


Abstract
The existing procedures for quantitative in vitro-in vivo clearance prediction can be significantly biased either by totally neglecting the existing variability and uncertainty by using mean parameter values or by implementing Monte Carlo simulation with statistical distribution of the parameters reconstructed from very small sets of data. The aim of the present study is to develop a methodology for the prediction of in vivo hepatic clearance in the presence of semiquantitative or qualitative data and accounting for the existing uncertainty and variability. The method consists of two steps: 1) transformation of the information available into fuzzy sets (fuzzification); and 2) computation of the in vivo clearance using arithmetic operations with fuzzy sets. To illustrate the approach, rat hepatocyte and microsomal data for eight benzodiazepine compounds are used. A comparison with a standard Monte Carlo procedure is made. The methodology proposed can be used when Monte Carlo simulation may be biased or cannot be implemented. The obtained fuzzy in vivo clearance can be used subsequently in fuzzy simulations of pharmacokinetic models.
The ability to predict pharmacokinetic events in vivo, especially in humans, from in vitro data and other relevant information is of great importance both to academia in providing a quantitative framework for identification and investigation of the key processes involved and to industry in facilitating drug selection and development. Of all the pharmacokinetic parameters, most work in this endeavor has concentrated on the prediction of hepatic (metabolic) clearance. It usually comprises a series of sequential steps, starting from the estimation of intrinsic clearance in the in vitro system, through the identification of appropriate scaling coefficients, and ending with the implementation of a liver model. The ultimate goal is to achieve as accurate a quantitative prediction of the in vivo hepatic clearance as possible.
The information used and generated throughout the in vitro-in vivo clearance prediction process is characterized by a large degree of uncertainty and significant variation in the experimental values (Houston and Carlile, 1997; Iwatsubo et al., 1997; Lavè et al., 1997; Obach et al., 1997) due to a number of highly related reasons. These include the complexity and sometimes unknown nature of the phenomena involved, the imperfect instrumentation and information processing tools, and the high inherent variability of the biological systems. The terms variability and uncertainty are used almost interchangeably in the pharmacokinetic and metabolism literature. It should be noted, however, that variability is an inherent property of the system of interest; it can be observed and recorded but not changed. In contrast, uncertainty relates to variations due to errors in assumptions, hypotheses, observations, experiments, and handling of the system studied. Accordingly, uncertainty in the information available can be decreased and theoretically eliminated by implementing “ideal” experiments and data-processing techniques. Consequently, there is significant variability and uncertainty in the parameters of the scaling models used and ultimately in the predicted in vivo clearance estimates.
Despite this situation, most researchers continue to develop, use, and report models with single, fixed value (mean) parameters, producing predictions in the form of single variables or single curves without even considering the influence of the variability and uncertainty involved on the reported results (Carlile et al., 1998;Matsui et al., 1999; Obach, 1999). This practice is a source of the following concerns. First, handling single values in the presence of significant variability and uncertainty is inappropriate and may be misleading (Farrar et al., 1989; Bois et al., 1991; Hattis et al., 1990; Gearhart et al., 1993; Krewski et al., 1995). Usually derived from a small sample, the mean may not be representative or even meaningful. Hence, modeling and predicting using mean parameter values are dubious. On the other hand, the scatter within the small sample gives some idea about the existing variability and uncertainty. Second, in most cases variability is one of the most important and interesting features of drug metabolism (Tooley et al., 1998a). Therefore, accounting for it is realistic; ignoring it may provide a skewed image of reality. Third, developing a model for uncertainty enables one to reduce it in a formal way by minimizing the errors introduced by the theoretical, experimental, and data processing methodologies. Various optimal experimental design methods show one of the ways to do that (Endrenyi, 1981). Fourth, the incorporated measure of uncertainty and variability in the scaled in vivo clearance value can easily be transformed into a measure of the confidence in the prediction, which could be useful, especially for decision-making purposes (e.g., in risk analysis or drug selection).
To accommodate the listed concerns, the aim of the current study is to develop and validate a methodology for incorporating measures of variability and uncertainty into the prediction of in vivo hepatic clearance from in vitro data. The approach, based on fuzzy set theory, handles virtually any form of in vitro-in vivo scaling information available.
Materials and Methods
Variability and Uncertainty in the in Vitro-in Vivo Scaling Procedure.
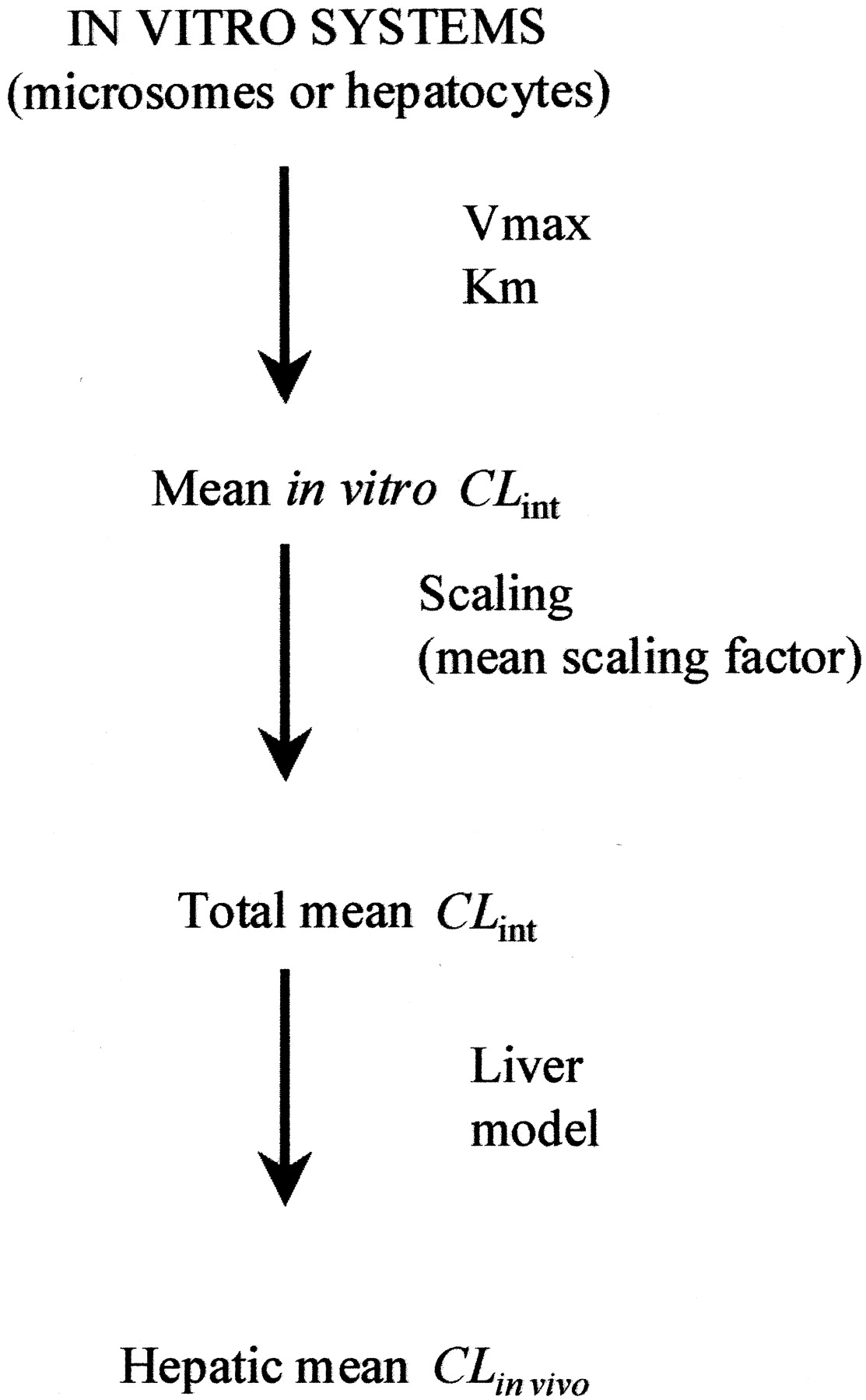
The standard, mean value-based procedure for predicting in vivo hepatic clearance from in vitro data is given in Fig.1. Scaling is done by using an appropriate scaling coefficient (Scaling factor). It relates metabolism determining properties (such as total microsomal protein or individual cytochrome P450 isoform content) of the in vivo liver to the particular in vitro system. Scaling factor is most often taken from literature but is ultimately determined from experimental data (Houston, 1994; Carlile et al., 1997; Obach et al., 1997). A mean value is usually used. Use a liver model, such as the well stirred model eq. 1 to predict the mean in vivo blood hepatic clearance.

Standard in vitro-in vivo scaling procedure (Houston, 1994).
Several typical features of the in vitro-in vivo clearance prediction exercise bear directly on the issue of variability and uncertainty. The in vitro experiment (metabolite formation or substrate depletion) is usually carried out on a small number of incubations (rarely more than five), with a limited number of sampling time points (Carlile et al., 1997; Obach et al., 1997). In such cases, the usual statistical processing procedures may not be relevant but are still widely applied, often without caution. Quite often, especially in early drug discovery, the only information derived is qualitative or semiquantitative (see Table 1 for examples).
Classification of common information types
A review of the existing in vivo clearance prediction literature shows that where such vague or limited information is available, no formal scaling techniques that incorporate it have been implemented. Sometimes attempts are made to extract quantitative values from the existing information, using empirical approaches, such as “best-guess estimates” or “trial-and-error”. The aim of this article is to fill this methodological gap.
Monte Carlo Simulation.
The usual procedure for incorporating measures of variability and uncertainty into pharmacokinetic modeling is Monte Carlo (MC1) simulation (Farrar et al., 1989; Bois et al., 1991;Hattis et al., 1990; Gearhart et al., 1993; Krewski et al., 1995). The core of this procedure is the specification of prior model parameter probability distribution functions (pdfs). These can either be defined directly from experimental observations or based on various assumptions and considerations (e.g., log-normality of clearances and rate constants). Once the pdfs are specified and defined quantitatively, the MC procedure involves their multiple sampling and subsequent computation of the model outputs.
Details of the MC simulation procedure adopted here are presented in detail in Nestorov et al. (2000). In summary, the procedure comprises the following steps:
Assignment of Distributions.
A prior probability distribution of CLint,in vitro, usually assumed lognormal, is specified. Distributions, usually assumed normal, are also assigned to the cytochrome P450 and/or protein contents of the in vitro system, the fraction unbound in plasmafU, and the fractions of cardiac output perfusing the hepatic artery qHA and hepatic portal vein qHP. Rat liver weight and cardiac output, assumed to be known and relatively stable, is not varied.
Sampling.
This took the form of the following steps. First, sample the in vitro intrinsic clearance distribution and then the distribution of the scaling coefficient factor (for microsomes and hepatocytes). Multiply the in vitro intrinsic clearance obtained in the sample by the scaling coefficient and the liver weight to calculate the predicted in vivo whole liver intrinsic clearance value, CLint. Finally, sample the liver model parameters (e.g., fraction unbound in plasma, blood-to-plasma ratio, and hepatic blood flow) and calculate the respective in vivo clearance value using Eq. 1. Repeat the sampling procedure to generate the statistical distribution of the scaled in vivo CLH,PRED. All calculations were programmed in MATLAB (MATLAB Manual, 1998; The Mathworks, Inc., Natick, MA).
Fuzzy Set-Based Simulation.
Fuzzy set theory (FST) incorporates measures of uncertainty and variability in model parameter values by representing them in the form of fuzzy sets (numbers) (Dubois and Prade, 1980;Ross, 1995; Berkan and Trubatch, 1997;Zimmerman, 1997). Unlike the MC simulations, no pdfs are assumed. Rather the information is taken directly from the experimental or reported data. A fuzzy number (Fig. 2) is described by an interval and a membership function. The membership function expresses the degree (of certainty) with which a parameter is considered to belong to the respective interval (set) of values, the maximum certainty being one and the minimum zero. The example in Fig. 2represents the fuzzy number “hepatic clearance of drug A”; its interpretation shows that the clearance value most certainly is between 5.8 and 7.8 and belongs to the intervals 2.1 to 5.8 and 7.8 to 12.9, with less certainty decreasing towards both ends. Thus, the fuzzy set attaches a measure of the respective uncertainty to each interval in which a parameter (e.g., clearance) is localized. The trapezoidal membership function, shown in Fig. 2 is only one of a variety of possible shapes (Ross, 1995; Berkan and Trubatch, 1997).

Fuzzy set representation of the hepatic clearance of a drug.
The clearance value most certainly is between 5.8 and 7.8 and belongs to the intervals 2.1 to 5.8 and 7.8 to 12.9, with less certainty decreasing toward both ends.
Application of FST to the current problem was achieved as follows. First, fuzzy numbers were generated from the available information for CLint,in vitro, the scaling factor,fU, R, qHA, and qHP. The liver weight and cardiac output were fixed because they are considered to be known and relatively stable. In each case, formal procedures exist to transform the respective information into fuzzy numbers (Ross, 1995;Berkan and Trubatch, 1997). Second, using the restricted DSW method for fuzzy arithmetic (Dong et al., 1985; Dong and Shah, 1987; Yager, 1986; Givens and Tahani, 1987; Ross, 1995), the intermediate fuzzy parameters needed for the liver model were calculated. For the well stirred model, these are the whole liver intrinsic clearance CLint, the fraction unbound in blood fUB, and the total hepatic blood flowQH. Third, using the well stirred liver model and the restricted DSW method, the in vivo hepatic clearance was calculated as a fuzzy number.
All computations were programmed in MATLAB. In any simulation, parameters can either be fixed (mean values), represented by probability distributions (mean + S.D.), or represented by fuzzy numbers. For illustrative purposes, in the current study all the parameters were represented by fuzzy numbers.
Method Implementation.
The fuzzy set methodology was applied throughout different stages of an in-house study of in vitro-in vivo prediction of rat hepatic clearance (using rat microsomal and hepatocyte data) for eight benzodiazepine drugs. The drugs used for the study were alprazolam, chlordiazepoxide, clobazam, clonazepam, diazepam, flunitrazepam, midazolam, and triazolam. The fraction of each compound unbound in blood is listed in Table 2.
Mean (S.D.) in vitro intrinsic clearance on normal scale estimated using rat microsomal and hepatocyte drug depletion data together with mean (S.D.) fraction unbound in blood (fUB)
All estimates of in vitro CLint (CLint,in vitro) were obtained from the fit of substrate depletion-time courses by exponential functions. Initially, with only one experiment completed for each of the drugs, the information for CLint,in vitro could only be presented in a linguistic form, such as “chlordiazepoxide is the most slowly cleared of the eight benzodiazepines. The value of CLint,in vitrosignificantly increases across the series. Triazolam is classified as a drug of high CLin vitro”. This linguistic information was then used to predict the hepatic CLin vivo for chlordiazepoxide.
Subsequently, as more CLint,in vitro data (but still of small sample size; n = 5–6) for each of the eight benzodiazepines were collected (Table 2), both MC and fuzzy simulations were performed. Finally, triazolam was selected for further study to yield more measurements of CLint,in vitro(n = 13) for this compound. The FST methodology was applied to this enlarged data base and compared with those made from the smaller sample size study (n = 6).
Results
Preliminary Data.
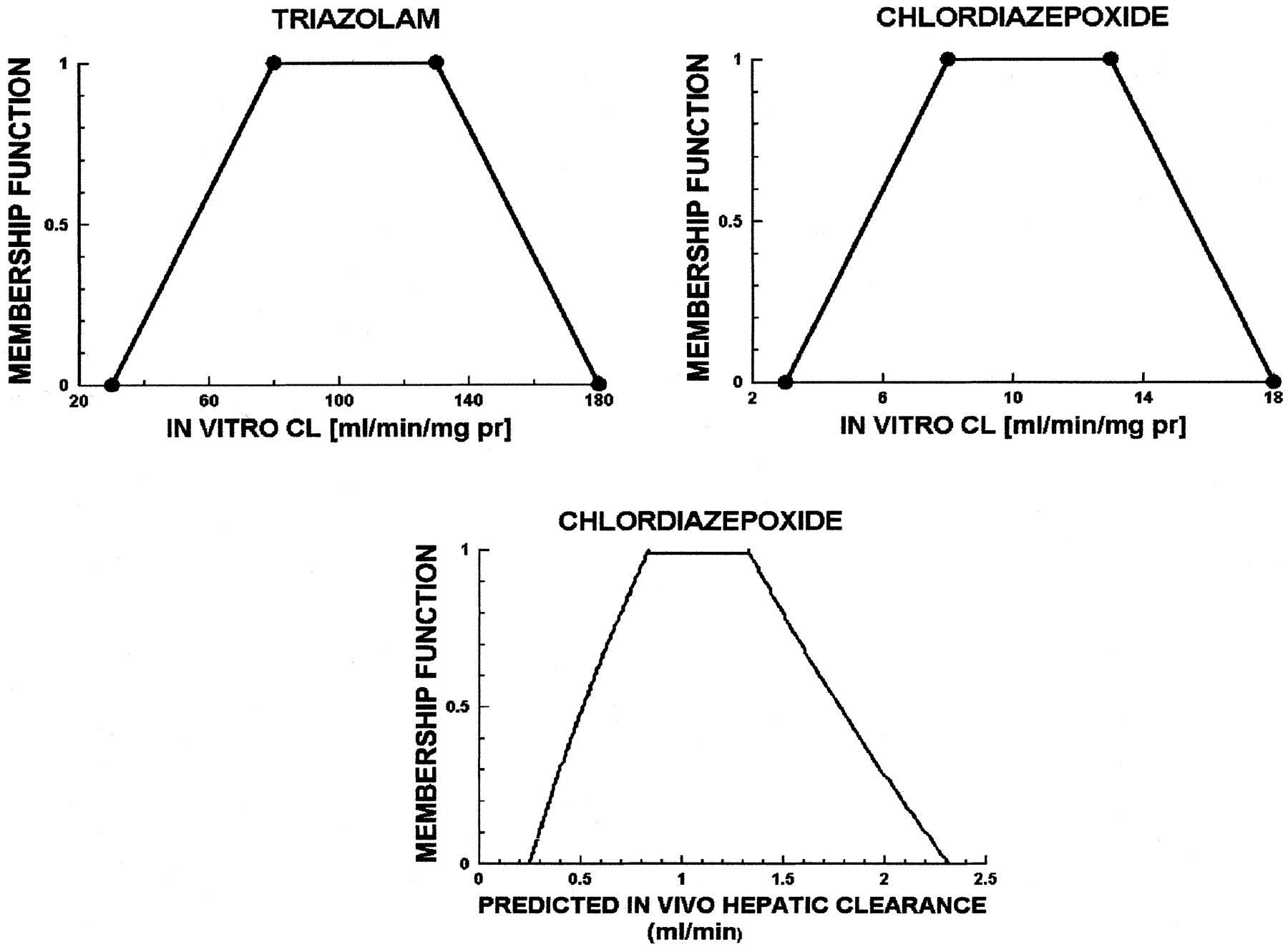
Figure 3 shows the CLint,in vitro fuzzy number for triazolam, a compound with a high CLint,in vitro. This, together with the linguistic grammar (see ), was used to calculate the fuzzy number of CLint,in vitro for chlordiazepoxide and then its predicted CLH,PRED fuzzy number (see Fig. 3, lower panel). It is seen that the membership function for the prediction of hepatic clearance is maximum (equal to 1) between 0.7 and 1.3 ml/min and belongs to the intervals 0.25 to 0.7 and 1.3 to 2.3 ml/min with less certainty, decreasing toward both ends.

Calculated in vivo hepatic clearance (in milliliters per minute) from a linguistic description.
Upper left panel, triazolam in vitro clearance (milliliters per minute per milligram of protein); upper right panel, chlordiazepoxide in vitro clearance (milliliters per minute per milligram of protein); lower panel, fuzzy predicted in vivo hepatic clearance (milliliters per minute) for chlordiazepoxide.
Monte Carlo and Fuzzy Set Predictions.
The MC-generated probability distribution histograms of the predicted in vivo hepatic clearance for several benzodiazepines using microsomal and hepatocyte data are given in Fig. 4. For each compound, the two distributions overlap substantially, although the values using hepatocytes yielded slightly higher clearance values than did those using microsomes. A more detailed discussion of the results from this exercise can be found in (Nestorov et al., 2000).

Monte Carlo-generated distribution histograms of the predicted in vivo hepatic clearance (in milliliters per minute) for diazepam, flunitrazepam, midazolam, and triazolam based on hepatocyte (black) and microsomal (gray) data.
The fuzzy numbers for fUB and CLint, derived from the microsomal and hepatocyte data, together with those for the respective scaling factors, are listed in Table 3. A fuzzy (total) hepatic blood flow was also calculated (Sasaki et al., 1971;Kuwahira et al., 1993) and is given in Table4. As examples, the resulting scaled fuzzy whole-liver CLint for alprazolam, chlordiazepoxide, diazepam, and midazolam are shown in Table5. The predicted fuzzy in vivo hepatic clearances for several of the benzodiazepines are shown in Fig.5. As with the MC case (Fig. 4), although the membership functions of the predicted clearances derived from microsomes and hepatocytes overlap, the range of values predicted from hepatocytes is usually shifted to the right of that for microsomes. In three out of four cases shown, this difference is substantial (e.g., alprazolam, chlordiazepoxide, and clonazepam). Examination of the prediction of hepatic clearance for chlordiazepoxide shows the membership function is maximum (equal to 1) between 0.5 and 0.7 ml/min and belongs to the intervals 0.2 to 0.5 and 0.7 to 2.3 ml/min with less certainty, decreasing toward both ends.
Fuzzy clearance scaling factors in rat
Fuzzy blood flows in standard rat
Fuzzy intrinsic clearance for alprazolam, chlordiazepoxide, diazepam, and midazolam derived from the in vitro measurement
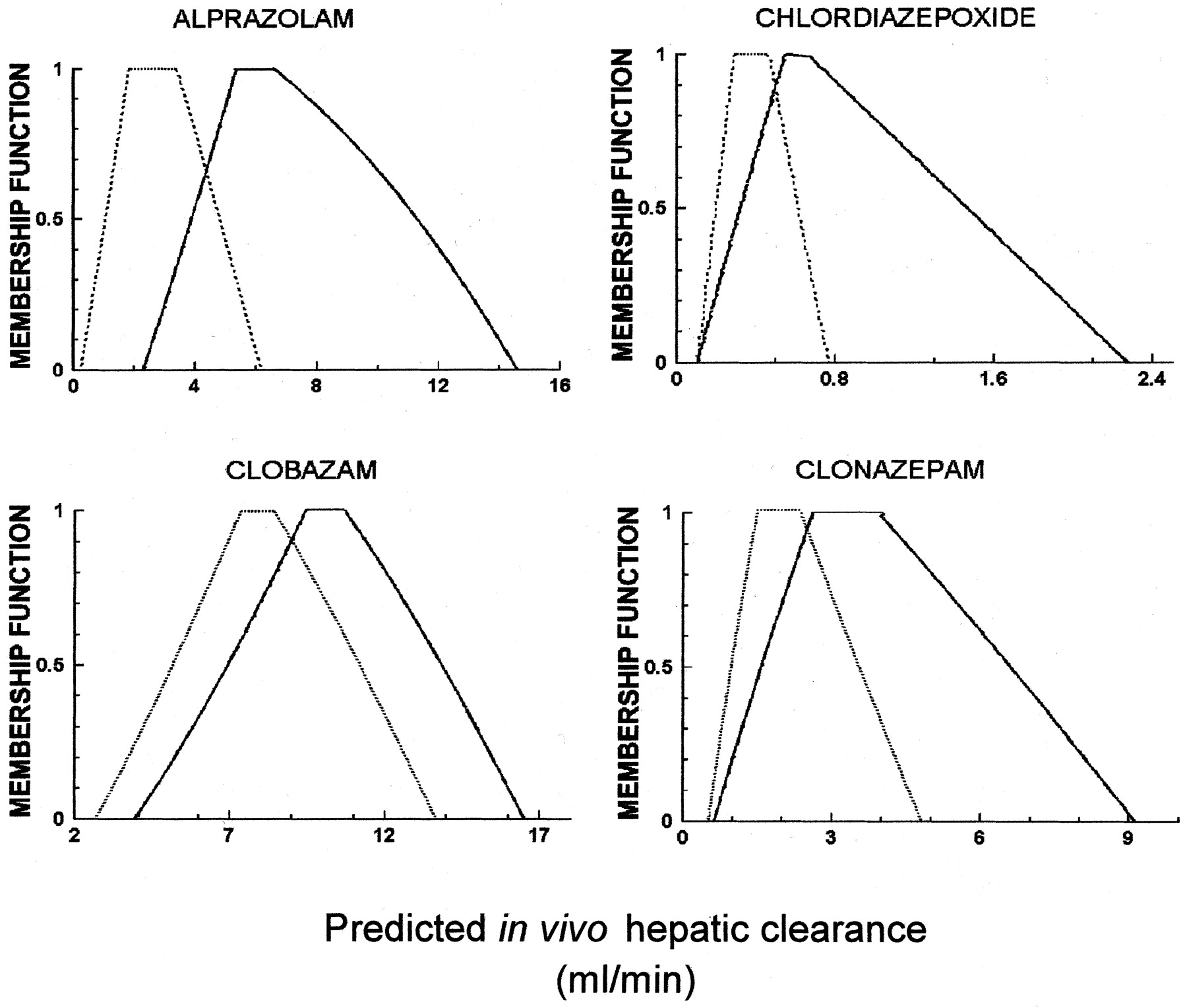
Calculated fuzzy in vivo hepatic clearance (in milliliters per minute) based on hepatocyte (black solid line) and microsomal (black dotted line) data for alprazolam, chlordiazepoxide, clobazam, and clonazepam.
Figure 6 compares the computed statistical distributions and fuzzy numbers of the in vivo hepatic clearance, predicted from hepatocytes, for all eight benzodiazepines. Also shown are the reported literature values of the blood clearances in rat. It is apparent that for all compounds there is a significant overlap between the statistical distribution and the respective fuzzy number. Furthermore, the reported in vivo clearance values are within the ranges of both the predicted fuzzy numbers and the MC-generated histograms but to the right of their centrums. Included in Fig. 6 is the fuzzy number for the predicted CLH for triazolam based on the larger data base (n = 13). As might be expected, the most typical values (membership function 1) cover the reported in vivo clearance range better than when fewer data (n = 6) are available.

Monte Carlo-generated distribution frequency histograms and the fuzzy numbers (continuous line) of the predicted in vivo hepatic clearance (in milliliters per minute) based on in vitro hepatocyte data for all eight benzodiazepines.
Also shown for triazolam is the predicted fuzzy number of the in vivo hepatic clearance (milliliters per minute) based on the larger sample size (black dotted line). Literature values for estimated in vivo blood hepatic clearances are also indicated by the small arrows; reported mean values—clobazam (Hoogerkamp et al., 1996); clonazepam (Hoogerkamp et al., 1996); midazolam (Lau et al., 1998; Aarons et al., 1991); triazolam reported mean and interval of individual clearance values (Gaudreault et al., 1996).
Discussion
The current work was prompted by the view that any approach leading to the incorporation of measures of the existing variability and uncertainty in the model makes better use of the prior information available and provides more informative results than the widely used mean value-based modeling practices. However, defining the theoretical dividing line between uncertainty and variability, as is done in the introduction, is of limited value in practice. Given an experimentally generated data set (e.g., a metabolite formation or a substrate depletion profile), it is practically impossible to separate completely the variability from the uncertainty contained within it. The best one can achieve is to identify the major sources of variability and uncertainty contributing in the particular case and characterize these as much as possible.
In a previous publication (Nestorov et al., 2000), formal incorporation of measures of the existing variability and uncertainty in the in vitro-in vivo prediction of hepatic clearance using MC simulation was suggested. The MC method is based on a reconstruction of the prior probability distributions of the parameters involved and produces the probability distribution of the scaled in vivo clearance, which includes a description of the parameter variability. This approach, although widely used in other pharmacokinetic modeling areas (Farrar et al., 1989;Hattis et al., 1990; Bois et al., 1991;Gearhart et al., 1993; Krewski et al., 1995), has several major drawbacks when applied to in vitro-in vivo clearance predictions. Reconstructing a prior statistical distribution from (as a rule) a small sample can often introduce a significant bias. The most important shortfall of the MC procedure, however, is that it cannot be applied when there is no quantitative data about the processes available, such that reconstruction of the prior distributions is not possible.
Application of FST addresses this identified gap in methodology. FST may best be applied in the following common cases: 1) to replace the current empirical approach to analysis, such as a best-guess estimate, when no quantitative information is available; and 2) when the existing quantitative information is insufficient (small sample size) to reconstruct reliable prior probability distributions of the model parameters needed for the classical MC simulation procedure.
The progression of analyses conducted in the present work follows that likely to arise in numerous situations in research and drug development. Initially, minimal data are collected on various compounds from which, as seen for chlordiazepoxide, predictions (of in vivo hepatic clearance) can still be made using FST. Subsequently, as more but still limited data on each of the compounds becomes available, FST can readily be used as a prediction tool, incorporating the observed uncertainty and variability, when the data are still insufficient to permit MC simulation, unless one wishes to assume a priori an underlying statistical distribution for the uncertainty and variability, which may be inappropriate and bias interpretation. Returning to the specific case of chlordiazepoxide, a comparison of the FST simulations indicates that the most typical values for predicted hepatic clearance (membership function equal to 1) are similar but, as expected, have narrowed in going from the preliminary data to the subsequent larger, albeit still limited, data base, although the entire range of predicted values has changed little. This analysis illustrates the value of applying FST to even preliminary data. Ultimately, however, enough data may be collected on a compound to enable selection of the appropriate statistical distributions to allow meaningful full-blown MC simulations to be made.
Both fuzzy and MC simulations have been applied to predict the rat in vivo hepatic clearances for eight benzodiazepine drugs based on the limited in vitro data to explore the intermediate situation of limited data. The predicted fuzzy hepatic clearances overlap with the respective statistical distributions generated from the MC simulation (Fig. 6). The mean value of the distributions in most cases is within the range of the most typical values (with membership function 1) given by the fuzzy numbers, showing the consistency of the two methods. The advantage of the FST method over the MC simulations is that, as shown, the former can also be applied in cases where no quantitative information is available and when no prior statistical distribution for the variability and uncertainty is assumed.
Both the MC and FST methods predict an overlap between the values of the in vivo clearance, scaled from microsomes and hepatocytes but with a shift of the hepatocyte predictions toward higher values. Although there is a tendency for slight underprediction in both cases, there is an overlap between both the Monte Carlo and the fuzzy predictions and the literature values for in vivo clearances, with values based on the hepatocyte data tending to be closer than microsomal data to observed in vivo observations. Although it is not the purpose of this article to discuss the predictive potential of the in vitro systems to the in vivo case, it should be noted that both the MC and the FST methods bring an improvement over the mean value approach by including a measure of variability and uncertainty in the clearance predictions. This conclusion is supported by reports of comparatively large interindividual variability in clearance for triazolam (Gaudreault et al., 1996).
The statistical distribution generated by the MC procedure is a complete quantitative characteristic of a random variable (in this case hepatic clearance), accompanying each parameter value with a probability measure of its incidence. The fuzzy prediction, resulting from the FST method, not only gives an interval for the likely clearance values but also assigns a measure of the certainty to each value from this interval. As it is an incomplete quantitative characteristic, the fuzzy number contains less information than the statistical distribution of a parameter and cannot form the basis for formal statistical hypothesis testing. Nonetheless, it can be argued that, when limited and vague parameter information is available, incorporating this information into fuzzy parameters and using the FST-based method is more natural and realistic than attempting to reconstruct the respective parameter probability distributions and implementing the full MC procedure. Humans base their thinking primarily on conceptual patterns rather than on numerical quantities (Ross, 1995). The FST method results are also based on concepts, such as “more or less certain” and “typical”. Hence, the FST method provides a natural framework for interpretation (of limited data). For example, the FST method predicts that the most typical values of the in vivo triazolam hepatic clearance (Fig. 6) lie between 9.5 and 12.5 ml/min (membership function equal to 1); the values between 7.5 and 15.5 ml/min are less typical (membership function equal to 0.5), whereas the whole predicted range of possible values is between 3.5 and 17.5 ml/min.
It should be noted that the well stirred liver model is only one of a number of possible liver models that can be used for in vivo clearance prediction (St-Pierre et al., 1992). Other models, such as the parallel tube and dispersion models, can equally be applied within the framework both of the proposed FST approach and the MC procedure without limitation.
With a reasonable dimensionality of the models used, the efforts and resources involved in the implementation of MC or fuzzy simulation are not significant both in terms of time or software. Both methods can easily be executed on most contemporary hardware platforms. Given the importance of the information these approaches generate, the related computational effort is negligible, and their introduction into routine pharmacokinetic and pharmacodynamic prediction practices is to be encouraged.
The most important advantage of the FST-based method is that it can be implemented at a very early stage of the drug discovery/development programs, when predominantly incomplete information is available. The formal integration of information and the implementation of models at such an early stage provide a basis for making more informed decisions. The resulting fuzzy in vivo clearances can also be used subsequently in simulations of pharmacokinetic models.
It would be wrong, however, to consider the MC and the fuzzy simulation techniques as mutually exclusive alternatives. It is our belief that fuzzy simulation should be applied when MC simulation cannot, should not, or need not be implemented. During drug development programs, the accumulation of quantitative information may result in the opportunity to specify reliable prior probability distributions for some of the parameters of interest, replacing the fuzzy set of these parameters. In this respect, the fuzzy simulation may be viewed as a precursor for a full-blown MC simulation, as more data becomes available. It should be realized that the random variables of stochastic processes resulting from the MC simulation are the ultimate description of the model outputs because they represent full quantitative characteristics. However, due to the extensive complexity and dimensionality of the pharmacokinetic and pharmacodynamic processes and the pressures on drug development programs, it is highly likely that even at the very late stages the available information about certain parameters will still be vague, qualitative, or semiquantitative. In such cases, a hybrid simulation scheme combining parameters specified as single values, statistical distributions, and fuzzy numbers will ultimately be needed.
Linguistic Description:
Grammar adopted in fuzzy sets theory terms to describe the following items: high clearance
 (milliliters per minute per milligram of protein) (see the pictorial representation in Fig. 2, upper left panel):
(milliliters per minute per milligram of protein) (see the pictorial representation in Fig. 2, upper left panel): significantly decreased
significantly decreased
 =
=
 /10.
/10.
Footnotes
- Abbreviations used are::
- MC
- Monte Carlo
- probability distribution functions
- FST
- fuzzy set theory
- CLH
- predicted in vivo blood hepatic clearance
- QH
- total hepatic blood flow
- QHA
- hepatic arterial blood flow
- QHP
- blood flow perfusing hepatic portal vein draining the splanchnic organs
- fUB
- fraction unbound in blood
- fU
- fraction unbound in plasma
- R
- blood/plasma drug concentration ratio
- CLint
- intrinsic clearance scaled to the whole liver
- CLint,in vitro
- intrinsic clearance not scaled
- qHA
- fraction of cardiac output perfusing the hepatic artery
- qHP
- fraction of cardiac output perfusing the hepatic portal vein
- Received September 28, 2001.
- Accepted November 30, 2001.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}