


Abstract
Since sulfation is the main metabolic pathway of troglitazone, accounting for about 70% of the metabolites detected in human plasma, we have aimed to identify human cytosolic sulfotransferases catalyzing the sulfation of troglitazone and to examine a possible role of the sulfation in the cytotoxicity observed in cell lines of human origin (HepG2 and Hep3B). Experiments using the recombinant sulfotransferases and human liver cytosols indicated that phenol sulfotransferase (ST1A3) and estrogen sulfotransferase (ST1E4) were the sulfotransferases most active toward troglitazone. Immunoblot analyses indicated that hepatic content of ST1A3 is about 13 times higher than that of ST1E4, suggesting that ST1A3 is mainly responsible for the sulfation of troglitazone in the liver. Lactate dehydrogenase (LDH) leakage was elicited by troglitazone in a concentration-dependent manner in the hepatoma cells. The troglitazone metabolites (the sulfate, glucuronide, and quinone forms) caused negligible LDH leakage. These findings suggest that accumulation of unmetabolized troglitazone causes the cytotoxicity in the hepatoma cells and may be responsible for toxicity in human liver.
Troglitazone (Rezulin, Warner-Lambert Co. or Noscal, Sankyo Co., Ltd.) has been used as an oral antidiabetic drug for the treatment of non-insulin-dependent diabetes mellitus. It lowers blood glucose levels through increasing glucose uptake at skeletal muscles, decreasing hepatic glucose production and increasing sensitivity to insulin (Ciaraldi et al., 1990; Fujiwara et al., 1995, 1998). A rare but serious idiosyncratic hepatotoxicity due to the troglitazone-treatment has been reported (Watkins and Whitcomb, 1998), leading to the withdrawal of this drug from the market and leaving the mechanism of toxicity obscure. The sulfate and quinone forms of troglitazone are the major metabolites, whereas the glucuronide form is a minor metabolite in humans. Troglitazone sulfate accounted for about 70% of the metabolites detected in human plasma (Shibata et al., 1993; Loi et al., 1997). Thus, sulfation is considered the major pathway in troglitazone metabolism, determining the exposure to this drug. Little information on enzymatic sulfation of troglitazone has, however, been reported. Despite its withdrawal, study of the metabolism would contribute to understanding the mechanism of toxicity caused by troglitazone.
Cytosolic sulfotransferases (STs or SULTs12) catalyze sulfation of various endogenous and exogenous chemicals (De Meio, 1975; Jakoby et al., 1980; Yamazoe and Kato, 1995; Nagata and Yamazoe, 2000). The reaction involves the transfer of a sulfonate group from 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to the substrate. STs are known to constitute a gene superfamily, which contains at least five subfamilies (ST1 to ST5) in mammals based on the similarities of their deduced amino acid sequences. There are differences in the substrate specificities and expression profiles among ST forms (Yamazoe et al., 1994; Weinshilboum et al., 1997; Dooley et al., 2000; Nagata and Yamazoe, 2000). In the present study, we have aimed to identify the human cytosolic sulfotransferases catalyzing the sulfation of troglitazone and to examine a possible role of sulfation in the toxicity of troglitazone using hepatoma cell lines of human origin.
Experimental Procedures
Materials.
Troglitazone (purity >99%), its sulfate (>98%) and glucuronide (>98%) conjugates and its quinone-type metabolite [(±)-5-(4-(2-hydroxy-2-methyl-4-(3,5,6-trimethyl-1,4-benzoquinon-2-yl)butoxy)benzyl)-2,4-thiazolidine-dion, purity >98%] were provided by Sankyo Co. Ltd. (Tokyo, Japan). Restriction endonucleases, DNA-modifying enzymes, and TaKaRaEx Taq were purchased from Takara Shuzo (Kyoto, Japan). Enterokinase was obtained from Biozyme Laboratories, Ltd. (Gwent, UK). Isopropyl-β-d-thiogalactopyranoside, dithiothreitol, alkaline phosphatase-conjugated goat anti-rabbit IgG, 5-bromo-4-chloro-3-indolylphosphate, and nitro blue tetrazonium were purchased from Sigma-Aldrich (St. Louis, MO). [35S]PAPS (2,000 mCi/mmol) was from PerkinElmer Life Sciences (Boston, MA). QIAexpress and nickel-nitrilotriacetic acid agarose were the products of QIAGEN (Valencia, CA). Bio-Rad Protein assay kit and SDS-polyacrylamide gel electrophoresis molecular weight standards (low range) were from Bio-Rad (Hercules, CA). All other chemicals used were of the highest grade available.
Female Japanese White rabbits (8-weeks old) were obtained from Japan SLC (Shizuoka, Japan). Hepatoma cells of human origin, HepG2 and Hep3B, were donated from the Cell Resource Center for Biomedical Research (Tohoku University, Sendai, Japan). Human liver samples provided by the SRI International Toxicology Laboratory (Menlo Park, CA) and Department of Anatomic Pathology (School of Medicine, Tohoku University) were used. Human small intestine (jejunum) samples were donated from the Human & Animal Bridge Discussion Group (Tokyo, Japan). Experiments with human samples were approved by the Tohoku University Ethical Committee. Animal experiments were done under the instruction of Tohoku University Animal Care and Use Committee.
Methods.
Construction of expression vectors, expression and purification of recombinant human ST proteins
Human ST cDNA fragments contained nucleotides encoding seven additional amino acid residues (GlySerAspAspAspAspLys), which include a sequence of the recognition site of enterokinase next to theN-terminal methionine of the native form. Human ST1A3, ST1A5, ST1B2, ST1C2, ST1E4, and ST2A3 cDNA fragments were obtained by polymerase chain reaction as described previously (Yoshinari et al., 1998b; Fujita et al, 1999a,b). The human ST cDNAs were ligated into a prokaryotic expression vector, pQE30 (QIAGEN). The constructed plasmid DNAs were transformed into Escherichia coli, M15 (pREP4) strain. Recombinant proteins, termed His-STs, were expressed and purified from bacterial cytosols by nickel-nitrilotriacetic acid affinity chromatography. The fused portion of His-STs was removed to yield ΔHis-ST proteins for standards of immunoblot analyses by use of enterokinase as described previously (Fujita et al., 1997). ST1A3*1 and ST1A3*2 proteins were expressed in E. coli and purified as described previously (Ozawa et al., 1999). The protein concentration was determined by the method of Bradford (1976) using bovine serum albumin as the standard.
Assay of sulfation.
Sulfating activities were determined by the measurements of radioactivity of the troglitazone sulfate obtained with [35S]PAPS as a sulfate donor after thin layer chromatography (Yoshinari et al., 1998a). A typical incubation mixture consisted of 50 mM Tris-HCl buffer (pH 7.4), 1 mM dithiothreitol, 20 mM MgCl2, 10 μM troglitazone, 5 μM [35S]PAPS (0.1–0.2 Ci/mmol), and 2 μg of cytosolic protein or 50 ng of His-ST in a final volume of 10 μl. The reaction was initiated by addition of [35S]PAPS and terminated by addition of 5 μl of chilled acetonitrile after incubation at 37oC for 10 min. A portion (10 μl) of the reaction mixture was applied to a thin layer plate (thin layer chromatography aluminum sheets 20 × 20 cm silica gel 60 F254; Merck, Darmstadt, Germany). Metabolites were developed with the solvent system of benzene (saturated with water)/ chloroform/ methanol (5:9:5). The radioactive spots on the chromatogram were analyzed by a BAS1000 Bioimaging Analyzer (Fuji Photofilms, Tokyo, Japan). Molecular weights of 36,174 for His-ST1A3, 36,160 for His-ST1A5, 36,862 for His-ST1B2, 36,535 for His-ST1C2, 37,089 for His-ST1E4, and 35,744 for His-ST2A3, calculated on the basis of their deduced amino acid sequences, were used for the determination of each sulfating activity in pmol-troglitazone sulfate/nmol-ST/min. The apparent kinetic parameters derived from analyses of Lineweaver-Burk plots were from the assays examined with several concentrations of troglitazone (0.5–15 μM for ST1A3, 5–40 μM for ST1B2, 0.5–20 μM for ST1E4, and 10–50 μM for ST2A3). Each reaction showed linearity within the concentration range examined.
Antibody preparation and immunoblot analysis.
Japanese white rabbits (2.5 kg, female) were immunized by intradermally injecting 20 to 50 μg of the purified each ΔHis-ST protein in complete Freund's adjuvant, and immunity was boosted by intravenously injecting 20 to 50 μg of the protein 3 weeks later. One week after the boost, anti-sera were obtained. Specific IgG was purified by the affinity column with Sepharose 4B-ΔHis-ST1A5, -ΔHis-ST1B2, -ΔHis-ST1E4, or -ΔHis-ST2A3 and stored frozen at −80oC until use. Cytosolic proteins were separated by 10.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose sheet. The sheet was immunostained with anti-ST IgG, alkaline phosphatase-conjugated goat anti-rabbit IgG, 5-bromo-4-chloro-3-indolylphosphate, and nitro blue tetrazonium as described previously (Blake et al., 1984). To determine the contents of ST1A3, we used the IgG raised against the purified ΔHis-ST1A5 protein. The stained band was scanned with Nikon AX-1200 (Nikon, Tokyo, Japan), and the intensity was quantified by use of the National Institutes of Health image (version 1.59) software (Bethesda, MD). The contents of each ST form in cytosols were determined using corresponding ΔHis-ST proteins as the standards. Molecular weights of 34,171 for ΔHis-ST1A3, 34,859 for ΔHis-ST1B2, 35,086 for ΔHis-ST1E4, and 33,741 for ΔHis-ST2A3, calculated on the basis of their deduced amino acid sequences, were used to determine the content of each ST form in cytosols in pmol-ST/mg of cytosolic protein.
Cell culture.
Hepatoma cells of human origin (HepG2 and Hep3B) were plated on 100 mm culture dish (Falcon Scientific Co., Oxnard, CA) with 8 ml of Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum. The dish was incubated at 37oC in an atmosphere of 5% CO2.
Assay of lactate dehydrogenase (LDH) and analysis of media.
HepG2 and Hep3B cells were suspended in serum-free DMEM and aliquots (1.5 × 106 cells/ml) were plated in 24-well tissue-culture plates (Falcon Scientific Co.). After incubation at 37oC in an atmosphere of 5% CO2 for 18 h, the DMEM was removed, and fresh serum-free DMEM and test compounds were added. All the chemicals were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO was 0.4% in each culture. After 20 h of incubation with test compounds, supernatants of the cell cultures were obtained by centrifugation (1,000 rpm, 5 min). LDH activities in the supernatant fractions were assayed with LDH-Cytotoxic Test Wako (Wako Pure Chemical Industries, Ltd., Osaka, Japan) as assessed by oxidation of NADH (Wroblewski and La Due, 1955). LDH leakage was expressed as a percentage of total LDH activity. Positive control (100%) was from supernatant of cells treated with 2% Tween 20 solution after 20 h incubation, and negative control (0%) was from supernatant incubated with 0.4% DMSO. Non-linear progression (sigmoidal curve) was assessed by GraphPad Prism (version 3) software (GraphPad Software, Inc., San Diego, CA). Analyses of the media for troglitazone sulfate contents were performed with a HPLC system (Jasco Gulliver system; Jasco Corp., Tokyo, Japan) by a modification of the method described by Kawai et al. (1997). The media were added with an equal volume of acetonitrile and testosterone as an internal standard. HPLC was performed using ChemcoPak (6.0 × 150 mm, Chemco Scientific Co., Ltd., Osaka, Japan) column and 50% acetonitrile containing 0.1% phosphoric acid as a mobile phase. The corresponding peak was monitored at 230 nm.
Results
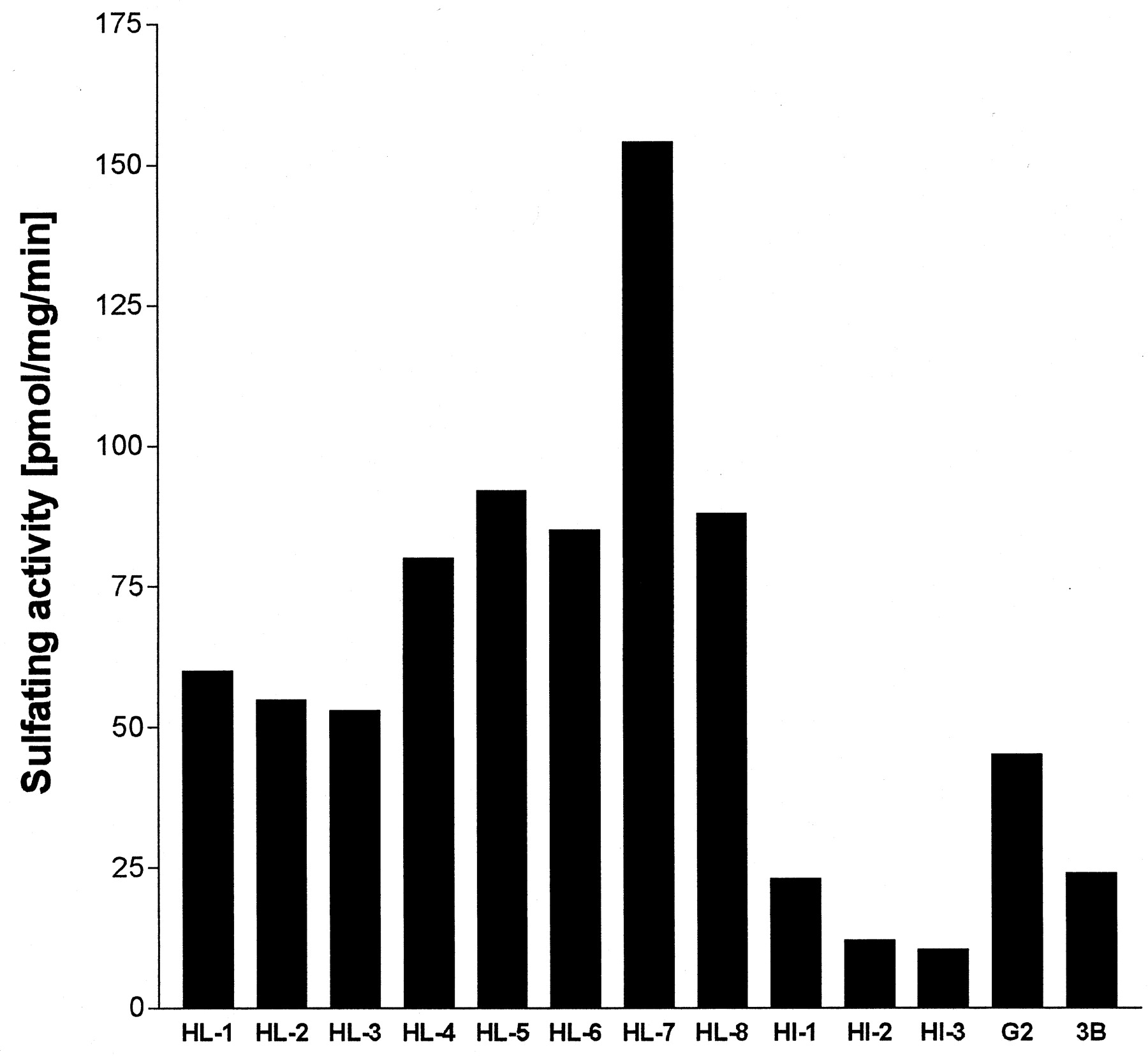
The sulfating activities toward troglitazone (10 μM) were determined using eight human liver cytosols (HL-1–HL-8) in the presence of 5 μM PAPS (Fig. 1). After incubation for 10 min, the activities were detected in all the liver samples examined with noticeable deviations (84.0 ± 30.0 pmol/mg/min). Human intestinal cytosols (HI-1–HI-3) catalyzed the reaction (15.0 ± 5.6 pmol/mg/min) with lower activities than the liver cytosols. The cytosols of hepatoma cells, HepG2 and Hep3B, also catalyzed the sulfation of troglitazone with activities of 45 and 24 pmol/mg/min, respectively.

Sulfating activities of cytosolic fractions toward troglitazone.
The assays were performed with 10 μM troglitazone as a substrate and 5 μM [35S]PAPS as a sulfate donor using cytosolic proteins (2 μg). HL, HI, G2, and 3B mean human hepatic, intestinal, HepG2, and Hep3B cell cytosols, respectively. After 10 min of incubation at 37oC, aliquots of the reaction mixtures were separated by thin layer chromatography. Radioactivity of the troglitazone sulfate was determined by means of BAS1000 Bioimaging Analyzer (Fuji Photofilms). Each column indicates the mean of duplicate determinations.
Different types of ST forms are known to be expressed in human liver and intestine. Thus, we examined which human ST forms mediate the sulfation of troglitazone (Fig. 2), using His-STs expressed in E. coli and purified by a nickel-chelate column chromatography. Human phenol sulfotransferase (ST1A3) and estrogen sulfotransferase (ST1E4) catalyzed the sulfation of troglitazone effectively compared with the other human ST forms examined (ST1A5, ST1B2, ST1C2, and ST2A3), which showed negligible or very low activity. ST1E4 had a higher activity (1.22 nmol/nmol/min) than did ST1A3 at 10 μM troglitazone (0.90 nmol/nmol/min). Even at the highest troglitazone concentration (20 μM), the sulfating activities of ST1A5 and ST1C2 were not detected (less than 1 pmol/mg/min), and those of ST1B2 and ST2A3 were low, about 9 to 17 times less than that of ST1E4 at 10 μM troglitazone.
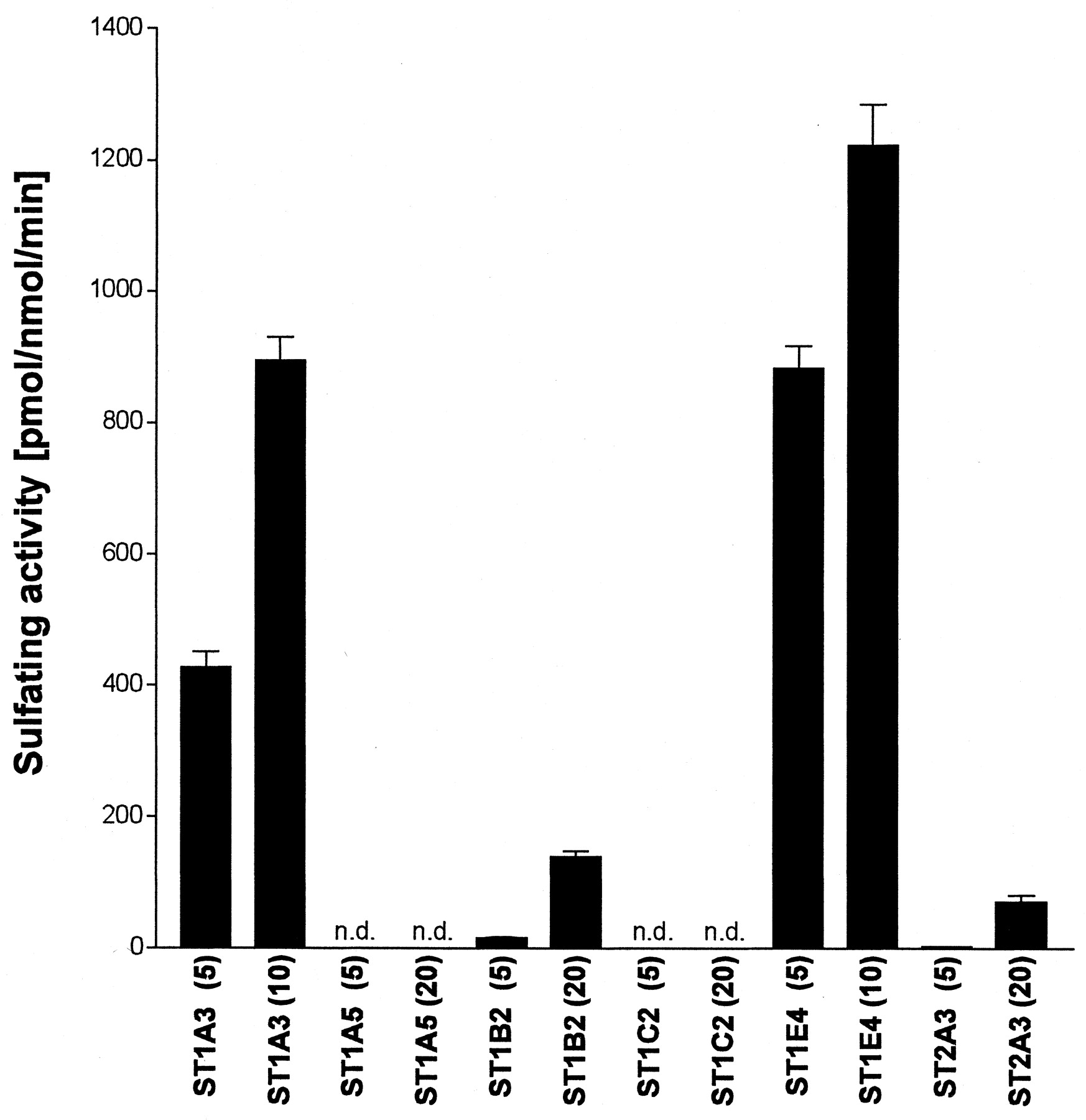
Sulfating activities of human recombinant ST forms toward troglitazone.
Assays were performed at pH 7.4 with 5, 10, or 20 μM troglitazone, 5 μM [35S]PAPS and 50 ng of His-ST proteins. The names of individual human ST forms are based on the proposed nomenclature system (Nagata and Yamazoe, 2000). The figures in parentheses indicate the concentration of troglitazone in μM. Each column indicates the mean ± S.D. of triplicate determinations. n.d., not detected (less than 1 pmol/mg/min).
The apparent kinetic parameters for troglitazone were determined among four ST forms with 5 μM PAPS at pH 7.4. As shown in Table1, Kmvalues of ST1A3, ST1B2, ST1E4, and ST2A3 were calculated to be 5.4, 17.0, 8.5, and 28.0 μM, respectively. The ratioVmax/Km, which is known to be specificity constant (Cornish-Bowden, 1995), was compared among these ST forms to elucidate their affinities for troglitazone. A greater specificity constant (Vmax/Km) was obtained with ST1A3 (0.15) and ST1E4 (0.21) than those of ST1B2 (0.016) and ST2A3 (0.0028).
Apparent kinetic parameters of STs for troglitazone
Contents of ST1A3, ST1B2, ST1E4, and ST2A3 in the cytosolic fraction of human liver (n = 30) and intestine (n = 3) and in hepatoma cells were determined by immunoblot analyses (Table2). Average hepatic content of ST1A3 (110.0 ± 35.0 pmol/mg of cytosolic protein) was about 13 times higher than that of ST1E4 (8.3 ± 5.1 pmol/mg). Immunodetectable ST1A3 and ST1E4 proteins were also observed in the human intestinal cytosols. The average contents of ST1A3 were about 5 times higher with liver (110.0 ± 35.0 pmol/mg) than intestine (23.0 ± 3.3 pmol/mg), although no clear difference was observed in the hepatic and intestinal content of ST1E4 (8.3 ± 5.1 in liver versus 6.3 ± 2.2 pmol/mg in intestine). Average contents of ST1B2 in liver and intestine were 28.0 ± 6.6 and 36.0 ± 1.7 pmol/mg, respectively. Hepatic content of ST2A3 (190.0 ± 50.0 pmol/mg) was about 1.8 times higher than that of ST1A3 (110.0 ± 35.0 pmol/mg). The hepatic average content of ST2A3 (190.0 ± 50.0 pmol/mg) was about 6.8 times higher than that of intestinal content (28.0 ± 9.5 pmol/mg). The specific contents of ST1A3 were 51.0 pmol/mg in HepG2 and 26.0 pmol/mg in Hep3B, respectively. ST1E4 content was 5.7 pmol/mg in Hep3B, but ST1E4 protein was not detected in HepG2 (less than 1 pmol/mg). These data were roughly consistent with those of human livers. ST1B2 and ST2A3 proteins were detected in HepG2 (9.8 for ST1B2 and 15.0 pmol/mg for ST2A3), whereas they were not detected in Hep3B (less than 1 pmol/mg).
Contents of human STs
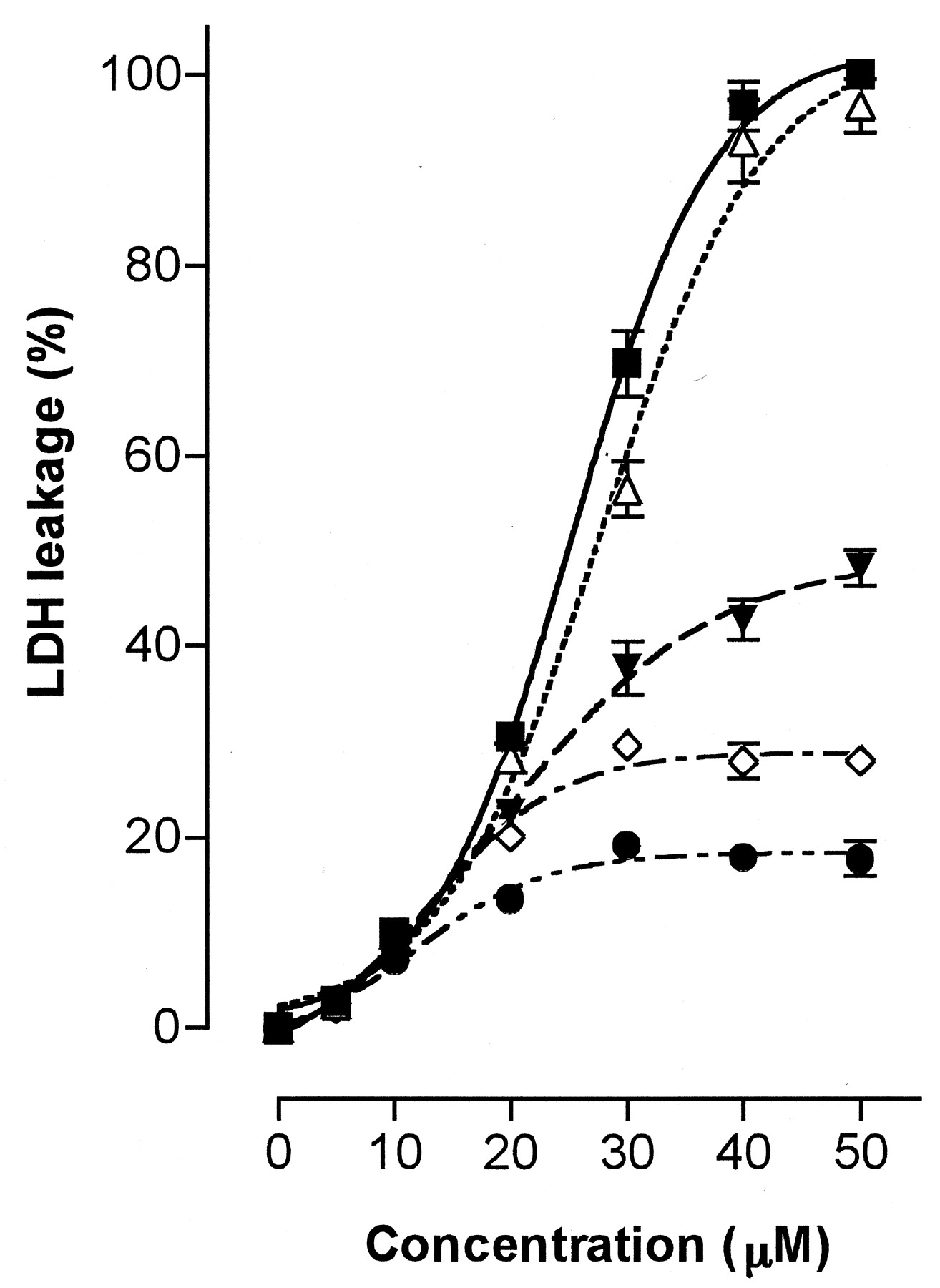
We examined the cytotoxicity of troglitazone in cultured hepatoma cells (HepG2 and Hep3B) as a leakage of LDH into the cultured medium and compared the results to effect of the troglitazone metabolites (the sulfate, glucuronide, and quinone forms). Because of the sulfating activities and the contents of ST1A3, we used hepatoma cells as a model to examine a tentative toxicity marker. As shown in Fig.3, troglitazone showed a marked cytotoxicity on these hepatoma cells after 20 h incubation. The apparent median lethal concentration, LC50 values (mean ± S.E., n = 4), were 24.6 ± 0.0 μM and 33.0 ± 1.6 μM in HepG2 and Hep3B cells, respectively. LDH leakage increased in a concentration-dependent manner at concentrations up to 40 μM of troglitazone but saturated more than 50 μM in HepG2 cells or 60 μM in Hep3B cells. The metabolites of troglitazone, however, caused negligible LDH leakage in both hepatoma cells (not more than 15% of the total leakage). As shown in Fig.4, the periods of exposure to troglitazone affected the magnitude of LDH leakage in HepG2 cells. The LDH leakages were observed already after incubation for 1 h, gradually increased with prolonged incubation time from 1 h to 3 and 5 h, and reached a plateau level after incubation for 10 and 20 h. Analyses of the culture media of HepG2 cells with incubation for 5 h revealed that additions of troglitazone decreased the sulfate formation in a concentration-dependent manner (Fig.5).

LDH leakage induced by troglitazone and its metabolites in HepG2 and Hep3B cells.
After 20 h incubation of HepG2 (a) and Hep3B (b) cultures with troglitazone (▪), its quinone (♦), sulfate (▴) and glucuronide (▾) forms, LDH leakages were measured. Each value represents the mean ± S.D. (n = 4).
Time-dependent increase of LDH leakage by troglitazone in HepG2 cells.
LDH leakages from HepG2 cells were measured after incubations with troglitazone for 20 (▪), 10 (▵), 5 (▾), 3 (⋄) and 1 (●) h. Each value represents the mean ± S.D. (n = 4).

Troglitazone sulfate content in the media.
Analyses of the media for the troglitazone sulfate contents were performed by HPLC as described under Experimental Procedures. Abscissa indicates concentration (micromolars) of troglitazone added to HepG2 cultures. Ordinate represents concentration of troglitazone sulfate in the media after 5-h incubation. Each column indicates the mean ± S.D. (n = 3).
A genetic polymorphism is known for cytosolic sulfotransferase. Four allelic variants accompanying amino acid exchange in ST1A3 have been identified. One of them, ST1A3*2 (R213H), is associated both with very low sulfating activities of several substrates such asp-nitrophenol and low thermal stability in platelets. (Raftogianis et al., 1997; Ozawa et al., 1998, 1999). Thus, we determined the sulfating activities for troglitazone (10 μM) using recombinant ST1A3*1 (wild type) and ST1A3*2. The activity of ST1A3*2 (0.51 ± 0.02 nmol/nmol/min) was 40% lower than the activity of ST1A3*1 (0.85 ± 0.04 nmol/nmol/min).
Discussion
In the present study, we examined the sulfation of troglitazone catalyzed by the cytosolic sulfotransferases. Experiments using recombinant sulfotransferases and tissue cytosols indicated the involvement of phenol sulfotransferase (ST1A3) and estrogen sulfotransferase (ST1E4) in troglitazone sulfation. Troglitazone showed a high affinity for both ST1A3 and ST1E4 as indicated by the lowKm values (5.6 μM for ST1A3 and 8.1 μM for ST1E4) compared with other ST forms (17.0 μM for ST1B2 and 28.0 μM for ST2A3). Concentrations of 3.6 and 6.3 μM were reported as the maximum plasma concentrations in humans taking troglitazone at therapeutic doses of 400 and 600 mg/day, respectively (Loi et al., 1999). As shown in Table 2, average hepatic content of ST1A3 was about 13 times higher than that of ST1E4. In addition to liver, the small intestine may possibly influence the disposition of troglitazone. Thus, we determined the absolute contents of ST forms in small intestine. The contents of ST1A3 and sulfating activities of intestinal cytosols were about 5 times lower than those of hepatic cytosols. Therefore, these data suggest that hepatic ST1A3 is mainly responsible for the sulfation of troglitazone in humans.
Despite a marked difference in the route of excretion between troglitazone (biliary excretion) and rosiglitazone (urinary excretion) in humans, their major metabolites are the sulfate forms, namely the 6-O-sulfate in the case of troglitazone, and theN-desmethyl-para-O-sulfate andpara-O-sulfate in the case of rosiglitazone (Shibata et al., 1993; Loi et al., 1997; Cox et al. 2000). Therefore, the sulfating reactions are considered to play an important role in the disposition of these thiazolidinediones. ST1A3 and ST1E4, the sulfotransferases involved in the troglitazone sulfation, are considered very likely to catalyze the sulfation of rosiglitazone as well.
Cytotoxicity of troglitazone, evaluated as the LDH leakage, was observed in HepG2 and Hep3B cells (Fig. 3). However, additions of the troglitazone metabolites (the sulfate, glucuronide, and quinone forms) were not cytotoxic in HepG2 and Hep3B cell cultures, suggesting that the unchanged troglitazone causes the cytotoxic effect. As in the case of acetaminophen, menadione, and diclofenac, quinone-type metabolites are generally believed to be the active intermediates in drug-induced hepatic toxicity (Pumford et al., 1997; Bort et al., 1999; Hojo et al., 2000). However, the quinone form of troglitazone did not show a noticeable cytotoxicity in the hepatoma cells. The present experiment did not exclude the possibility that polar metabolites of troglitazone generated within cells cause the toxic event, because of the possible low permeability. The precise mechanism by which the troglitazone causes the cytotoxicity toward the hepatoma cells is presently unknown.Kostrubsky et al. (2000) reported that the troglitazone-treatment of the cultured human hepatocytes caused a decreased total protein synthesis, and the cytotoxicity correlated with accumulation of unmetabolized troglitazone. Therefore, the metabolism of troglitazone to the quinone form as a causal factor for the cytotoxicity is quite unlikely.
Metabolic activation of troglitazone in vitro has recently been suggested to proceed through the oxidation of the substituted chromane ring to a reactive o-quinone methide derivatives and the oxidative cleavage of the thiazolidinedione ring by human cytochrome P450 3A4 (Kassahun et al, 2001). There are other reports that troglitazone induces apoptosis in human gastric cancer cells and in rat vascular smooth muscle cells (Takahashi et al., 1999; Gouni-Berthold et al., 2001). All these observations point to the importance of sulfation as the major detoxicating pathway of troglitazone. Thus, the reduction and inhibition of the sulfation pathway may lead to enhanced hepatotoxicity.
As shown in Fig. 5, addition of troglitazone to HepG2 cultures caused a concentration-dependent decrease of the sulfate formation. Similar substrate inhibition was also observed in the kinetic study in vitro using ST1A3 and ST1E4. Troglitazone added to the system at concentrations of more than 25 μM inhibited markedly the activities of ST1A3 and ST1E4 (data not shown). These results suggest that increasing concentrations of troglitazone lead to the decreasing metabolic clearance of troglitazone and to increasing exposure to this cytotoxic compound. Consistent with this idea, there is a recent report that the toxic effect of troglitazone in primary cultures of rat hepatocytes was decreased by reducing the free concentration of troglitazone by addition of bovine serum albumin (Toyoda et al., 2001). Although the ST1A3 content in normal human liver was about double that of HepG2 cell (Table 2), a high dose of troglitazone may cause the accumulation of troglitazone as a consequence of inhibiting this major eliminating pathway.
In the clinical situation, a combination of factors rather than a single reason are likely to cause the so-called idiosyncratic hepatotoxicity of troglitazone. As described above, decreased activity of the sulfation either by inhibition or by genetic polymorphism of ST1A3 is likely to be one of the possible risk factors. Allele frequencies of ST1A3*1 and ST1A3*2 were reportedly 0.67 and 0.31 in populations of Caucasians (Raftogianis et al., 1997). ST1A3*2 showed 40% lower activity for troglitazone sulfation than did ST1A3*1. Therefore, this polymorphism may cause an increased exposure to troglitazone in certain individuals.
The findings presented here suggest that accumulation of the unmetabolized troglitazone stimulated the cytotoxicity in the hepatoma cells rather than the quinone form. Since sulfation by the sulfotransferases is considered as the major elimination pathway for troglitazone, the importance of the sulfotransferases, especially ST1A3, from the toxicological viewpoint is also suggested in the present study.
Footnotes
-
This study was supported in part by grant-in-aids from the Ministry of Education, Science and Culture and the Ministry of Health and Welfare, Japan; the Japan Health Sciences Foundation, Smoking Research Foundation, Japan; and Human & Animal Bridge Discussion Group, Japan.
-
↵2 STs or SULTs are as follows: ST1A3, ST1A5, ST1B2, ST1C2, ST1E4, and ST2A3 correspond to SULT1A1, SULT1A3, SULT1B1, SULT1C2, SULT1E1 and SULT2A1, respectively.
- Abbreviations used are::
- ST or SULT
- cytosolic sulfotransferase
- PAPS
- 3′-phosphoadenosine-5′-phosphosulfate
- His-ST
- recombinant ST protein that has additional amino acid residues at theN-terminal
- ΔHis-ST
- fused portion removed from His-ST by digestion with enterokinase
- DMEM
- Dulbecco's modified Eagle's medium
- DMSO
- dimethyl sulfoxide
- HPLC
- high-performance liquid chromatography
- LDH
- lactate dehydrogenase
- Received November 2, 2001.
- Accepted May 15, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}