


Abstract
This article reports on a symposium sponsored by the American Society for Pharmacology and Experimental Therapeutics and held at the April 2001 Experimental Biology meeting. Current developments in molecular-based studies into the structure and function of cholinesterases, carboxylesterases, and paraoxonases are described. This article covers mechanisms of regulation of gene expression of the various esterases by developmental factors and xenobiotics, as well as the interplay between physiological and chemical regulation of enzyme activity.
This article summarizes four presentations (by P.T., W.F.B., M.H., and B.N.L.D.) at a symposium chaired by T.S. and sponsored by the American Society for Pharmacology and Experimental Therapeutics at the April 2001 Experimental Biology meeting in Orlando, FL. These presentations highlight the importance of structure in delineating overall function, substrate specificity, and localization of the esterase enzymes. Structural considerations emerge from the genes encoding the family of enzymes. Sequence homology typically yields insights into the evolutionary relationships between the members and the conserved and divergent areas of sequence. Diversity in the structure and ultimately function and cellular localization of the gene product is achieved through gene doubling and divergence, alternative mRNA processing, and post-translational modifications.
The cholinesterases and carboxylesterases belong to a protein superfamily termed the α,β-hydrolase-fold family (Cygler et al., 1993) in which members may have highly specialized functions, as is the case for acetylcholinesterase and juvenile hormone esterase. These members show a high degree of selectivity for a neurotransmitter or a hormone, respectively. Other members of the family, such as butyrylcholinesterase or the wide variety of carboxylesterases found in tissues and plasma, show greater promiscuity in substrates for which they catalyze. Hence, they serve a protective and clearing function for foreign substances encountered through the diet or other routes of exposure. This family of enzymes also shows great differences in the cells in which they are expressed; some are found in multiple cell types, whereas others show highly selective expression. Finally, within the cell itself, we observe distinctive localizations; some of the enzymes are destined for export into the plasma, others associate with the cell membrane with its catalytic function directed extracellularly. Others may be retained within subcellular organelles, such as the endoplasmic reticulum, whereas still others are found in the cytoplasm. This article details the signal sequences and splice options that govern cellular localization.
Although we know less about the structures of the paraoxonases, it is clear that they are not serine or cysteine esterases. Rather, they belong to a discrete family of esterases, most likely ones in which a divalent metal is required for catalysis. Considerable progress has been made recently in their purification and structural elucidation. Importantly, inroads also have been made in detecting the natural substrates for the paraoxonases, and as alluded to in this article, lactones emerge as a prime candidate across several species.
Expression profiles of genes encoding esterases are highly regulated during development by nutritional status, hormonal factors, and xenobiotics. Although the consequences of regulation of esterases by drugs and chemicals have been intensively studied, relatively little is known about the mechanisms by which esterases are regulated by physiological factors. This regulation has several potential consequences for pharmacological and toxicological actions of drugs and chemicals in humans and animals in different developmental stages and nutritional states.
α,β-Hydrolase-Fold Proteins: A Unique Superfamily of Proteins with a Diversity of Catalytic and Noncatalytic Functions (P.T.)
Proteins conforming to the structure of the α,β-hydrolase-fold family (Cygler et al., 1993) have emerged as a growing superfamily of proteins with a wide range of functional properties. That cholinesterase was to define a unique family of proteins became evident 16 years ago (Schumacher et al., 1986) when it was noted that the sequence of acetylcholinesterase bore no resemblance to sequences of other known serine hydrolases but rather showed sequence homology to thyroglobulin, a protein whose cDNA was cloned at nearly the same time (Swillens et al., 1986).
The idea that an enzyme designed to hydrolyze rapidly a neurotransmitter would show identity with the protein thyroglobulin, which functions as a substrate for tyrosine iodination and conjugation ultimately leading to thyroxin formation, made little sense at the time. As this protein superfamily grew, three global functions could be ascribed to its members: 1) hydrolytic catalysis of a wide variety of substrates; 2) heterologous cellular recognition as seen for the tactins and between neuroligin and neurexin; and 3) a matrix for post-translational modifications of amino acid residues as found for thyroglobulin. At present, the database for this family contains over 450 orthologous or homologous members with 44 distinctive functions (http://www.ensam.enra.fr/cgi-bin/ace/index). Figure1 shows the major structural similarities of several distinctive proteins of the α,β-hydrolase-fold family. It is also apparent that the family has a variety of means for attaching the catalytic or functional domain to the membrane. Although most activities involve catalysis of hydrolysis or hydrolytically coupled reactions, annotations of ongoing databases are probably not only going to increase the number of members of this superfamily but also identify several new members, perhaps with unique noncatalytic functions.

The α,β-hydrolase-fold family of proteins.
Shown in this listing of representative proteins is the common α,β-hydrolase-fold domain that is responsible for catalysis, recognition in the formation of heterologous cell contacts, and other specialized functions. A common disulfide bond pattern is also found in this region of the molecule. Those proteins with catalytic function contain the members of a catalytic triad: Asp/Glu (D/E), His (H), and Ser (S) in the same sequence order. Typically one or more of these residues is lost in those proteins lacking hydrolase function: thyroglobulin, neuroligin, and the tactins. Several splice options exist for assembling the subunit and tethering the proteins to the membrane or producing a transmembrane span. The region of homology contains variable percentages of sequence identity. The transmembrane span crosses the membrane giving the protein an extracellular and cytoplasmic exposure, whereas the glycophospholipid link tethers the protein to the extracellular membrane face. Splicing, giving variable coding sequences of acetylcholinesterase at its carboxy-terminal region, gives rise to monomeric, homomeric and heteromeric oligomeric forms containing up to 12 subunits. The PEST sequence denotes a proteolytic signal targeting the protein for rapid destruction. Approximate lengths of the nonhomologous sequences are shown in white. The homologous region of the thyroglobulin begins at residue 2229. Modified from Taylor et al. (2000).
Dual functions in a single protein, such as hydrolytic catalysis and forming heterologous cell adhesive contacts for certain family members, remain an attractive but unproven possibility. Moreover, interactions between heterologous-neighboring proteins for members of this family may offer a means of delimiting catalysis to the context of associated proteins within the cell or subcellular organelle.
Since neuroligin is a mammalian protein and possesses the greatest sequence identity with the cholinesterases (Ichtchenko et al., 1995), our initial studies have characterized neuroligin's interactions with its presynaptic partner neurexin. The extracellular portion of neuroligin, a region homologous to other α,β-hydrolase-fold proteins, contains the recognition domain(s) for its Ca2+-dependent interactions with neurexin (Tsigelny et al., 2000). This extracellular domain of the protein can be expressed as a soluble entity, and its interaction with neurexin can be studied by surface plasmon resonance. Similar to the behavior of the developmental protein notch (Molaney et al., 2000), glycosylation processing at certain positions on neuroligin inhibits its associations with neurexin.
Basis for Catalytic Diversity of the α,β-Hydrolase-Fold Proteins.
The α,β-hydrolase-fold proteins show great diversity not only in their substrate recognition but also in catalytic parameters. For example, acetylcholinesterase is one of the most efficient enzymes known with rates of catalysis occurring at or near the diffusion limitation. By contrast, juvenile hormone esterase shows akcat that is 4 orders of magnitude slower but with a higher affinity (lowerKm) for the substrate (Thomas et al., 1999). The juvenile hormone esterases haveKm values ranging between 1 and 102 nM, whereas the Km value for acetylcholinesterase is about 50 μM. Since juvenile hormone esterase substrates contain an extended hydrophobic acyl group and small alcohol moiety (methanol) and acetylcholinesterase requires a small acyl group for efficient catalysis, it is probably the substrate orientations in the active center of two enzymes that differ with respect to the entry portal and the position of the catalytic serine in the active center.
The availability of crystal structures of acetylcholinesterase andCandida lipase has provided invaluable templates for the overall study of the structure of the hydrolase family (Cygler et al., 1993). However, the organization of residues in the crystal may not reveal details on dynamic motion or conformational changes essential for ligand recognition and catalysis. Molecular dynamics calculations allow for theoretical predictions of molecular motion and low-energy conformations, and solution-based methods to investigate protein conformation will provide increasingly important means for examining the dynamics of the structure. Quenching of intrinsic protein fluorescence by nonresonance energy transfer mechanisms and the behavior of fluorescence side chains conjugated with selected cysteines on acetylcholinesterase provide two means for directly examining changes in conformation associated with ligand binding (Boyd et al., 2000; Radic and Taylor, 2001).
In the case of acetylcholinesterase, an ω-loop extending between the disulfide-linked cysteines residues 69 and 96 is in a homologous position with the activation flap on the fungal lipase (Cygler et al., 1993). Interfacial activation through binding of the hydrophobic substrate results in opening of the active center to the substrate for the lipase. Although the smaller loop on acetylcholinesterase does not seem to open and close with substrate activation, steady-state and time-resolved studies with substituted fluorescent (acrylodan) side chains indicate that bound ligands influence gorge dimensions. Segmental motion of this ω-loop may be critical to the catalytic cycle of acylation and deacylation, and the binding of ligands seems to distort the loop structure, as revealed by the fluorescence characteristics of the acrylodan conjugated at particular cysteines in the molecule (Shi et al., 2001).
Finally, sequence diversity in a region carboxy terminus to the catalytic triad in the α,β-hydrolase-fold proteins arises from alternative mRNA splicing. This region governs the cellular disposition of the acetylcholinesterases but does not affect their catalytic properties. The most prevalent splice option encodes for a carboxyl terminus containing a disulfide for linkage to other catalytic subunits or to either a collagen-like or lipid-linked structural subunit. Other splice options give rise to a glycophospholipid-linked enzyme or a soluble monomeric species. Tissue-specific splicing has been examined in several systems.
Identification and Substrate Specificity of Human Carboxylesterases, hCE-1, and hCE-2 (S.P.S. and W.F.B.)
Mammalian liver carboxylesterases belong to a family of proteins encoded by multiple genes. The isoenzymes were initially classified by their substrate specificity and pI. However, this classification is now ambiguous because of overlapping substrate specificities and imprecise because the carboxylesterases as glycoproteins exhibit multiple bands with different pI values upon electrophoresis. Satoh and Hosokawa (1998) recommended an improved classification based on sequence alignments of the encoding genes.
The isoenzymes were classified into four main carboxylesterase (CES1) groups and several subgroups. In general, carboxylesterases exhibit about 80% sequence identity within a CES group. The two major human liver isoenzymes hCE-1 (GI: 119576) and hCE-2 (GI: 4504565) belong to classes CES1 and CES2, respectively. In this article, we summarize the tissue-specific expression and substrate specificity of the hCE-1 and hCE-2 isoenzymes.
Tissue-specific expression of human carboxylesterases hCE-1 and hCE-2 was examined by Northern blots of adult tissues. As shown in Fig.2, a single band of approximately 2.1 kb was seen for hCE-1 (Riddles et al., 1991) and three bands of approximately 2, 3, and 4.2 kb were seen with hCE-2 (Schwer et al., 1997). Both mRNAs are highly expressed in liver. For hCE-1, the intensities of the 2.1-kb band was liver ≫ heart > stomach > testis = kidney = spleen > colon > other tissues (Fig. 2). For hCE-2, the abundance of the 2-kb message in adult tissues was liver > colon > small intestine > heart, and 3-kb message was liver > small intestine > colon > heart (Fig. 2). A 4.2-kb message was detected in brain, testis, and kidney only. hCE-2 expression was essentially absent in other tissues.

Distribution of carboxylesterases in adult human tissues by Northern blot analysis.
A multitissue Northern blot was purchased from Origene Technologies (Rockville, Maryland). Specific cDNA probes were designed for hCE-1 and hCE-2 (Kroetz et al., 1993; Pindel et al., 1997). The sequence homology of the individual probe with the other isoenzyme was <55%. ASapI fragment (425 bp) for hCE-1 and a 340-bpEcoRI-NcoI fragment for hCE-2 were radiolabeled as probes. The multitissue Northern (Origene Technologies) blot was probed for α-actin as a loading control. The tissues corresponding to the numbered lanes are 1, brain; 2, colon; 3, heart; 4, kidney; 5, liver; 6, lung; 7, muscle; 8, placenta; 9, small intestine; 10, spleen; 11, stomach; 12, testis.
It is not known why the relative intensity of the 2- and 3-kb bands for hCE-2 vary in different tissues, but it could be due to alternate splicing, use of different start sites, or expression of different but highly homologous genes. The implications for drug metabolism are that both hCE-1 and hCE-2 are important for systemic clearance of esters from blood through the liver. hCE-1 seems more important for clearance through the kidney, whereas hCE-2 is important for clearance of orally administered drugs through the small intestine and colon.
A rapid spectrophotometric assay examining the hydrolysis of 4-methylumbelliferyl acetate to 4-methylumbelliferone (λmax, 350 nm) was routinely used during purification and characterization of hCE-1 and hCE-2 (Brzezinski et al., 1994; Humerickhouse et al., 1997; Pindel et al., 1997). The catalytic efficiency of 4-methylumbelliferyl acetate is about 1000 times that of the drug esters investigated in this study (Fig.3).
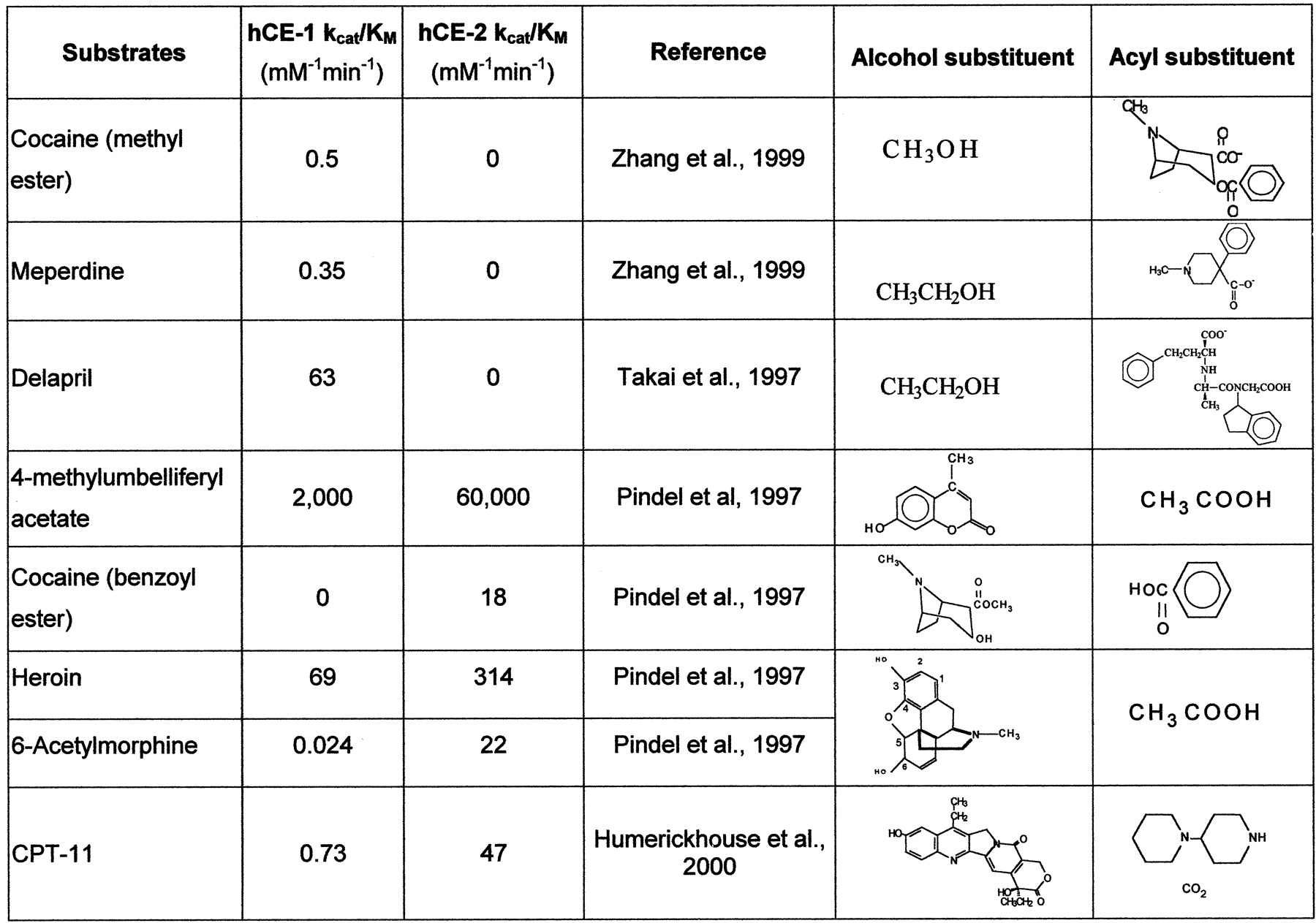
Structure-activity relationships of substrates with hCE-1 and hCE-2.
The catalytic efficiencies of hCE-1 and hCE-2 for several pharmaceuticals were examined (Fig. 3). For drugs of abuse, heroin shows the highest rates of catalysis by both enzymes. hCE-1, but not hCE-2, hydrolyzed the methyl ester of cocaine and the ethyl esters of meperidine and delapril. In contrast to the specificity of hCE-1 for the methyl ester of cocaine, only hCE-2 hydrolyzed the benzoyl ester of cocaine. The benzoyl group of cocaine is also hydrolyzed by serum butyrylcholinesterase (Mattes et al., 1996; Lynch et al., 1997). For the remaining substrates that could be hydrolyzed by both enzymes, hCE-2 exhibited higher catalytic efficiency than hCE-1 for heroin; enzymatic conversion of 6-acetylmorphine to morphine was not known before the isolation and characterization of hCE-2 (Kamendulis et al., 1996). Takai et al. (1997) also reported that hCE-2 (a form with pI 4.5) had higher activity with irinotecan (CPT-11) than hCE-1 (form with pI 5.3).
Analysis of substrate structure versus catalytic efficiency for the ester or carbamate substrates reveals that the two carboxylesterases recognize different structural features of the substrate. The catalytic mechanism for carboxylesterases involves formation of an acyl-enzyme intermediate on an active site serine. If we divide the enzyme active site into “alcohol” and “acyl or acid” recognition subsites, we find the following relationships for hCE-1 and hCE-2 reactively. For those substrates in which hCE-1-catalytic efficiency is greater than hCE-2, such as cocaine (methyl ester), meperidine (Zhang et al., 1999), and delapril (Fig. 3), the alcohol site recognizes the smaller groups (e.g., methanol and ethanol), and the acyl (acid) site recognizes the larger multiring substitutes of these drugs. For those substrates in which hCE-2 catalytic efficiency is greater than hCE-1, such as 4-methylumbelliferyl acetate, cocaine (benzoyl ester), heroin, and 6-acetylmorphine (Fig. 3), the alcohol site recognizes the bulky multiring groups, and the acyl (acid) site recognizes the relatively smaller carboxylate groups. CPT-11 follows a similar relationship in which hCE-2 hydrolyzes the bulky multiring alcohol SN-38 group better than hCE-1 even though the bulky piperidine/bicarbonate products would presumably occupy the acyl (acid) site of hCE-2.
Takai et al. (1997) compared hydrolysis of many drugs catalyzed by hCE-1 (pI 5.3) and hCE-2 (pI 4.5). Several angiotensin-converting enzyme (ACE) inhibitors, such as benazepril, cilazapril, quinapril, temocapril, delapril, and imidapril were studied. All of these drugs are ethyl esters with large acyl group (Fig. 3), and they are better substrates for hCE-1. Delapril and imidapril were hydrolyzed only by hCE-1 and not hCE-2. In contrast, Takai et al. (1997) reported that the local anesthetic drug procaine and the anticholinergic drug oxybutynin with large alcohol substitutes are substrates for hCE-2 but not hCE-1. Enzyme preference is less selective for substrates, such as camostat and dilazep, that have both large alcohol and acyl substituents (Takai et al., 1997).
In general, the alcohol binding site in hCE-1 accepts small groups relative to the acyl (acid) site (Fig. 3). This relationship is also seen with the increasing Ki to inhibitors and for longer chain alkylesters of cocaine for hCE-1 [e.g., benzoylecgonine methyl ester (cocaine) < benzoylecgonine ethyl ester < benzoylecgonine propyl ester (Brzezinski et al., 1997)]. As the size of the group in the alcohol binding site increases, hydrolase activity of hCE can be completely lost (e.g., procaine and oxybutynin) (Takai et al., 1997).
In contrast to hCE-1, hCE-2 can efficiently hydrolyze compounds with large alcohol groups (Fig. 3). However, there is a limit to the size of the group that can be accommodated in the acyl binding site. For example, meperidine and delapril are not substrates for hCE-2. We conclude that although these two human carboxylesterases exhibit broad substrate specificity for ester, carbamate, or amide hydrolysis, they do exhibit distinct catalytic efficiencies that correlate with the relative size of the substrate substituents versus that of the enzyme active sites. Knowledge of these substrate structure-activity relationships and the tissue distribution of carboxylesterase isoenzymes is critical to predicting the metabolism and pharmacokinetics of drug esters in humans.
Molecular Cloning and Expression of Liver, Intestine, and Brain Carboxylesterase Isozymes: The Role in Metabolic Activation of Drugs (M.H. and T.S.)
Mammalian carboxylesterases (EC 3.1.1.1) compose a multigene family, the gene products of which are localized in the endoplasmic reticulum of many tissues (Hosokawa et al., 1987, 1990, 1995, 2001;Maki et al., 1991; Satoh et al., 1994; Yamada et al., 1994; Satoh and Hosokawa, 1995, 1998; Takayama et al., 1998; Mori et al., 1999). These enzymes efficiently catalyze the hydrolysis of a variety of drugs or prodrugs containing ester- and amide-bonds to the respective free acids and alcohol. Since ester derivatives of therapeutic agents have been in use as prodrugs, carboxylesterases are major determinants of the pharmacokinetic behavior of most prodrugs, and the activity can be influenced by direct interactions with a variety of compounds, either directly or by enzyme regulation (Hosokawa et al., 1993, 1994,1998).
The mammalian carboxylesterases are localized in the endoplasmic reticulum of many tissues. Among various tissues of animals, the highest substrate hydrolase activity is typically found in the liver (Hosokawa et al., 2001), but activities can be found in several other tissues, such as testis, kidney, and plasma (Morgan et al., 1994). Since a significant number of drugs are metabolized by carboxylesterases, altering activities of these enzymes expressed in each tissue has important clinical implications. However, little is known about the differences in structure and hydrolytic capabilities of carboxylesterase isozymes expressed in tissues of humans. Structural characterizations of carboxylesterase isozymes, their expression in various tissues, and their substrate specificities provide important insights into the molecular basis for their functional differences in humans.
Recombinant DNA and expression techniques have been introduced to the field of carboxylesterases as a means to clarify the substrate specificity of each isozyme, elucidate the catalytic mechanism of carboxylesterases, and delineate the role of carboxylesterases in the endoplasmic reticulum. Several proteins of endoplasmic reticulum lumen have the common carboxy-terminal recognition sequence KDEL-COOH, a structural motif essential for retention of the protein in the luminal site of the endoplasmic reticulum (Satoh and Hosokawa, 1998). Proteins with this carboxy-terminal sequence bind the KDEL receptor and associate with the endoplasmic membrane. Our group showed that the endoplasmic reticulum retention motif of carboxylesterase is HXEL-COOH (Satoh and Hosokawa, 1995). The recognition sequence dictates that proteins are primarily found on the luminal side of the endoplasmic reticulum. The three amino acid residues of the catalytic triad (Ser, Glu, and His) and four cysteines that may be involved in specific disulfide bonds are similarly positioned in each carboxylesterase, and some N-linked glycosylation sites are also conserved. It is interesting that human liver and brain carboxylesterase (HU1 and hBr1) have a free cysteine residue that is not involved in a disulfide bond. Recently, Satoh and Hosokawa (1998) proposed a novel classification and nomenclature of carboxylesterase isozymes. Considering the high-residue identity and similarity of the characteristics, they proposed that carboxylesterase isozymes be classified into four families, CES1, CES2, CES3, and CES4, a classification based on phylogenetic analysis. According to the classification, human liver HU1, and human brain hBr1, hBr2, and hBr3 belong to the CES1 family. Human liver HU3 and intestine HSICE are classified into the CES2 family.
Ester and amide derivatives of therapeutically active agents have been in use as prodrugs for many years to improve solubility, absorption, and bioavailability, and to extend duration of action of the drug, camptothecin derivatives, such as topotecan and CPT-11, are used as antineoplastic agents and possess a novel mechanism of action involving inhibition of DNA topoisomerase I. CPT-11, a carbamate derivative of 10-hydroxy-7-ethylcamptothecin, can be classified as a prodrug because we and others found conversion of CPT-11 to its active metabolite 10-hydroxy derivatives by liver carboxylesterase in various mammals and humans (Satoh and Hosokawa, 1993; Satoh et al., 1994; Takayama et al., 1998). The Km value of human intestine HSICE is lower than liver HU1, and theVmax value of HSICE is higher than HU1. Therefore, human intestine carboxylesterase has a much higher capability of CPT-11 hydrolase activity than human liver carboxylesterase. Temocapril is a prodrug type of the ACE inhibitor that is rapidly hydrolyzed by carboxylesterase at its 2′-ethyl ester group and converted into the pharmacologically active diacid metabolite temocaprilat, a potent inhibitor of ACE. The comparison of the temocapril hydrolase activity in human liver and brain carboxylesterase was expressed in V79 cells. Human brain carboxylesterase hBr2 was found to have the highest specific activity toward temocapril. However, human intestinal carboxylesterase poorly catalyzes the hydrolysis of temocapril. These results suggest that the therapeutic prodrug was metabolically activated by human carboxylesterases isozymes, although the capability of metabolic activation was different for each isozyme.
In conclusion, recent studies demonstrate that liver, small intestine, and brain carboxylesterase isozymes in humans are intimately involved in metabolic activation of therapeutic prodrugs. The multiplicity of carboxylesterase structures enable this family of enzymes to hydrolyze a diverse range of esters.
Evolution and Enzymatic Properties of Mammalian Paraoxonases (B.N.L.D.)
Augustinsson (1968) proposed that arylesterases arose as an offshoot from the carboxylesterase but differ in having cysteine rather than serine as the key component of their active centers. Although this hypothesis remained unchanged for many years, we now know that the mammalian paraoxonase/arylesterase (PON1) requires calcium and does not need a free cysteine residue for paraoxonase activity. Three such esterases (PON1, PON2, and PON3) have been named in the order of their discovery. The corresponding three genes are closely aligned on chromosome 7 in humans and on chromosome 6 in mice. Since there is no structural similarity in the amino acid sequences of the PONs with sequences of the cholinesterases or the carboxylesterases, it is now quite evident that they are not related, and ancestral relatives of the PONs must be found elsewhere.
A recent, very relevant report showed that a lactonase of the fungusFusarium oxysporium (Kobayashi et al., 1999) has appreciable structural homology with human serum PON1. Furthermore, these two enzymes share a number of substrates, such as dihydrocoumarin and homogentisic acid lactone. Interestingly, the fungal lactonase does not catalyze the hydrolysis of paraoxon.
Our laboratory has recently characterized the lactonase activity of purified human PON1 (Billecke et al., 2000) and found it to hydrolyze a large number of both aromatic and aliphatic lactones. Its substrates also include a number of drugs, such as several statin lactones (lovastatin and simvastatin) and the diuretic agent spironolactone. The rates of hydrolysis are different for some of these substrates with the two purified human PON1 Q and R isozymes and show stereospecificity for some of the lactone enantiomers. Since lactones are commonly found in plants, natural flavoring agents, and many food products, we may find that PON1 lactonase activity represents the common theme of this family of enzymes. Protection against dietary and environmental lactones could be the selective forces that have been responsible for maintaining the balanced polymorphisms in the PON enzymes in mammals.
Support for the key role of the lactonase activity comes from the recent findings in our laboratory that neither human PON2 nor PON3 have any paraoxonase activity. They have very little if any arylesterase activity, but they share the lactonase activity of PON1 (D. Draganov, unpublished results). Rabbit serum contains PON3 in its HDL fraction, and it has a very high statinase activity (Draganov et al., 2000). A recent report identifies human serum PON3 as a minor component of the HDL fraction, but its enzymatic activity still must be examined (Reddy et al., 2001).
On the basis of the structural homology and predicted evolutionary distance between them, it seems that PON2 is the oldest member of the family, PON3 arose from it, and finally PON1 appears. All three PONs have nine similar exons and a high degree of identity in their structural characteristics, such as number and location of the cysteine residues. Based on their respective cDNA structures and the deduced amino acid sequences, there is over 80% identity in amino acid residues in human, mouse, and rabbit PON1s and at least 60% identity between the PONs 1,2, and 3 of each of these species (Primo-Parmo et al., 1996). Polymorphic variants are common in at least the human and rabbit PONs. A reasonable conclusion from these observations is that this family of enzymes probably has some important physiological role, which is protected by the redundancy and polymorphic forms of the enzymes produced.
An important clue to a physiological function is provided by the studies of mice lacking PON1 that were produced by Shih et al. (1998). The knockout mice developed atherosclerosis when fed an atherogenic diet, and their HDL fraction lacking PON1 was not protective against oxidative damage produced by low-density lipoprotein, as was found for the HDL fraction from wild-type mice. Purified PON1 showed the expected protection against oxidative damage from low-density lipoprotein, and purified rabbit serum PON3 was even more protective than rabbit purified PON1 (Draganov et al., 2000). Although lactonase activity with model compounds and protection against oxidative damage seem to be parallel activities, it is not clear how directly they are related. Perhaps protection involves the hydrolysis of some potentially toxic endogenous lactone that would produce oxidative damage if not hydrolyzed. Such questions will be answered by future research. A useful recent review on the relationship between PON1 and atherosclerosis has been published by Durrington et al. (2001).
Footnotes
- Abbreviations used are::
- CES
- carboxylesterase
- kb
- kilobase
- CPT-11
- irinotecan
- SN-38
- 7-ethyl-10-hydroxycamptothecin
- ACE
- angiotensin-converting enzyme
- PON
- paraoxonase
- HDL
- high-density lipoprotein
- Received November 28, 2001.
- Accepted January 22, 2002.
- The American Society for Pharmacology and Experimental Therapeutics
References




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- α,β-Hydrolase-Fold Proteins: A Unique Superfamily of Proteins with a Diversity of Catalytic and Noncatalytic Functions (P.T.)
- Identification and Substrate Specificity of Human Carboxylesterases, hCE-1, and hCE-2 (S.P.S. and W.F.B.)
- Molecular Cloning and Expression of Liver, Intestine, and Brain Carboxylesterase Isozymes: The Role in Metabolic Activation of Drugs (M.H. and T.S.)
- Evolution and Enzymatic Properties of Mammalian Paraoxonases (B.N.L.D.)
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters