), the intestinal cytosolic scaling factor was obtained from cytosols prepared by mucosal scraping. Use of this method rather than enterocyte elution may influence activity and the protein yield of cytosolic enzymes, as was reported previously for a range of P450s (Galetin and Houston, 2006), and hence the estimate of intestinal cytosolic scaling factor. Distribution of sulfation activity along the length of the small intestine has been reported to be highly variable and was also associated with substrate and donor differences (Chen et al., 2003). However, the section of intestine used in Gibbs et al., 1998 was not reported; hence, the use of a single intestinal cytosolic scaling factor and lack of any information on the regional differences in these factors may affect the analysis of the contribution of intestinal sulfation relative to hepatic. Therefore, further refinement of cytosolic scaling factors in the liver and intestine is required using larger numbers of individuals and a more consistent protocol and analysis for covariates.
In the current study, scaled CLint, SULT per gram of organ was generally higher in the liver than in the intestine, with raloxifene and salbutamol showing the largest difference (>4-fold). This trend was also seen for most compounds in the glucuronidation dataset (Cubitt et al., 2009) with the exception of raloxifene and troglitazone. If CLint, SULT were corrected for protein abundance of the main SULT enzyme responsible for the sulfation of each drug (e.g., SULT1E1 for troglitazone and SULT1A3 for salbutamol) (Honma et al., 2002; Riches et al., 2009b), an increased apparent importance of intestinal sulfation relative to that in the liver was observed (data not shown). Protein abundance data for a range of SULTs reported by Riches et al. (2009) in various human tissues were obtained using cDNA-expressed protein as a positive control (abundance units of nanograms of SULT per milligram of cytosolic protein). However, SULT abundance data obtained using purified SULT as a positive control (units of picomoles of SULT per milligram of cytosolic protein) (Honma et al., 2002) are limited, in contrast to the data for P450s. In addition, corresponding UGT absolute abundance data in the liver and intestine are currently lacking, affecting the assessment of the relative contribution of conjugation pathways in the liver and intestine to the overall clearance.
The in vitro fraction metabolized by each metabolic pathway was calculated from mean scaled CLint, SULT, CLint, UGT, and CLint, CYP for each drug in both intestine and liver (Fig. 3). Intestinal mean fmUGT values were within 20% of hepatic for only one of the four drugs analyzed (quercetin). The difference between hepatic and intestinal fmCYP was consistently >40% for all compounds. Consideration of the sulfation clearance provided a more accurate estimate of the contribution of the individual pathways to the total clearance in comparison with the previous analysis performed using only glucuronidation and P450 metabolism data for the drugs investigated (Cubitt et al., 2009). In addition, the discrepancy between high enterocytic concentrations estimated in vivo for some drugs in the dataset (≥9 μM for quercetin and troglitazone, see Materials and Methods) and current in vitro conditions (at 1 μM) may affect the estimated extent of intestinal glucuronidation and sulfation. Finally, any potential contribution of renal elimination is not considered when fractions metabolized in vitro are estimated, and hence their interpretation needs to be done with caution; in the current study this would only affect salbutamol.
The relative role of different metabolic pathways (glucuronidation, sulfation, and P450) in the clearance prediction was investigated using either in vitro data corresponding to the primary metabolic pathway in isolation or a combined approach in which all contributing metabolic pathways were taken into account. In each case the ability to predict intravenous and oral clearance was used to delineate the role of intestinal conjugation processes relative to hepatic (Fig. 4). The use of combined SULT, UGT, and P450 data significantly improved accuracy of predicted in vivo clearance for raloxifene and troglitazone and resulted in predictions within 2.5-fold of observed. Raloxifene has a low and variable bioavailability (approximately 2%) and intravenous clearance was indirectly estimated from oral data (obtained in >1000 healthy subjects); therefore, any inaccuracy in this method will affect the assessment of prediction accuracy. In the case of quercetin, the inclusion of additional hepatic pathways resulted in negligible differences in prediction accuracy, considering the predominant contribution of glucuronidation (>90%) to the total hepatic clearance. Overprediction of quercetin clearance is partially associated with a large variability of its clearance in vivo (20- and >1000-fold range for intravenous and oral in vivo clearance, respectively) (Table 3), in contrast to other drugs. Data from cellular systems for the drugs investigated are not available to allow any direct comparison; the only exception is salbutamol for which depletion clearance reported in human hepatocytes is approximately 10-fold lower when expressed per gram of liver in comparison with the estimate obtained by adding different metabolic pathways from different systems performed here. Overestimation of in vivo clearance by sulfation may also be associated with differences in PAPS concentration in cytosolic in vitro systems and in vivo. In vivo PAPS concentrations have been reported to range from 20 to 30 μM (Coughtrie, 2002), and rapid depletion of the cofactor may limit the capacity of SULT enzymes; in particular, this might be the case in the enterocytes during the absorption phase.
High intestinal extraction of troglitazone, raloxifene, and quercetin (for the latter two, FG < 0.1) has been estimated from in vivo data, supporting indirectly the extent of intestinal conjugation seen in vitro for these compounds. The estimated enterocytic concentrations of raloxifene and troglitazone are less than the reported Km for intestinal UGTs contributing to their metabolism, so saturation of intestinal UGTs is not expected. However, potential saturation of SULTs, in particular for troglitazone, cannot be ruled out. Accounting for intestinal extraction using the corresponding FG significantly improved the prediction accuracy of oral clearance data and reduced the underprediction trend reported previously for raloxifene and troglitazone. In addition, incorporation of FG in the oral clearance resulted in prediction accuracy comparable to that of intravenous data, as expected (Fig. 4). These findings are consistent with recent analysis of a large range of CYP3A substrates (Gertz et al., 2010), illustrating indirectly the importance of intestinal metabolism for some drugs in the current dataset. In contrast with CYP3A, the current ability to predict FG by conjugation from in vitro data is confounded by a number of factors, including differential contribution of UGT and SULT enzymes, lack of absolute enzyme abundance data, and its variability along the intestine.
In conclusion, the current study has illustrated integration of cytosolic and microsomal metabolic data for the assessment of multiple metabolic pathways. Appropriate cytosolic scaling factors have been identified for both liver, and intestine and limitations were discussed. The need to account for all metabolic processes and sites of metabolism in the in vitro-in vivo extrapolation was evident, in particular for raloxifene and troglitazone. Extensive intestinal conjugation contributes to the underprediction of oral clearance for some drugs and emphasizes the need for the incorporation of extrahepatic processes into clearance prediction using appropriate physiologically based pharmacokinetic modeling approaches.
Authorship Contributions
Participated in research design: Cubitt, Houston, and Galetin.
Conducted experiments: Cubitt.
Performed data analysis: Cubitt.
Wrote or contributed to the writing of the manuscript: Cubitt, Houston, and Galetin.
Acknowledgments
We thank Sue Murby and Dr. David Hallifax (University of Manchester) for valuable assistance with the LC-MS/MS, Professor Brian Lake (University of Surrey) for useful discussions on the scaling of in vitro cytosolic data, and Dr. Michael Gertz (University of Manchester) for helpful comments on the manuscript.
Footnotes
The work was supported by a consortium of pharmaceutical companies (GlaxoSmithKline, Lilly, Novartis, Pfizer, and Servier) within the Centre for Applied Pharmacokinetic Research at the University of Manchester.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.036566.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- P450
- cytochrome P450
- UGT
- UDP-glucuronosyltransferase
- SULT
- sulfotransferase
- PAPS
- 3′-phosphoadenosine-5′-phosphosulfate
- HLC
- human liver cytosol
- HIC
- human intestinal cytosol
- HLM
- human liver microsome(s)
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry.

- Received September 30, 2010.
- Accepted February 8, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}