


Abstract
For the first time the human intestinal effective permeability, estimated from the luminal disappearance and intestinal metabolism of phytochemicals, sulforaphane and quercetin-3,4′-glucoside, as well as the simultaneous changes in gene expression in vivo in enterocytes, has been studied in the human jejunum in vivo (Loc-I-Gut). Both compounds as components of an onion and broccoli extract could readily permeate the enterocytes in the perfused jejunal segment. At the physiologically relevant, dietary concentration tested, the average effective jejunal permeability (Peff) and percentage absorbed (± S.D.) were 18.7 ± 12.6 × 10-4 cm/s and 74 ± 29% for sulforaphane and 8.9 ± 7.1 × 10-4 cm/s and 60 ± 31% for quercetin-3,4′-diglucoside, respectively. Furthermore, a proportion of each compound was conjugated and excreted back into the lumen as sulforaphane-glutathione and quercetin-3′-glucuronide. The capacity of the isolated segment to deconjugate quercetin from quercetin-3,4′-diglucoside during the perfusion was much higher than the β-glucosidase activity of the preperfusion jejunal contents, indicating that the majority (79–100%) of the β-glucosidase capacity derives from the enterocytes in situ. Simultaneously, we determined short-term changes in gene expression in exfoliated enterocytes, which showed 2.0 ± 0.4-fold induction of glutathione transferase A1 (GSTA1) mRNA (p < 0.002) and 2.4 ± 1.2-fold induction of UDP-glucuronosyl transferase 1A1 (UGT1A1) mRNA (p < 0.02). The changes in gene expression were also seen in differentiated Caco-2 cells, where sulforaphane was responsible for induction of GSTA1 and quercetin for induction of UGT1A1. These results show that food components have the potential to modify drug metabolism in the human enterocyte in vivo very rapidly.
Mean (± S.D.) values of the absorption markers and perfusion parameters in the control and perfusion with the extract.
The primary biological functions of the gastrointestinal tract are to absorb nutrients from food and to serve as a barrier against toxins and bacteria. Besides nutrients, vitamins and minerals, the human diet also contains a large number of highly biologically active phytochemicals. Some phytochemicals regulate phase I and II enzymes, especially in the liver (Hecht, 1999), and affect the development of chronic diseases (Waladkhani and Clemens, 1998; Kelloff et al., 2000). In unprocessed plant material, many phytochemicals occur as glycosides, which are generally biologically inert. Deglycosylation may occur at different stages of food processing, mastication, digestion, and metabolism, leading to the formation of active compounds. The chemical form (glycoside or aglycone) of phytochemicals affects intestinal absorption and gut wall extraction, modifying their pharmacokinetics (Hollman et al., 1999). Sulforaphane, an isothiocyanate derived from a glucosinolate found in broccoli, has been proposed as a potent dietary anticarcinogen (Kelloff et al., 2000). Very little is known about its intestinal absorption, since there are no specific methods for measuring sulforaphane and its metabolites in plasma. It is found in the urine as a mercapturate, but the total yield is low (Shapiro et al., 2001). The absorption and gut wall extraction of quercetin-3,4′-diglucoside, a flavonoid glycoside naturally occurring in onion, involves deglycosylation by β-glucosidases (Day et al., 1998, 2000) and conjugation with glucuronic acid, methyl or sulfate groups (Gee et al., 2000); although the sites of these modifications are not clear, the conjugated forms and not the glycosides are found in plasma (Day et al., 2001).
For direct investigations of small intestinal absorption and gut wall extraction in humans, only a limited number of methods are available. As a “gold standard” for studying the effective intestinal permeability and metabolism, an intestinal perfusion technique (Loc-I-Gut) has been developed, validated, and widely applied to investigate drug absorption and intestinal, presystemic metabolism (Lennernas et al., 1992; Lindahl et al., 1996; Lennernas, 1997, 1998). A good correlation exists between the measured jejunal Peff2 and the fraction dose absorbed of drugs in humans determined by pharmacokinetic studies.
Using this technique, the majority of shed human enterocytes collected from an intestinal perfusion were still functionally active and did not show signs of apoptosis (Ahrenstedt et al., 1991; Glaeser et al., 2002). Therefore we not only investigated the transport and metabolism of phytochemicals in the human jejunum but also their effect on the short term changes in mRNA expression of phase II enzymes in shed enterocytes after perfusion.
Materials and Methods
In Vivo Jejunal Perfusion. After an overnight fast of 10 h, a regional single-pass perfusion of the proximal jejunum (hereafter called perfusion) was performed in six volunteers using a Loc-I-Gut perfusion tube (Fig. 1) (Synectics Medical, Stockholm, Sweden). Having applied a local anesthetic (lidocaine) to the throat, the Loc-I-Gut tube was introduced through the mouth. During insertion, a Teflon coated guide wire inside the instrument was used to facilitate the passage of the tube into the small intestine. The position of the tube was checked by fluoroscopy until the perfused segment was located in the proximal part of the jejunum. Along with the Loc-I-Gut instrument, another tube was positioned in the stomach for drainage of gastric juice during the experiment. Once the perfusion tube was in place, two balloons as part of the perfusion instrument were inflated with air creating an isolated 10-cm long intestinal segment. A vacuum pump was connected to a proximal drainage channel to drain off any intestinal fluid above the perfused segment. A more extensive description of this intestinal perfusion technique is published elsewhere (Knutson et al., 1989; Lennernas et al., 1992). All solutions (37°C) were pumped into the jejunal segment at a flow rate (Q) of 2.0 ml/min using a calibrated syringe pump. The segment was first rinsed with isotonic saline (37°C) for 10 to 20 min as a prewash and the intestinal contents collected. The segment was then perfused with further isotonic saline (30 min) in a preirrigation step during which the perfusate (solution leaving the intestinal segment) was collected and used to isolate cells (see below). After administration of the perfusion solution (onion and broccoli extract), the perfusate was quantitatively collected on ice in 10 min fractions and immediately frozen at -20°C pending analysis. The segment was then perfused again with isotonic saline, and the perfusate in this postirrigation step was used to isolate cells. The pre and post perfusates for subsequent cell preparation were collected on ice and immediately centrifuged at 4°C, 500g for 5 min. The supernatant was removed and the pellet instantly frozen with liquid nitrogen. The pellet was kept at -70°C pending analysis.
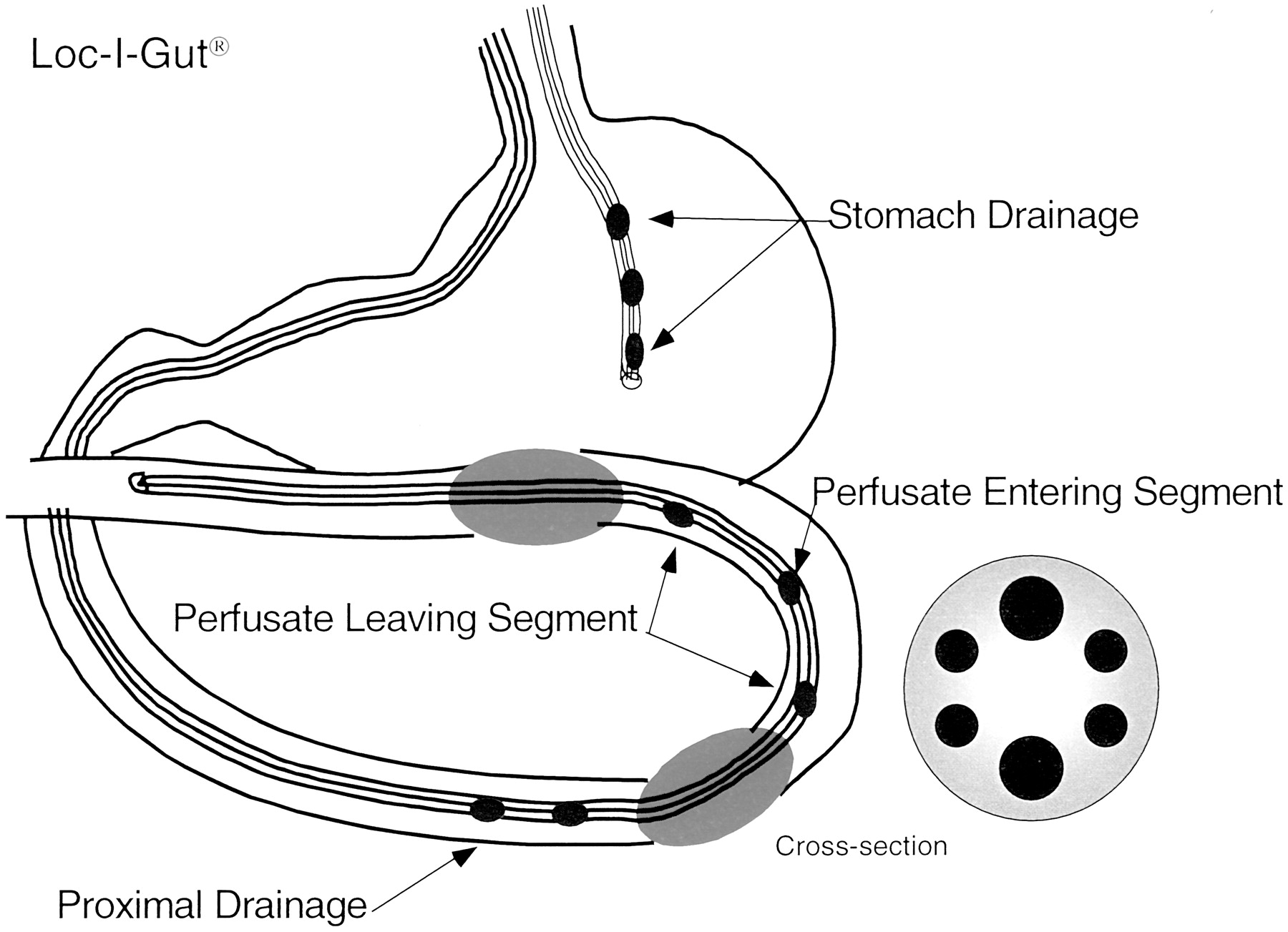
Loc-I-Gut is a perfusion technique for the proximal region of the human jejunum.
The multichannel tube is 175-cm long and is made of polyvinyl chloride with an external diameter of 5.3 mm. It contains six channels and is provided distally with two 40-mm long, elongated latex balloons, placed 10 cm apart, each separately connected to one of the smaller channels. The two wider channels in the center of the tube are for infusion and aspiration of perfusate. The two remaining peripheral smaller channels are used for administration of marker substances and/or for drainage. At the distal end of the tube is a tungsten weight attached to facilitate passage of the tube into the jejunum. The balloons are filled with air when the proximal balloon has passed the ligament of Treitz. A separate tube obtains gastric suction. 14C-PEG 4000 is used as a volume marker to detect water flux across the intestinal barrier.
To confirm that changes in mRNA expression were only due to compounds in the onion and broccoli extract, a control perfusion was performed in five healthy volunteers using isotonic buffer solution containing volume marker but no plant extracts. As we studied relative changes due to the perfusion, independently from interindividual variations, we did not perform the control study in the same volunteers. Cells were collected as described above. The present study was performed at Uppsala University and was approved by the Ethics Committee of the Medical faculty, Uppsala University.
Perfusion Solution (Onion and Broccoli Extract). For the preparation of the plant extract, all glassware used in the preparation of the extract was autoclaved. The water used was obtained from a Millex Q water filtration system and further filtered through a 0.45-μm filter.
Florets of broccoli (Brassica oleracea var. botrytis subvar. cymosa cv. Marathon F1) and red onions (Allium cepa) were obtained from a local supermarket (Norwich, UK). The broccoli florets were washed, stem tissue removed and cut into pieces of approximately 4 to 5 cm in diameter. The outer skins and dead tissue of the onion were removed and the onions cut into quarters. The samples were frozen at -20°C for 16 h and lyophilized at -60°C and 10-1 mbar. The dried samples were milled to fine powders.
A combined extract was prepared by adding 300 g of onion and 167 g of broccoli powder to 5 liters of 2 mM Na2HPO4 (pH 6.4) in water. After stirring at 20°C for 30 min, the extract was filtered through a muslin cloth and centrifuged (5000g, 4°C, 30 min). The clarified extract was sterile filtered through a Millipore “Pellicon” membrane with a 10 kDa cut-off to remove the majority of proteins in the extract. Two subsamples of the final extract were analyzed by LC/MS for sulforaphane, quercetin-3,4′-diglucoside, and possible metabolites. The remaining extract was aliquoted and frozen at -20°C.
The perfusion buffer was isotonic (about 270–300 mOsm l-1) composed of Na+ (147 mM), (Cl- 156 mM), K+(4 mM), phosphate buffer, pH 6.5, 70 mM. 14C-Labeled polyethylene glycol ([14C]PEG 4000) (Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK) was added to the perfusion buffer as a nonabsorbable volume marker (2.5 μCi/l). A low concentration of antipyrine (Astra Läkemedel AB, Södertälje, Sweden) was used as a marker for passive transcellular diffusion in all perfusion experiments.
Immediately before the perfusion experiments, the samples were thawed and diluted 1 in 5 with the perfusion buffer.
Data Analysis. Calculations of the net water flux (NWF), the fraction absorbed (fabs) and Peff, during the perfusion, were made from the mean of five measurements of concentrations during steady state in the perfusate fractions. The NWF (ml/cm) in the perfused jejunal segment was calculated according to eq. 1: 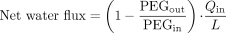 in which PEGin and PEGout are the concentrations of [14C]PEG 4000 (dpm/ml) entering and leaving the segment, respectively. Qin is the flow rate of the perfusion solution (2 ml/min) and L is the length of the perfused jejunal segment (10 cm). The concentration of the analytes in the perfusate was corrected for the water flux before any other calculations were performed.
in which PEGin and PEGout are the concentrations of [14C]PEG 4000 (dpm/ml) entering and leaving the segment, respectively. Qin is the flow rate of the perfusion solution (2 ml/min) and L is the length of the perfused jejunal segment (10 cm). The concentration of the analytes in the perfusate was corrected for the water flux before any other calculations were performed.
The amount that disappeared during the perfusion through the jejunal segment was assumed to have been absorbed (fabs) and was calculated according to eq. 2: 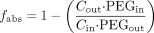 in which Cin and Cout are the concentrations of the analyte entering and leaving the jejunal segment, respectively. The Peff was calculated according to a well-mixed tank model as shown in eq. 3 (Lennernas et al., 1992; Lennernas, 1997):
in which Cin and Cout are the concentrations of the analyte entering and leaving the jejunal segment, respectively. The Peff was calculated according to a well-mixed tank model as shown in eq. 3 (Lennernas et al., 1992; Lennernas, 1997): 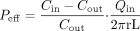 in which the surface of the cylinder (2πrL) of the jejunal segment was calculated using the intestinal radius and length of the segment. The human jejunal radius is 1.75 cm as determined by perfusion of the jejunal segment with barium and X-ray analysis (L. Knutson, personal communication)
in which the surface of the cylinder (2πrL) of the jejunal segment was calculated using the intestinal radius and length of the segment. The human jejunal radius is 1.75 cm as determined by perfusion of the jejunal segment with barium and X-ray analysis (L. Knutson, personal communication)
We assessed if metabolites or conjugates were excreted back into the lumen during the perfusion. For this purpose, the appearance ratio (Ar) was calculated, using eq. 4. 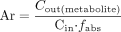 The small amount of sulforaphane-glutathione in the perfusion solution (formed by spontaneous reaction of sulforaphane with plant glutathione) was subtracted, based on the assumption that glutathione conjugates are hydrophilic and therefore poorly absorbed by the gut wall.
The small amount of sulforaphane-glutathione in the perfusion solution (formed by spontaneous reaction of sulforaphane with plant glutathione) was subtracted, based on the assumption that glutathione conjugates are hydrophilic and therefore poorly absorbed by the gut wall.
Statistical Analysis. Gene expression data were normalized against GAPDH, and significance of fold-changes (pre-against postperfusion) was analyzed by Student's t test. All data in the present study are presented as mean and standard deviation (± S.D.).
Reference Compounds. The sulforaphane-glutathione, sulforaphane-l-Cys conjugate, and sulforaphane-mercapturic acid were synthesized according to the method of Kassahun et al. (1997) with minor modifications. Reduced GSH, l-Cys, or N-acetyl-l-Cys were first dissolved in distilled water and 100% ethanol was added. dl-Sulforaphane was dissolved in 80% ethanol, and this was added to each of the sulfur compounds. The solutions were left to react at 20°C for 3 days in sealed round-bottomed flasks. The dithiocarbamates, formed from the reaction between sulforaphane with either GSH or l-Cys, precipitated during the reaction. The precipitates were collected by centrifugation and the pellets were washed with 100% ethanol. The mercapturic acid formed by the reaction of sulforaphane and N-acetyl-l-Cys did not precipitate, and the mixture was rotary evaporated and finally purified chromatographically. A purity of >95% was assessed by 13C and 1H NMR, Maldi-Tof MS and LC-positive ion electro-spray MS.
Standards of quercetin glucuronides and of quercetin glucosides were either synthesized or extracted from plants as described previously (Price et al., 1998; Day et al., 2001). All polyphenolic standards were >97% pure; the identities, including position of attachment of the conjugate, have been determined using chemical synthesis and NMR (O'Leary et al., 2001).
Perfusate Analysis. After thawing at 20°C, each perfusion sample was vortex mixed for 30 s and centrifuged (4000g, 10°C, 10 min) to yield a clear solution.
All the quantification was performed by selected ion monitoring (SIM) LC/MS. Calibration curves were obtained for dl-sulforaphane (from ICN Pharmaceuticals, Biochemicals Division, Aurora, OH), sulforaphane-GSH, sulforaphane-cysteine, sulforaphane-mercapturic acid, quercetin-4′-glucoside, quercetin-3,4′-diglucoside, quercetin-3-glucuronide, quercetin-3-sulfate, glucocheirolin [3-(methylsulfonyl)-propyl glucosinolate; LKT Laboratories, Inc., St. Paul, MN] and quercetin-3-rhamnoglucoside (rutin). All standards were >95% pure.
The concentrations of antipyrine in the perfusion solutions were analyzed by HPLC/UV using a previously validated method (Lindahl et al., 1996; Sandstrom et al., 1999). The total radioactivity of [14C]PEG 4000 in the perfusion solutions were determined by liquid scintillation counting (Mark III; Searle Analytic Inc., Des Plaines, IL) and the osmolality by the vapor pressure method (Vescor osmometer 5500, Wescor Inc., Des Plaines, IL).
SIM-LC/MS Analysis. All SIM-LC/MS analyses were conducted using a Micromass Quattro II triple quadrupole mass spectrometer (Micromass UK Ltd., Manchester, UK) equipped with a Z-spray electrospray ion source, which was coupled to either an Hewlett-Packard 1050 quaternary pump HPLC system (Agilent Technologies, Stockport, UK) or to a Jasco PU-1585 triple pump HPLC equipped with an AS-1559 cooled autoinjector, CO-1560 column oven and UV-1575 UV detector [Jasco (UK) Ltd., Great Dunmow, UK].
HPLC conditions were as follows: solvent A was a 0.1% (v/v) trifluoroacetic/water solution, solvent B was 0.1% (v/v) trifluoroacetic/methanol. The HPLC gradient was 100% A, 0% B at 0.0 min; 80% A, 20% B at 10.0 min; 50% A, 50% B at 25.0 min; 0.0% A 100% B at 40 min; 100% A, 0.0% B at 45 min; equilibrate for 10 min at this composition before next injection. The HPLC column temperature was maintained at 25°C and the autoinjector at 4°C. The 1 ml/min mobile phase flow exiting the HPLC column was split using an ASI 600 fixed ratio splitter valve (Presearch, Hitchin, UK) so that approximately 200 μl/min entered the mass spectrometer (the optimal flow for the Z-spray source); the remainder of the flow was diverted to the diode array detector (UV data were plotted at 227 and 370 nm). A Phenomenex (Phenomenex UK Ltd., Macclesfield, UK) Luna C18 (2) reverse-phase column (250 × 4.6 mm, 5 μM), in combination with a Phenomenex SecurityGuard guard column, was used for all HPLC analyses. All water used in extractions and analyses had been distilled, de-ionized, and filtered (0.45 μm) before use.
The mass spectrometer electrospray source capillary voltage was set to 3.3 to 3.5 kV, cone voltage to 29 V, source block temperature to 120°C and desolvation temperature to 300°C. The nitrogen nebulizing and desolvation gas flows were set to 15 l/h and 450 l/h, respectively. All SIM experiments were performed using dwell times of 0.1 s per mass channel and an inter channel delay of 0.03 s. In most cases, both [M + H]+ and [M + Na]+ ions were monitored. Peak areas were measured, after Savitzky-Golay smoothing, using MassLynx 3.4 (Micromass UK Ltd.) acquisition and processing software.
Caco-2 Cells. Caco-2 cells (human adenocarcinoma cells), obtained from the European Collection of Cell Culture, were maintained in Eagle's minimal essential medium supplemented with 10% fetal calf serum, 1% (v/v) nonessential amino acids and 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were grown in an incubator at 5% C02, 95% air, 37°C and passaged every 7 days. The cells were used from passage numbers 41 to 47. Cells were seeded at a density of 2 × 104 cells per cm2 in 55 cm2 dishes and allowed to adhere overnight. Undifferentiated cells were grown to confluency over a period of 7 days, and the medium was changed every second day. After this time, the cells were allowed to differentiate over a period of 14 days, and the medium was changed every day. On day 21, the medium was removed and replaced with Eagle's minimal essential medium supplemented with 10% fetal calf serum containing the following treatments (2.5 μM quercetin, 25 μM quercetin, 10 μM sulforaphane, 1 in 5 dilution of onion/broccoli extract and 1 in 5 dilution of onion/broccoli extract with 25 μM quercetin). The samples were incubated for a further 2 h. Control samples not treated with a flavonoid or sulforaphane were incubated with the equivalent volume of carrier vehicle (methanol <0.1% v/v final concentration, or water).
Measurement of β-Glucosidase Activity. The β-glucosidase activity of the intestinal contents (collected prior to the test perfusion by elution with saline) was determined according to a method described by Day et al. (2000) by incubating samples (20–50 μl) with quercetin 3,4′-diglucoside (197 μM final concentration, 100 μl of final volume) in 50 mM sodium phosphate buffer (pH 6.0) at 37°C. Incubations were terminated by the addition of an equivalent volume of stop solution (0.01% trifluoroacetic acid in 50% aqueous methanol), filtered and analyzed by HPLC with diode array detection as described elsewhere (Day et al., 2000). Peaks were confirmed by matching retention times and spectra with those of authentic standards (quercetin-3,4′-diglucoside, quercetin-3-glucoside, quercetin-4′-glucoside, quercetin, see above).
Preparation of RNA. Total RNA was extracted from cells using a Qiagen RNeasy mini kit according to the protocol described by the manufacturer (QIAGEN, Dorking, Surrey, UK) after homogenization using a QIAshredder column. RNA was eluted from a binding column with RNase-free water, and RNase inhibitor was immediately added (20 U per preparation) and stored at -70°C. The yield of total RNA was determined using Ribogreen RNA Quantitation kit (R11490; Molecular Probes, Eugene, OR) against a standard curve of ribosomal RNA (16S and 23S rRNA from Escherichia coli).
TaqMan Real-Time RT-PCR Assay. Target mRNA levels were determined by real-time RT-PCR using the ABI prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). The threshold cycle (Ct) was defined by when the fluorescence signal reached to 10 times of noise level (baseline). The TaqMan GAPDH-glyceraldehyde-3-phoshate dehydrogenase control reagents kit (402869) was purchased from Applied Biosystems. Forward and reverse primers, and the fluorogenic TaqMan probes for target genes, were designed using Primer Express Software (Applied Biosystems). Sequence homology of selected oligomers was checked using a National Center for Biotechnology Information BLAST search to ensure that sequences were specific to target genes. Primer and probe sets (Sigma-Genosys, The Woodlands, TX and Applied Biosystems, respectively) were as follows:
GST-α-Human glutathione S-transferase [GST A1–1 (GenBank accession no. M14777), A1–2 (GenBank accession no. M16594)].
Forward Primer: CAG CAA GTG CCA ATG GTT GA
Reverse Primer: TAT TTG CTG GCA ATG TAG TTG AGA A
Probe: 5′-FAM-TGG TCT GCA CCA GCT TCA TCC CAT C-3′–TAMRA
UGT1A1 UDP-glucuronosyltransferase (Basten et al., 2002).
Forward Primer: GGT GAC TGT CCA GGA CCT ATT GA
Reverse Primer: TAG TGG ATT TTG GTG AAG GCA GTT
Probe: 5′-FAM-ATT ACC CTA GGC CCA TCA TGC CCA ATA TG-3′-TAMRA
Real-time RT-PCR reactions were carried out in 96-well plates using TaqMan one-step RT-PCR master mix reagent kit (Applied Biosystems) in a total of 25 μl per well, consisting of 100 to 200 nM probe, 200 to 400 nM primers, and 1 to 20 ng of total RNA. TaqMan RT-PCR conditions were as follows: 48°C 30 min, 95°C 10 min then 40 cycles of 95°C for 15 s and 60°C for 1 min. Data from reactions in triplicate were analyzed with software provided with the TaqMan and quantified against a standard curve over a 4-fold concentration range (correlation coefficients for all quantification assays were >0.985). Ct values were normalized using -fold induction = 2Ct(control)-Ct(treatment). Ct values were converted to an estimate of relative copy numbers of each mRNA/ng total RNA based on a universal standard curve used for TaqMan assays. Expression was normalized by eq. 5: 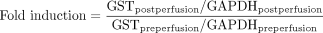
Results
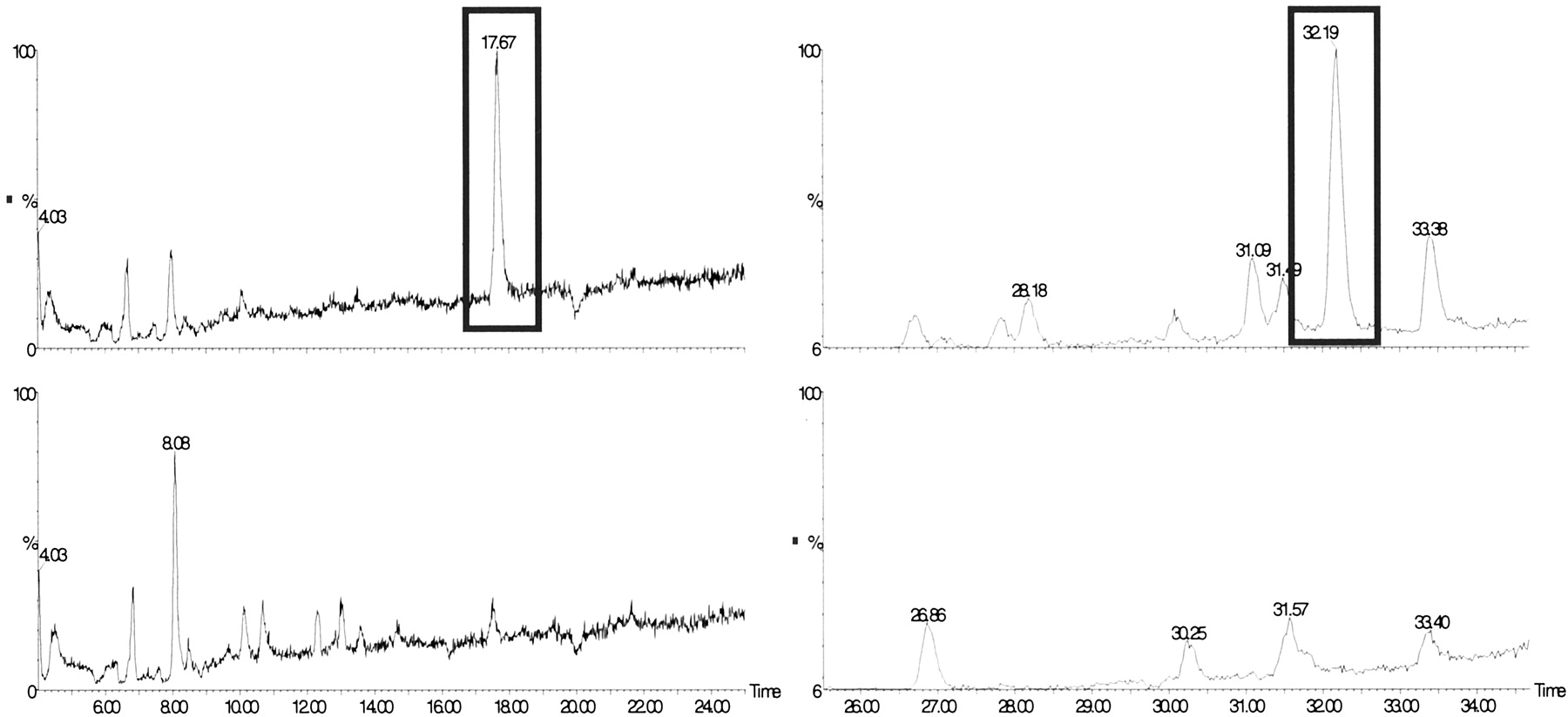
Intestinal Transport and Metabolism of Sulforaphane and Quercetin-3,4′-diglucoside in Human Enterocytes in Vivo. The jejunal perfusion of six healthy volunteers with an extract of onion and broccoli showed that sulforaphane, derived from the broccoli in the extract, was well absorbed from the jejunum as evidenced by a high disappearance rate from the perfused jejunal segment. The concentration of sulforaphane in the segment reached a steady state after 50 min. The average (± S.D.) Peff and fabs values were 18.7 ± 12.6 × 10-4 cm/s and 74 ± 29% for sulforaphane (Table 1). LC/MS analysis of the perfusate (Fig. 2) revealed the presence of sulforaphane-glutathione conjugate but no conjugates with cysteine or with N-acetyl cysteine; the latter is the major urinary metabolite in humans (Conaway et al., 2000). This indicates a phase II metabolism of sulforaphane in the enterocytes. The sulforaphane conjugate appearance ratio (Table 1) suggests that a high proportion of this metabolite is effluxed back into the jejunal lumen.
Absorption parameters of sulforaphane and quercetin-3,4′-diglucoside (mean ± S.D.) based on concentrations for sulforaphane-glutathione and quercetin-3′-glucuronide.
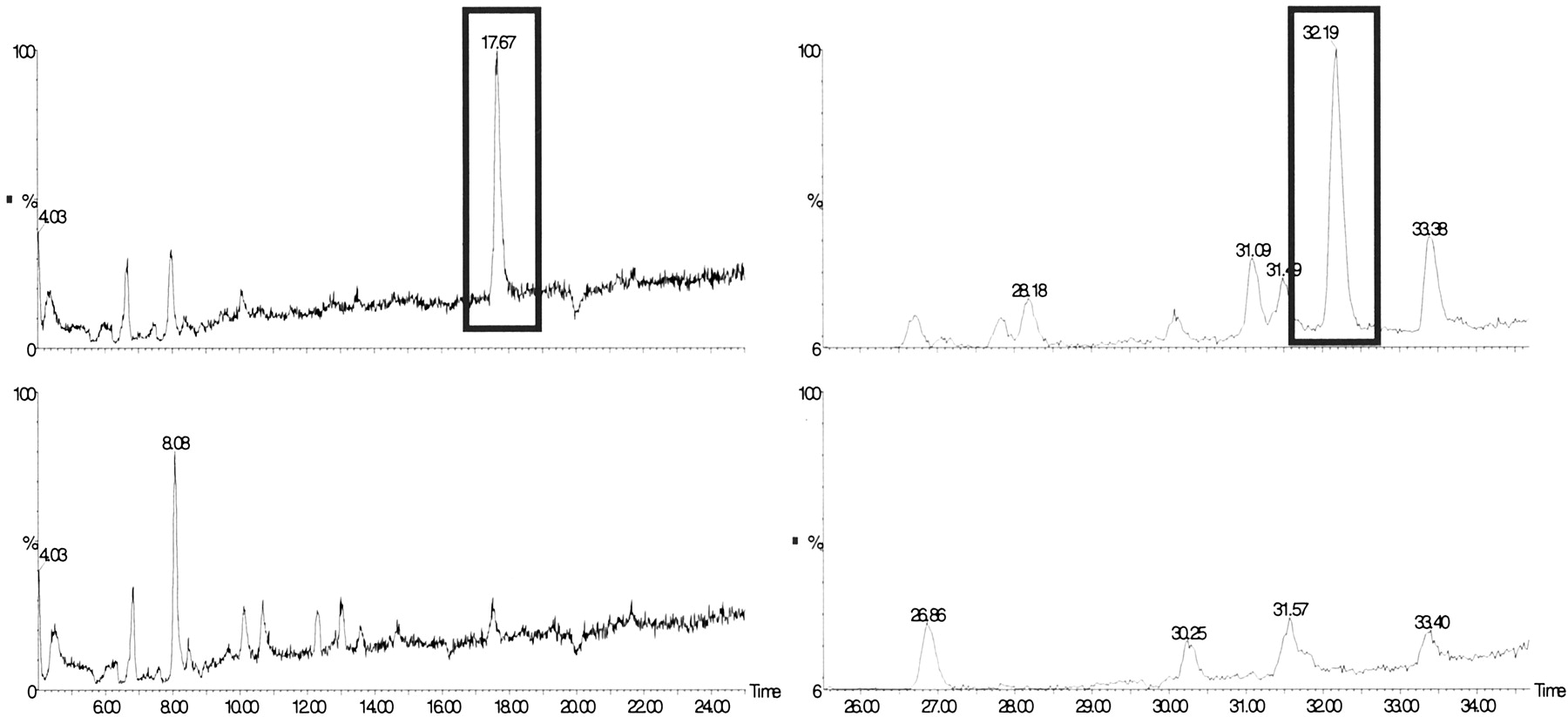
LC/MS analysis of perfusion solutions.
SIM LC/MS traces of the [M + H]+ ion (m/z 485) of the sulforaphane-glutathione conjugate (left) and the [M + Na]+ ion (m/z 501) of quercetin-3′-glucuronide (right) pre- (lower traces) and post- (upper traces) perfusion. The pre and postperfusion traces have been normalized to the same scale. x scale, LC retention time; y scale, abundance of ion (arbitrary units)
Quercetin-3,4′-diglucoside, derived from the onion in the extract, also rapidly disappeared from the perfused jejunal segment (Table 1). We have previously shown that the glucoside is metabolized to an aglycone by lactase phlorizin hydrolase (LPH) before it is absorbed by the intestine (Nemeth et al., 2002). Therefore the permeability for this compound is perhaps better described as a disappearance rate, which is composed of both brush-border metabolism mediated by LPH and cellular permeability for the aglycone. LC/MS analysis of samples obtained at 70 min for quercetin-7-, quercetin-4′-, quercetin-3-, and quercetin-3′-glucuronides revealed that quercetin-3′-glucuronide was the only metabolite effluxed back into the lumen. As indicated by the appearance ratio, the relative amount of metabolite (quercetin-3′-glucuronide) that was effluxed was lower than for the sulforaphane conjugate (Table 1). The excretion of the conjugate indicates that at least part of the glucuronidation of quercetin takes place in the enterocyte. No quercetin aglycone was found in the effluxed samples, indicating that after deglycosylation, quercetin enters the cells, probably by passive diffusion, and is conjugated within the enterocyte. We cannot rule out the possibility that some changes (degradation and other conjugation reactions) occur to the aglycone, which would prevent detection by our mass spectrometry methods. However, at the pH of the perfusion (6.5), quercetin is relatively stable, although it is unstable at pH 7.4 (Boulton et al.1999; Day, 2000).
β-Glucosidase Activity of the Luminal Contents. In the jejunal perfusion described here, the contents of the jejunum were washed out before the perfusion began. To determine whether there were any enzymes in the lumen capable of deglycosylating quercetin-3,4′-diglucoside, we collected the lumen contents and assayed them for β-glucosidase activity (Table 2). This was compared with the capacity of the perfused segment to deglycosylate quercetin-3,4′-diglucoside as calculated from the change in quercetin-3,4′-diglucoside concentration during the perfusion. The results show that the enterocytes are responsible for the majority (79–100%) of the β-glucosidase activity and that the lumen contents account for <20% of the activity.
β-Glucosidase activities in the isolated small intestinal segments
β-Glucosidase activities were assayed using samples of the luminal contents recovered from the isolated segment prior to the test perfusion and quercetin-3,4′-diglucoside as substrate (197 μM final concentration). The capacity of the isolated segment to deglycosylate the quercetin-3,4′-diglucoside component of the onion-broccoli extract was calculated from the change in quercetin-3,4′-diglucoside concentration occurring during the perfusion (mean concentration in perfusate = 57 μM) using the segment volumes estimated from 14C-PEG 4000 radio-counting and a flow rate of 2.0 ml/min.
Modulation of Gene Expression in Human Enterocytes as the Result of the Perfusion. Shed human enterocytes were collected before and after the perfusion.
Table 3 shows the changes in relative mRNA expression of GSTA1 and of UGT1A1 normalized against GAPDH, when compared with cells collected before and after the perfusion. In both cases, perfusion with the onion/broccoli extract led to a significant increase in expression of mRNA encoding for both phase II enzymes. A successful perfusion with buffer alone was carried out in five volunteers. No significant change in expression of mRNA of GST or UGT relative to expression of GAPDH mRNA was seen in these experiments (Table 3).
Relative changes in mRNA expression of GSTA1 and UGT1A1 in enterocytes collected postperfusion with perfusion solution or perfusion buffer (control) compared to enterocytes collected preperfusion.
Data are normalized against GAPDH mRNA expression. Perfusion solution, six individuals; buffer control, five individuals.
Metabolism of Quercetin-3,4′-diglucoside in Caco-2 Cells. Differentiated Caco-2 cells were extremely slow in metabolizing quercetin-3,4′-diglucoside, an activity which we have previously shown to be carried out by LPH (Day et al., 2000), the brush border β-glucosidase. We therefore measured LPH activity in Caco-2 cells and found that the levels are extremely low compared with human small intestine, which is in accordance with a recent report showing significant differences in gene expression levels in Caco-2 cells compared with cells of the human duodenum (Sun et al., 2002). Differentiated Caco-2 cells contained 0 to 0.12 mU/mg LPH activity in agreement with published data (0.1–0.3 mU/mg even in the “high LPH” PD7 Caco-2 clone) (Chantret et al., 1994), whereas human small intestine from lactose tolerant and intolerant individuals contained 20 to 80 and 2 to 10 mU/mg, respectively (Rossi et al., 1997).
Induction in Caco-2 Cells. Since Caco-2 cells do not metabolize quercetin-3,4′-diglucoside, unlike the human jejunum in vivo, we substituted quercetin-3,4′-diglucoside with quercetin (the product of active LPH) in the cell culture medium and measured induction of UGT and GST in Caco-2 cells. Differentiated Caco-2 cells were treated with the onion/broccoli extract, with quercetin and with sulforaphane, for the same length of time (2 h) as the in vivo perfusion (Fig. 3). The onion/broccoli extract showed a significant induction of both GST and UGT mRNA of 1.5- to 1.6-fold. Quercetin alone induced UGT (up to 2.6-fold) but not GST, whereas sulforaphane alone induced GST (3.1-fold) but not UGT.

Modulation of expression of GSTA1 (open bars) and UGT1A1 (black bars) mRNA in differentiated Caco-2 cells following an incubation period of 2 h.
Control, carrier vehicle (water or methanol); OB, onion/broccoli extract diluted 1/5; Q 2.5, 2.5 μM quercetin; Q 25, 25 μM quercetin; sulforaphane 10, 10 μM sulforaphane. Results are the mean value ± S.D. from six monolayers obtained from two-independent experiments. For experimental details, see Materials and Methods section on Caco-2 cells.
Discussion
The questions that we have addressed in this paper are as follows: 1) Are sulforaphane and quercetin-3,4′-diglucoside absorbed and metabolized in vivo in the human small intestine? 2) Are their conjugates subjected to intestinal efflux in vivo? 3) Does intestinal exposure cause short-term changes in gene expression of GST1A1 and UGT1A in enterocytes? 4) Are Caco-2 cells a good model for examination of transport, metabolism, and gene regulation for these compounds? To answer these questions, we have used the Loc-I-Gut system as a validated experimental setup to determine in vivo Peff-values (Lennernas et al., 1992; Lennernas, 1998), intestinal transport, and gut wall metabolism in vivo in humans (Sandstrom et al., 1999). The major advantage of the current perfusion method is that it is possible to measure absorption, metabolism, and secretion without influence of other gastrointestinal factors such as transit time and regional pH-differences. The total amount of broccoli and onion administered in a perfusion to each volunteer was equivalent to a dry weight of 2.2 g onion and 1.2 g broccoli.
Because of its lipophilicity [log P (octanol/water) = 0.72] (Cooper et al., 1997) and molecular size (mol. wt. = 177), sulforaphane is likely to passively diffuse into the enterocytes (Winiwarter et al., 1998). In other experimental systems, sulforaphane was conjugated (in hepatic cells) with glutathione by GSTs leading to maintenance of the concentration gradient, thus a fast passive absorption into the cell and a build up of intracellular sulforaphane-glutathione (Zhang and Callaway, 2002). In the perfused small intestine, we have shown that a significant proportion of the sulforaphane absorbed into enterocytes was effluxed back into the lumen as a sulforaphane-glutathione conjugate. Based on previous studies in cultured human cells, P-glyco-protein is likely to be responsible for the efflux of the sulforaphane conjugate (Dietrich et al., 2001; Zhang and Callaway, 2002). These results suggest that the excretion of conjugated sulforaphane could determine the extent of absorption of isothiocyanates following the mechanism proposed in Fig. 4. Under nonperfusion conditions, GSH is supplied to the intestinal lumen with the bile and reaches luminal concentrations in rats of about 250 μM (Samiec et al., 2000). In the jejunal perfusion model, this glutathione is removed during washing of the segment in the preperfusion phase, and the two balloons prevent leakage of intestinal contents into and out of the segment, as indicated by the high recovery of the nonabsorbable marker PEG 4000 (Table 1). Although during perfusion conjugation is most likely to occur inside the enterocyte where the concentration of glutathione is likely to be in the millimolar range (Baillie and Slatter, 1991), under nonperfusion conditions, nonenzymatic conjugation of sulforaphane probably also occurs in the lumen in addition to the conjugation in the enterocytes. Taking together the luminal conjugation and the high extent of efflux into the lumen, the transmucosal transport of sulforaphane and conjugate is not expected to be high, indicating that the bioavailability might be substantially lower.

Suggested mechanism for sulforaphane absorption.
Quercetin-3,4′-diglucoside disappeared readily from the perfused jejunal segment in humans. However, as quercetin-3,4′-diglucoside is hydrophilic (log P = -1.3; A. J. Day, personal communication) and relatively large (mol. wt. = 626), it is expected that the passive permeability across the apical enterocyte membrane would be low. Unlike quercetin monoglucosides, quercetin-3,4′-diglucoside only interacts very poorly with the sugar transporter SGLT1 (Gee et al., 2000), which means that carrier-mediated transport is unlikely.
However, it is a substrate for β-glucosidases, which will release the aglycone from quercetin 3,4′-diglucoside. Based on a log P value of 1.8 and a mol. wt. of 302, the aglycone is predicted to have a high passive membrane permeability (Murota et al., 2000). The intestinal contents may contain β-glucosidases originating from the microbial population (very low in the proximal jejunum; ∼104 pfu/ml), food, bile, shed cells from the upper gastrointestinal tract, or cellular secretions. Enterocytes contain β-glucosidases that are capable of deglycosylating quercetin glucosides, evidenced by deglycosylation of quercetin monoglucosides during absorption in rats (Day et al., 1998; Gee et al., 2000; Nemeth et al., 2002). Our data demonstrate that the majority of the β-glucosidase activity is derived from intact enterocytes (Table 2). A minimum of 79% of the deglycosylation capacity observed during perfusion was due to intact enterocytes. LPH is expressed on the outside surface of enterocytes allowing direct contact with luminal contents. Thus, in the perfusion experiments, it is the most likely enzyme to be responsible for hydrolysis of quercetin-3,4′-diglucoside. Once inside the enterocyte, quercetin will be subject to glucuronidation. In the human perfusion experiment, quercetin-3′-glucuronide was the only metabolite that was effluxed back into the jejunal lumen. Thus we speculate that the chemical position of intracellular conjugation of quercetin with glucuronic acid determines the subsequent metabolic route [i.e., that conjugates in the B ring (i.e., 3′ and 4′) are excreted back into the lumen, and conjugates at other positions (3 and 7) are absorbed into the body]. This is supported by studies in isolated rat everted intestinal sacs, in which quercetin glucosides were deglycosylated and appeared at the serosal site as quercetin-3- and 7-glucuronides (Gee et al., 2000). In addition, Boersma et al. (2002) reported glucuronidation of quercetin in four positions, when they incubated quercetin with human small intestine microsomes. In their study, the 3′-position was the preferential site of glucuronide conjugation, followed by the 4′-, 7- and 3-positions. Using supersomes containing individual UGTs, they also demonstrated that UGT1A8 (found mainly in the small intestine) selectively glucuronidated quercetin in the 3′-position. This further indicates that the glucuronidation of quercetin in the human small intestine is regioselective for B ring conjugates, causing them to be excreted back into the lumen. A summary of the proposed mechanism is given in Fig. 5.
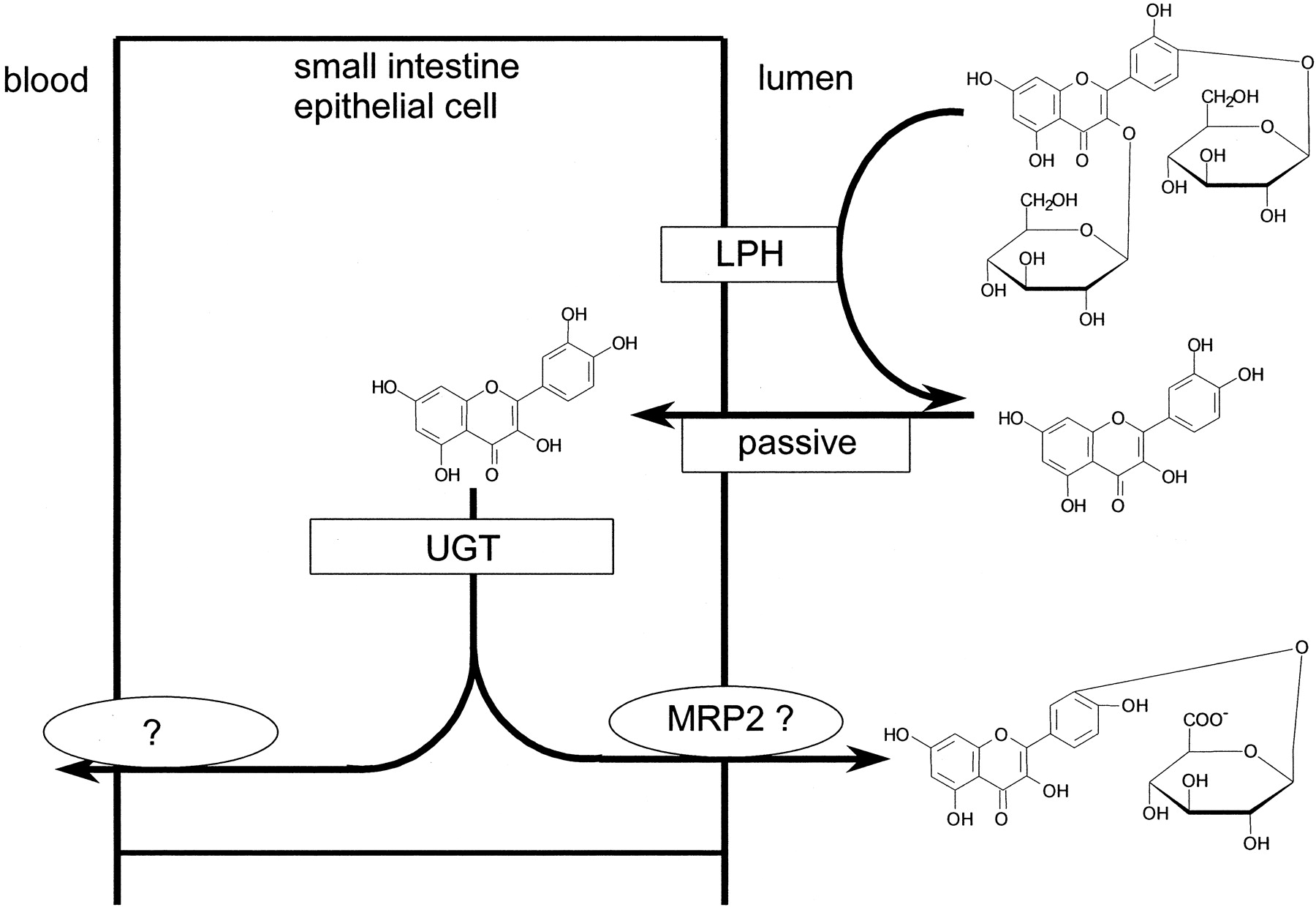
Suggested mechanism for quercetin absorption.
It is well known that there are significant differences in gene expression levels between Caco-2 cells and human enterocytes (Sun et al., 2002). It has also been reported that these differences in gene expression are consistent with observed differences in carrier-mediated cellular processes. In this study we showed additionally that the LPH activity was significantly lower in the Caco-2 model compared with human jejunum, which leads to pronounced differences in the absorption/metabolism capability for a range of compounds and shows the limitations of the Caco-2 cell model.
A similar experimental setup to the Loc-I-Gut has been used to collect and study shed human enterocytes (Glaeser et al., 2002). The investigators found that the majority of shed human enterocytes collected were still functionally active, expressed drug-metabolizing enzymes and transporters, and did not show signs of apoptosis. Based on these findings, for the first time we exploited a human perfusion system to study cellular response of human enterocytes to compounds applied with the perfusion solution in vivo. Therefore this is the first report of mRNA modulation encoding metabolic enzymes in human enterocytes in vivo. GST1A and UGT1A1 were chosen because they are the major enzymes involved in the metabolism of the compounds considered in the permeability study (van Lieshout et al., 1998), they are major phase II enzymes involved in first pass metabolism in enterocytes, and they are known to be inducible in hepatocytes (Manson et al., 1997). Control perfusions of buffer alone showed no induction of either GSTA1 or UGT1A1. After deglycosylation by β-glucosidases, quercetin-3,4′-diglucoside is metabolized by UGT1A1 and also induces UGT1A1 mRNA levels. In Caco-2 cells, sulforaphane increases GSTA1 mRNA levels, and this is the enzyme that conjugates sulforaphane. Other compounds have also been reported to modulate GST in the gastrointestinal tract of animals and in human biopsies. Thus phenylethylisothiocyanate and the sulforaphane analog compound-30 induced rGSTT1–1 protein levels in the rat gastrointestinal tract (van Lieshout et al., 1998). Cisapride, diuretics, cortisol, analgesics, or a high consumption of vegetables increased duodenal GST π and GST α in biopsies from 202 human volunteers (Hoensch et al., 2002). On the other hand, continuous exposure to 5-fluorouracil led to a decrease in the activities of drug-metabolizing enzymes [e.g., GSTs and UGTs in the rat intestinal mucosa (Yoshisue et al., 2001)]. There is increasing evidence for a coordinated regulation of drug-metabolizing enzymes and the export pump MRP2 via common cellular mechanisms, most likely to be related by nuclear hormone receptors [e.g., the pregnane X receptor (Payen et al., 2001)]. Thus we speculate that MRP2 may be responsible for efflux of quercetin conjugates from the enterocyte, especially since this transporter is active on quercetin-4′-glucoside (Walgren et al., 2000). The basolateral transporter is unknown. In the future, a combination of the Loc-I-Gut technique with genomic approaches would enable us to study gene regulation and to discover common regulatory mechanisms involved in intestinal absorption and defense.
Conclusions
For the first time, the human intestinal permeability and metabolism of phytochemicals as well as resulting changes in gene expression in human enterocytes in vivo have been studied. Our results demonstrate that food components induce phase II enzyme-encoding mRNA in the human enterocyte very rapidly. These new in vivo data provide further evidence of the contribution of the diet to a large interindividual variability seen in pharmacokinetics of drugs. It also demonstrates a potential pathway for direct food-drug interaction at the gastrointestinal level.
Footnotes
-
↵ 1 Current address: Toxicology Unit, Section on Clinical Pharmacology, Imperial College, Hammersmith Campus, DuCane Road, London, W12 ONN, UK.
-
↵ 2 Abbreviations used are: Peff, effective jejunal permeability; Q, flow rate; LC/MS, liquid chromatography/mass spectrometry; PEG; polyethylene glycol; NWF, net water flux; fabs, fraction absorbed; L, length; C, concentrations of the analyte; GSH, glutathione; l-Cys, l-cysteine; SIM, selected ion monitoring; HPLC, high performance liquid chromatography; EMEM, Eagle's minimal essential medium; RT-PCR, reverse transcription-polymerase chain reaction; Ct, threshold cycle; LPH, lactase phlorizin hydrolase; UGT, UDP-glucuronosyltransferase; GST, glutathione transferase.
-
Part of this work was supported by funding from the Biotechnology and Biological Science Research Council, UK.
- Received November 1, 2002.
- Accepted February 27, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}