


Abstract
The Michaelis constant (KM) for cytochrome P450-mediated drug biotransformation reactions can be an important parameter in understanding the potential for a drug to exhibit saturable metabolism in vivo and nonlinear dose-exposure relationships. KM values were measured for several drug biotransformation reactions using recombinant heterologously expressed human enzymes. These determinations were made using an approach of monitoring substrate loss (“in vitrot1/2” method) at multiple substrate concentrations, with the objective of comparingKM values determined by this approach withKM values determined using the conventional approach of measuring product formation rates at several substrate concentrations. The reactions examined were CYP2C9-catalyzed diclofenac 4′-hydroxylation, CYP2D6-catalyzed dextromethorphanO-demethylation and thioridazineS-oxidation, CYP2C19-catalyzed imipramineN-demethylation, CYP3A4-catalyzed midazolam 1′-hydroxylation, and CYP1A2-catalyzed tacrine 1-hydroxylation.KM values spanned an 80-fold range from 0.12 μM (CYP2D6-catalyzed thioridazine S-oxidation) to 9.8 μM (CYP2C19-catalyzed imipramine N-demethylation). On average, KM values determined by the substrate depletion approach were within 1.54-fold of those determined by measuring product formation. Thus, KMvalues can be determined for drug metabolism reactions without requiring knowledge of metabolite structures or requiring authentic standards of metabolites for use in construction of standard curves for quantitative bioanalysis. The in vitro t1/2approach of determining KM values should be useful in early drug discovery efforts to identify those compounds with low KM values and, hence, a greater probability of exhibiting supraproportional dose-exposure relationships.
The determination of enzyme kinetic parameters,KM,Vmax, and CL
The KM value (Michaelis constant) is a primary descriptor of the enzyme kinetic behavior of a biotransformation reaction. In simple kinetic systems, it represents the substrate concentration that will yield a reaction velocity that is half of the theoretical maximum reaction velocity that would occur at an infinite substrate concentration (Vmax). In a practical sense, theKM value can be reflective of the potential of a drug to saturate, or partially saturate, an enzyme involved in its metabolic clearance. The lower theKM value, the greater the possibility that the drug concentrations attained in vivo will saturate the metabolic clearance pathway. When this phenomenon occurs, it can be problematic because the drug will exhibit a nonlinear, supraproportional dose-exposure relationship (Ludden, 1991). For drugs with low-therapeutic indices, small increases in dose can potentially introduce toxicity as drug concentrations in vivo will increase to a much greater extent than that expected from the increase in dose. Such pharmacokinetic behavior has been observed for numerous compounds spanning several therapeutic indications, some of which are listed in Table 1. TheKM value will not solely be responsible for supraproportional dose-exposure relationships, as other factors such as dose, absorption rate (for orally administered compounds), extent of binding to blood components, and rate and extent of uptake into clearing organs (such as liver) will also contribute to the potential for this phenomenon. However, while not a sole determinant, a low KM value is frequently associated with this pharmacokinetic behavior.
Examples of drugs that demonstrate supraproportional dose-exposure relationships in humans proposed to be due to saturation of metabolism
Conventional determinations of KMvalues are made by assessing the rate of product (metabolite) formation at several substrate concentrations. To accomplish this, methods are required for measurement of metabolite concentrations in in vitro matrices. Such analytical methods themselves require that metabolites have been definitively identified and authentic standards prepared. Alternately, one can use radiolabeled materials for such analysis. While these materials and methods are feasibly obtained during drug development programs, they are not readily obtainable for each of the thousands of compounds typically studied in early drug discovery programs. In drug discovery, overall intrinsic clearance estimates can be obtained by monitoring substrate loss versus time using the in vitrot1/2 approach (Obach et al., 1997; Obach, 1999). This approach involves measurement of substrate consumption at a single low concentration (<KM). This simple experimental design will yield an estimate of intrinsic clearance but notKM.
In this report, we describe experiments conducted with the objective of assessing whether accurate determinations ofKM values can be made with the in vitro t1/2 approach carried out at multiple substrate concentrations. Such an approach would be useful in drug discovery programs in efforts to determine whether a compound would have the potential to exhibit nonlinear, supraproportional dose-exposure relationships. Well characterized drug metabolism reactions catalyzed by several different recombinant heterologously expressed human cytochrome P450 enzymes were used in this examination (Fig. 1).

Drug metabolism reactions examined in this study.
Experimental Procedures
Materials.
The following substrates, metabolites, and internal standards were obtained from Sigma-Aldrich (St. Louis, MO): tacrine, 1-hydroxytacrine, diclofenac, ibuprofen, ketoprofen, imipramine, chlorpromazine, desipramine, amitriptyline, thioridazine, clozapine, alprazolam, and NADPH. Sources of other reagents were as follows: dextromethorphan and dextrorphan (Sigma/RBI, Natick, MA), mesoridazine (BIOMOL Research Laboratories, Plymouth Meeting, PA), midazolam, 1′- and 4-hydroxymidazolam (Gentest Corp., Woburn, MA). 4′-Hydroxydiclofenac, deuterated 1-hydroxytacrine, 2-hydroxytacrine, 4-hydroxytacrine, levallorphan, and 8-chlorotacrine were from the Pfizer in-house chemical sample bank. Recombinant heterologously expressed cytochrome P450 enzymes were generated in-house using a baculovirus/Sf9 cell expression system (Christopherson et al., 1995).
In Vitro Incubations.
All incubations were conducted by shaking reaction mixtures under air in a heated water bath at 37°C. Incubations were carried out in 100 mM KH2PO4, pH 7.4, containing 3.3 mM MgCl2. Substrates, buffer, and microsomes were premixed, and incubations were commenced by addition of NADPH (1.3 mM). In substrate saturation experiments in which product formation was measured, incubation times were set that had provided a linear reaction velocity in preliminary experiments. In substrate consumption experiments, this was intentionally not the case.
CYP1A2-Catalyzed Tacrine Metabolism.
In product formation experiments, tacrine 1-hydroxylase was measured. Tacrine (0.1–100 μM) was incubated with rhCYP1A2 microsomes (0.1 mg/ml; 0.038 nmol of P4501/mg) in a total volume of 0.2 ml. The incubation time was 10 min at which time the reactions were terminated by addition of 20 μl of CH3CN containing 10 ng of [2H3]1-hydroxytacrine and placed on ice. The terminated reaction mixtures were filtered through a Millipore MultiScreen-HA 0.45-μm-mixed cellulose ester 96-well membrane vacuum filtration module (Millipore Corporation, Bedford, MA). The filtrate was analyzed by HPLC-MS without further processing.
In substrate depletion experiments, tacrine (0.30–100 μM) was incubated with rhCYP1A2 microsomes (0.92 mg/ml) in a total incubation volume of 1.4 ml. Aliquots (0.2 ml) were removed at 0, 2, 5, 10, 20, and 30 min and terminated by addition to 20 μl of CH3CN on ice containing 20 ng of 8-chlorotacrine as internal standard. Terminated reaction mixtures were filtered as above and filtrates analyzed directly by HPLC-MS.
CYP2C9-Catalyzed Diclofenac Metabolism.
In product formation experiments, diclofenac 4′-hydroxylase was measured. Diclofenac (0.5–50 μM) was incubated with rhCYP2C9 microsomes (0.075 mg/ml; 0.35 nmol of P450/mg) in a total volume of 0.2 ml. The incubation time was 10 min at which time the reactions were terminated by addition of 20 μl of CH3OH containing 400 ng of ketoprofen and placed on ice. The terminated reaction mixtures were filtered through a Millipore 96-well membrane vacuum filtration module (Millipore Corporation). The filtrate was analyzed by HPLC-MS without further processing.
In substrate depletion experiments, diclofenac (0.10 to 30 μM) was incubated with rhCYP2C9 microsomes (0.2 mg/ml) in a total incubation volume of 1.5 ml. Aliquots (0.2 ml) were removed at 0, 2, 5, 10, 20, and 30 min and terminated by addition to 20 μl of CH3OH on ice containing 300 ng of ibuprofen as internal standard. Terminated reaction mixtures were filtered as above and filtrates analyzed directly by HPLC-MS.
CYP2C19-Catalyzed Imipramine Metabolism.
In product formation experiments, imipramine N-demethylase activity was measured. Imipramine (1.0–100 μM) was incubated with rhCYP2C19 microsomes (0.2 mg/ml; 0.16 nmol of P450/mg) in a total incubation volume of 0.2 ml, under conditions as described above, for 5 min. Reactions were terminated by the addition of 25 μl of CH3OH containing 100 ng of amitriptyline as an internal standard. Samples were alkalinized (0.1 ml of 0.1 M NaOH), followed by extraction with MTBE (3 ml). The organic fraction was evaporated under N2; the residue was reconstituted in 0.2 ml of mobile phase for analysis of desipramine by HPLC.
In substrate depletion experiments, imipramine (0.60–200 μM) was incubated with rhCYP2C19 microsomes (0.4 mg/ml) in a total volume of 0.4 ml. At times of zero, 2, 5, 10, 20, and 30 min postcommencement of the incubation, 50 μl of aliquots were removed and added to 0.1 ml of NaOH (0.1 M). Internal standard (100 ng of amitriptyline in 20 μl of CH3OH) was added and the samples were extracted with MTBE (3 ml). The organic fraction was evaporated under N2, and the residue was reconstituted in 1 ml of mobile phase for HPLC analysis of imipramine.
CYP2D6-Catalyzed Dextromethorphan Metabolism.
In product formation experiments, dextromethorphanO-demethylase activity was measured. Dextromethorphan (0.1–10 μM) was incubated with rhCYP2D6 microsomes (0.01 mg/ml; 0.19 nmol of P450/mg) in a total incubation volume of 0.2 ml, under conditions as described above, for 5 min. Reactions were terminated by the addition of 0.2 ml of NaHCO3 (1 M, pH 9.5), followed by the addition of internal standard (20 ng of levallorphan in 20 μl of CH3OH). Samples were extracted with CH2Cl2 (3 ml), the aqueous fraction was aspirated off, the organic fraction was evaporated under N2 at 30°C, and the residue was reconstituted in 0.2 ml of mobile phase for analysis of dextrorphan by HPLC.
In substrate depletion experiments, dextromethorphan (0.015 to 5 μM) was incubated with rhCYP2D6 microsomes (0.02 mg/ml) in a total volume of 1.5 ml. At times of zero, 2, 5, 10, 20, and 30 min postcommencement of the incubation, 0.2 ml of aliquots were removed and added to 0.2 ml of NaHCO3 (1 M, pH 9.5) to terminate the reaction. Internal standard (20 ng of levallorphan in 20 μl of CH3OH) was added and the samples were extracted with CH2Cl2 and processed as above. The residue was reconstituted in 1 ml of mobile phase for HPLC analysis of dextromethorphan.
CYP2D6-Catalyzed Thioridazine Metabolism.
In product formation experiments, thioridazine S-oxidase activity was measured. Thioridazine (0.001 to 40 μM) was incubated with rhCYP2D6 microsomes (0.004 mg/ml; 0.19 nmol of P450/mg) in a total incubation volume of 0.2 ml, under conditions as described above, for 10 min. Reactions were terminated by the addition of 0.1 ml of NaOH (0.1 M), followed by the addition of internal standard (5 ng clozapine in 20 μl CH3OH). Samples were extracted with MTBE (3 ml), the organic fraction was evaporated under N2 at 35°C, and the residue was reconstituted in 0.03 ml of mobile phase for analysis of mesoridazine by HPLC.
In substrate depletion experiments, thioridazine (0.0032–1.0 μM) was incubated with rhCYP2D6 microsomes (0.004 mg/ml) in a total volume of 1.5 ml. At times of zero, 2, 5, 10, 20, and 30 min postcommencement of the incubation, 0.2 ml of aliquots were removed and added to 0.1 ml of NaOH (0.1 M) to terminate the reaction. Internal standard (20 ng of imipramine in 20 μl of CH3OH) was added, the samples were extracted with MTBE (3 ml) and processed as above. The residue was reconstituted in 0.1 ml of mobile phase for HPLC analysis of thioridazine.
CYP3A4-Catalyzed Midazolam Metabolism.
In product formation experiments, midazolam 1′- and 4-hydroxylase activity was measured. Midazolam (0.2–50 μM) was incubated with rhCYP3A4 microsomes (0.025 mg/ml; 48 nmol of P450/mg) in a total incubation volume of 0.2 ml, under conditions as described above, for 2 min. Reactions were terminated by the addition of 0.1 ml of NaOH (0.1 M), followed by the addition of internal standard (5 ng of alprazolam in 20 μl of CH3OH). Samples were extracted with MTBE (3 ml), the organic fraction was retained and evaporated under N2 at 30°C, and the residue was reconstituted in 0.1 ml of mobile phase for analysis of 1′- and 4-hydroxymidazolam by HPLC.
In substrate depletion experiments, midazolam (0.03–10 μM) was incubated with rhCYP3A4 microsomes (0.1 mg/ml) in a total volume of 1.5 ml. At times of zero, 2, 5, 10, 20, and 30 min postcommencement of the incubation, 0.2 ml of aliquots were removed and added to 0.1 ml of NaOH (0.1 M) to terminate the reaction. Internal standard (5 ng of alprazolam in 20 μl of CH3OH) was added, the samples were extracted with MTBE (3 ml) and processed as above. The residue was reconstituted in 0.1 ml of mobile phase for HPLC analysis of midazolam.
Analytical Instrumentation and General Aspects of Bioanalysis.
Analyses of substrates and metabolites were done by HPLC-MS. The system consisted of a Sciex API100 single quadrupole mass spectrometer with Turbo Ionspray source (Applied Biosystems/Sciex, Thornhill, Ontario, Canada). The flow was split 1:1 prior to entry into the source of the mass spectrometer. The HPLC consisted of an Agilent model 1100 quaternary gradient pump with membrane degasser (Agilent Technologies Inc., Palo Alto, CA). Injection was accomplished with a CTC PAL autosampler (LEAP Technologies, Carrboro, NC). HPLC columns used were a Phenomenex Luna C18 (2.1 × 50 mm; 3-μm particle size; Phenomenex, Torrance CA) or Polaris C18 (4.6 × 250 mm; 5 μm particle size; MetaChem, Torrance CA). Mobile phase compositions were mobile phase A, 95% 20 mM acetic acid (pH adjusted to 4.0 with NH4OH)/5% CH3CN; mobile phase B, 95% CH3CN/5% 20 mM acetic acid (pH adjusted to 4.0 with NH4OH); mobile phase C, 10 mM ammonium acetate containing 1% isopropanol; and mobile phase D, CH3CN.
Analysis of Desipramine.
Desipramine was quantitated from a standard curve ranging from 0.15 to 15 μM. Reconstituted extracts were injected (10 μl) onto a Phenomenex Luna C18 column equilibrated in a mobile phase of 70% A/30% B and maintained at an isocratic flow rate of 0.5 ml/min. The HPLC effluent was introduced into the mass spectrometer operated in the positive ion mode monitoringm/z 267.3 (desipramine) andm/z 278.0 (amitriptyline). The orifice voltage was 35 V, and the ionspray temperature was 450°C. Desipramine and amitriptyline internal standard eluted at 1.1 and 1.5 min, respectively.
Analysis of Dextromethorphan.
Reconstituted extracts were injected (10 μl) onto a Phenomenex Luna C18 column equilibrated in a mobile phase of 79% A/21% B and maintained at an isocratic flow rate of 0.5 ml/min. The HPLC effluent was introduced into the mass spectrometer in the positive ion mode, with the orifice voltage at 40 V and the ionspray temperature at 450°C. Ions monitored were m/z 272 (dextromethorphan), m/z 274 (natural abundance [13C2]dextromethorphan for high-substrate concentration samples), andm/z 284 (levallorphan internal standard). Retention times were 2.4 and 1.1 min for dextromethorphan and levallorphan, respectively.
Analysis of Dextrorphan.
Dextrorphan was quantitated from a standard curve ranging from 0.01 to 1.0 μM. Reconstituted extracts (10 μl) were injected onto a Phenomenex Luna C18 column equilibrated in 87% A/13% B at an isocratic flow rate of 0.5 ml/min. The effluent was introduced into the mass spectrometer operated at an orifice voltage of 40 V and a source temperature of 450°C. Protonated molecular ions ofm/z 258 (dextrorphan) andm/z 284 (levallorphan internal standard) were monitored. The retention times for dextrorphan and levallorphan were 1.5 and 2.1 min, respectively.
Analysis of Diclofenac.
Filtered incubation mixtures were injected (20 μl) onto a Phenomenex Luna C18 column equilibrated in mobile phase of composition 73% C/27% D at a flow rate of 0.5 ml/min. The mass spectrometer was operated in the negative ion mode at an orifice voltage of −25 V and an ionspray temperature of 400°C. Ions monitored were m/z 297.2 (diclofenac) andm/z 205.0 (ibuprofen internal standard). Retention times for diclofenac and ibuprofen were 1.6 and 1.3 min, respectively.
Analysis of 4′-Hydroxydiclofenac.
Filtered incubation mixtures were injected (20 μl) onto a Phenomenex Luna C18 column equilibrated in mobile phase of composition 78% C/22% D at a flow rate of 0.5 ml/min. The mass spectrometer was operated in the negative ion mode at an orifice voltage of −25 V and an ionspray temperature of 400°C. Ions monitored were m/z 310.2 (4′-hydroxydiclofenac),m/z 253.2 (ketoprofen). Retention times for 4′-hydroxydiclofenac and ketoprofen were 2.1 and 1.4 min, respectively. The dynamic range of the assay was 0.045 to 15 μM.
Analysis of 1′- and 4-Hydroxymidazolam.
1′- and 4-hydroxymidazolam were quantitated from a standard curve ranging from 0.003 to 1.0 μM. Reconstituted extracts (10 μl) were injected onto a Phenomenex Luna C18 column equilibrated in 70% A/30% B at an isocratic flow rate of 0.5 ml/min. The effluent was introduced into the mass spectrometer operated at an orifice voltage of 35 V and a source temperature of 450°C. Protonated molecular ions of m/z 342.4 (hydroxymidazolam isomers) and m/z 309.4 (alprazolam internal standard) were monitored. The retention times for 1′-hydroxymidazolam, 4-hydroxymidazolam, and alprazolam were 1.1, 2.0, and 2.2 min, respectively.
Analysis of 1-Hydroxytacrine.
Filtered incubation mixtures were injected (20 μl) onto a MetaChem Porasil C18 column equilibrated in mobile phase of composition 92% A/7% B at a flow rate of 0.8 ml/min. The mass spectrometer was operated in the positive ion mode at an orifice voltage of 25 V and an ionspray temperature of 500°C. Ions monitored were m/z 215.3 (hydroxytacrine isomers) andm/z 218.3 ([2H3]1-hydroxytacrine internal standard). Retention times for 1-, 2-, and 4-hydroxytacrine regioisomers were 11.5, 10.4, and 12.7 min, respectively. Quantitation was done from a standard curve with a dynamic range of 5.0 to 500 ng/ml.
Analysis of Imipramine.
Reconstituted extracts were injected (10 μl) onto a Phenomenex Luna C18 column equilibrated in a mobile phase of 70% A/30% B and maintained at an isocratic flow rate of 0.5 ml/min. The HPLC effluent was introduced into the mass spectrometer in the positive ion mode, with the orifice voltage at 35 V and the ionspray temperature at 450°C. Ions monitored were m/z 281.5 (imipramine), m/z 283.5 (natural abundance [13C2]imipramine for high-substrate concentration samples), and m/z278.5 (amitriptyline internal standard). Retention times were 1.2 and 1.4 min for imipramine and amitriptyline, respectively.
Analysis of Mesoridazine.
Reconstituted extracts (20 μl) were injected onto a Phenomenex Luna C18 column equilibrated in 75% A/25% B at a flow rate of 0.5 ml/min operated isocratically. The effluent was introduced into the TurboIon spray source operated with an orifice voltage of 25 V and a temperature of 400°C. Protonated molecular ions of mesoridazine (m/z 387 and 389;Rt = 1.3 min) and clozapine internal standard (m/z 326;Rt = 2.0 min) were monitored. Quantitation was done using a standard curve range of 0.05 to 500 ng/ml, in which the lower end of the curve used the signal atm/z 387, and the high end of the curve used the signal at m/z 389.
Analysis of Midazolam.
Reconstituted extracts were injected (10 μl) onto a Phenomenex Luna C18 column equilibrated in a mobile phase of 70% A/30% B and maintained at an isocratic flow rate of 0.5 ml/min. The HPLC effluent was introduced into the mass spectrometer in the positive ion mode, with the orifice voltage at 35 V and the ionspray temperature at 450°C. Ions monitored were m/z 326.4 (midazolam), m/z 330.4 (natural abundance [13C2/37Cl]midazolam for high-substrate concentration samples), andm/z 309.4 (alprazolam internal standard). Retention times were 1.5 and 2.2 min for midazolam and alprazolam, respectively.
Analysis of Tacrine.
Incubation mixture filtrates were injected (10 μl) onto a Phenomenex Luna C18 column equilibrated in a mobile phase of 91% A/9% B and maintained at an isocratic flow rate of 0.5 ml/min. The HPLC effluent was introduced into the mass spectrometer in the positive ion mode, with the orifice voltage at 25 V and the ionspray temperature at 500°C. Ions monitored were m/z199.2 (tacrine), m/z 201.2 (natural abundance [13C2]tacrine for high-substrate concentration samples), and m/z233.3 (8-chlorotacrine internal standard). Retention times were 1.1 and 2.7 min for tacrine and 8-chlorotacrine, respectively.
Analysis of Thioridazine.
Reconstituted extracts were injected (10 μl) onto a Phenomenex Luna C18 column equilibrated in a mobile phase of 78% A/22% B and maintained at an isocratic flow rate of 0.5 ml/min. The HPLC effluent was introduced into the mass spectrometer in the positive ion mode, with the orifice voltage at 25 V and the ionspray temperature at 400°C. Ions monitored were m/z 371 (thioridazine), m/z 373 (natural abundance [13C2]thioridazine for high-substrate concentration samples), and m/z280 (imipramine internal standard). Retention times were 3.7 and 1.2 min for thioridazine and imipramine, respectively.
Data Analysis.
Substrate saturation data were analyzed using DeltaGraph version 4.0.3 (SPSS Inc., Chicago, IL). Data were initially transformed and plotted on Eadie-Hofstee plots to assess linearity and diagnose the appropriate kinetic model with which to fit the data.KM values for product formation were determined by nonlinear regression of the reaction velocity versus substrate concentration data.
In substrate depletion experiments, analyte/internal standand peak height ratios were determined and normalized to the value obtained at t = 0. The percentage remaining versus time at each substrate concentration was fitted to first order decay functions to determine initial substrate depletion rate constants (kdep). If substrate decline demonstrated nonlinearity on log percentage remaining versus time curves, only those initial timepoints wherein log-linearity was observed were used to determine depletion rate constants.
KM values from substrate consumption experiments were determined by plotting thekdep versus the substrate concentration on a linear-log plot using the following equation:
Results and Discussion
The objective of these experiments was to determine the reliability with which Michaelis constants could be estimated by measuring substrate depletion rates for drug metabolism reactions. Six drug metabolism reactions, catalyzed by five different human P450 enzymes, were selected for this examination (Fig. 1). The enzymes represent five common human P450 enzymes that are most frequently responsible for drug metabolism reactions. Recombinant P450 expression systems were used in this study to simplify the reactions being examined to test this approach. Finally, to make this comparison and to assess whether the substrate depletion approach can yield accurateKM values, it was necessary to remove as many variables as was possible. To this end, it was important to redetermine KM values for these reactions, by measuring product formation, in the exact system and source of enzyme as used in the substrate depletion experiments, despite the fact that enzyme kinetic data have been previously reported for many of these metabolic reactions and enzymes. Michaelis constants reported in the scientific literature can vary substantially between laboratories and experimental systems used (e.g., liver microsomes, recombinant systems, etc.); thus, such previously reported values could not be used in the determination of the accuracy/inaccuracy ofKM values measured by the substrate depletion approach.
The enzyme kinetics for these six P450-catalyzed reactions were determined using the standard approach of measuring product formed under conditions of initial reaction velocity linearity (Fig.2). The Michaelis constants for each of these reactions are listed in Table 2. The enzyme kinetics observed for these reactions were observed to possess simple, monophasic, Michaelis-Menten behavior, and were fit to simple hyperbolic functions. Some tendency toward biphasic character could be observed for thioridazine S-oxidation and midazolam 4-hydroxylation; however, the data were not compelling enough to merit fitting to complex kinetic equations. Furthermore, for the objectives of these experiments, the differences inKM values obtained by fitting to more complex functions were not great enough to change the overall interpretation of the findings. In three cases, more than one reaction was observed for a given P450 and substrate. CYP1A2 catalyzed the conversion of tacrine into three products: 1-, 2-, and 4-hydroxytacrine. However, the rates of 2- and 4-hydroxylation were much lower than the rate of 1-hydroxylation. Both 2- and 4-hydroxy metabolites were detectable, but concentrations of 2-hydroxytacrine were all below the lower limit of quantitation of the analytical method, and 4-hydroxytacrine was able to be quantitated only at the highest two substrate concentrations examined, precluding determination of enzyme kinetic constants. Midazolam was converted into both 1′- and 4-hydroxy products. The intrinsic clearance for 1′-hydroxylation was over 10-fold higher than for 4-hydroxylation and theKM values were 1.0 and 15 μM for these two reactions, respectively. Thus, the 1′-hydroxylation enzyme kinetics were the only data considered for comparison of Michaelis constants determined by product formation versus substrate depletion approaches. Thioridazine appeared to form both mesoridazine (theS-oxidation product) and a second, unidentified metabolite arising via an addition of 16 mass units (presumably oxygen). This latter metabolite was not measured in enzyme kinetic determinations.

Substrate saturation plots, from product formation data, for (A) CYP1A2-catalyzed tacrine 1-hydroxylation; (B) CYP2C9-catalyzed diclofenac 4′-hydroxylation; (C) CYP2C19-catalyzed imipramine N-demethylation; (D) CYP2D6-catalyzed dextromethorphan O-demethylation; (E) CYP2D6-catalyzed thioridazineS-oxidation; and (F) midazolam 1′-hydroxylation.
Michaelis constants determined using product formation measurements and substrate depletion measurements for six P450 substrates
In theory, when substrate concentrations are well belowKM, the depletion of substrate should follow first-order decay kinetics (Segel, 1975). In fact, when there is no time-dependent loss of enzyme activity,KM values can be determined from measuring substrate depletion over time using an integrated form of the Michaelis-Menten equation. The measurement of substrate depletion, or in vitro t1/2 approach, is commonly used to estimate in vitro intrinsic clearance for the purpose of estimating in vivo intrinsic clearance (Obach et al., 1997; Obach, 1999). In the experiments reported in this paper, we attempt to take this approach one step further; to obtain estimates ofKM, a parameter that can be important in predicting nonlinear pharmacokinetic behavior (see Introduction).
The relationships between the first-order rate constants for substrate depletion (termed kdep) and the substrate concentrations for the six substrates examined in these studies are shown in Fig. 3. As the substrate concentration is elevated through theKM value, the measured values forkdep decline and become, in theory, more zero-order in character. The inflection point of this relationship represents the KM value and should occur at a substrate concentration that yields akdep value that is half of the theoretical maximum kdep at an infinitesimally low-substrate concentration. TheKM values obtained by measuring substrate depletion are listed in Table 2.
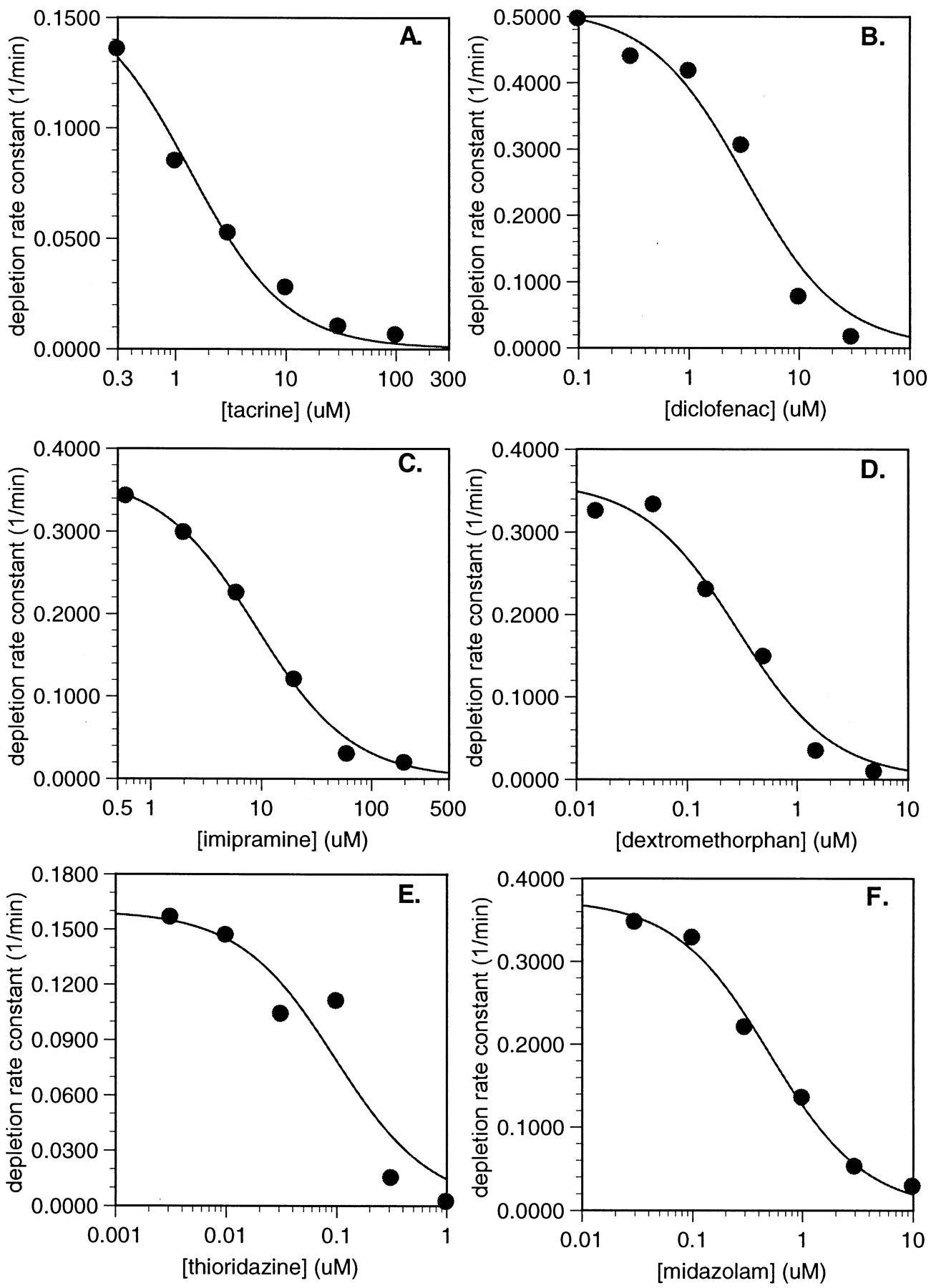
Plots of in vitro depletion rate constants versus substrate concentration for (A) CYP1A2-catalyzed tacrine metabolism; (B) CYP2C9-catalyzed diclofenac metabolism; (C) CYP2C19-catalyzed imipramine metabolism; (D) CYP2D6-catalyzed dextromethorphan metabolism; (E) CYP2D6-catalyzed thioridazine metabolism; and (F) CYP3A4-catalyzed midazolam metabolism.
A comparison between the KM values determined using the conventional product formation approach and the substrate depletion approach is shown in Fig.4. The differences between theKM values measured using the two approaches ranged from no measurable difference (thioridazine) to just over a 2-fold difference (imipramine). In two cases, the values were nearly identical (thioridazine and diclofenac), and in three other cases the KM values measured using substrate depletion ranged up to 2-fold lower (tacrine, dextromethorphan, and midazolam). Overall, the substrate depletion approach appears to provide estimates ofKM that are close to those obtained by following the formation of product(s). The mean fold-error of this approach (calculated as in Obach et al., 1997) for measurement ofKM values for these six substrates is 1.54.

Correlation between Michaelis constants determined using product formation and substrate depletion experiments.
The dashed line represents a line of unity and the solid lines represent the range of 2-fold error. The individual points are 1-CYP2D6 (thioridazine), 2-CYP2D6 (dextromethorphan), 3-CYP3A4 (midazolam), 4-CYP1A2 (tacrine), 5-CYP2C9 (diclofenac), and 6-CYP2C19 (imipramine).
The capability to measure KM values without requiring measurement of metabolite formation rates, and hence, without requiring authentic standards of metabolites for construction of calibration curves, is of use in an early drug discovery setting. In early drug discovery efforts, hundreds of compounds must be characterized in as brief and efficient a manner as possible. Determination of exact chemical structures of metabolites and synthesis of authentic standards are resource-intensive tasks and are not feasible in high-throughput. However, it is important to identify those compounds that have very low KM values since these will be potentially more prone to exhibiting supraproportional dose-exposure relationships, a negative attribute in drugs. Furthermore, in examinations of metabolic stability using high-throughput approaches, one must be assured that the substrate concentration used does not exceed theKM, or else artificially low-intrinsic clearance estimations may be made. Concentrations of 1 μM or greater are frequently used in high-throughput liver microsomal metabolic stability screens (to permit ease of sample processing and analysis); however, such concentrations can be in excess ofKM (e.g., as measured for thioridazine and dextromethorphan in this examination) and thus partially saturating. Identification of compounds with lowKM values can also be important in the planning of initial dose escalation studied in humans, as the size of incremental increases of dose can be reduced if it is predicted that the test compound will exhibit supraproportionality in exposure with increases in dose.
The substrate depletion approach for measuringKM possesses some practical limitations. Compounds that have low-intrinsic clearance will be difficult to examine, since application of the depletion approach requires that a substantial percentage of the initial substrate concentration be consumed during the incubation period. Additionally, to generate substantial substrate depletion, concentrations of enzyme may need to be increased, increasing the likelihood of nonspecific binding to incubation components (e.g., microsomes), which would increase the measured value for apparentKM (Obach, 1997; Kalvass et al., 2001). Also, by measuring substrate depletion, the enzyme kinetics of formation of individual products are missed, and theKM obtained would represent a conglomeration of all KM values for individual metabolic pathways. This shortcoming may not be important when one high capacity, low KMactivity predominates in the overall consumption of substrate (e.g., midazolam and tacrine examples in this work). However, if a compound is metabolized by both a low capacity, lowKM activity, and a high capacity, highKM activity, the latter may influence the overall estimate of the KM value obtained using the substrate depletion approach and result in missing a low-saturable KM value.
Future efforts will require that the method of estimatingKM values by measuring substrate depletion be tested in more complex in vitro systems, such as liver microsomes or hepatocytes, and be tested for other drug-metabolizing enzymes beside P450. Furthermore, gaining a better understanding of the relationship between KM measured in vitro and supraproportionality of exposure-dose relationships is warranted. In conclusion, an experimental approach has been devised in which KM values for drug metabolism reactions can be estimated to within two-fold of values measured using standard techniques. This approach should be of value in early drug discovery settings in which standard enzyme kinetic experiments are not feasible or possible.
Footnotes
- Abbreviations used are::
- P450
- cytochrome P450
- HPLC-MS
- high-performance liquid chromatography-mass spectometry
- MTBE
- methyl tertiary butyl ether
- kdep
- depletion rate constant
- [S]
- substrate concentration
- Received February 21, 2002.
- Accepted April 11, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}