


Abstract
To study mechanisms behind the interindividual variability in CYP3A expression and the relative contribution of the different CYP3A enzymes to the overall CYP3A activity, we have analyzed CYP3A4, CYP3A5, CYP3A43, and PXR mRNA and CYP3A4 and CYP3A5 protein expression, catalytic activity, and polymorphism in the CYP3A5 gene in a panel of 46 Caucasian human livers. Protein quantification was performed by Western blotting using enzyme-specific antibodies directed to the C termini of CYP3A4 or CYP3A5, and carrier protein-coupled peptides as standards. The mRNA levels were determined by quantitative real-time PCR. CYP3A activity was measured by analysis of the rate of testosterone 6β-hydroxylation. A correlation existed between all CYP3A and PXR mRNA transcripts measured. The interindividual variability in CYP3A4 and CYP3A5 mRNA expression was higher than that of CYP3A protein and activity. The CYP3A5 protein was expressed at quantifiable levels in 5 (10.9%) of the livers. Four of those were heterozygous for the CYP3A5*1 allele and had CYP3A5 protein at a mean level of 17% of that of total CYP3A, whereas one liver sample was from a CYP3A5*3 homozygote individual having lower amounts of CYP3A5. In total, CYP3A5 only contributed 2% of the overall CYP3A protein among all samples. In conclusion, our data indicate that CYP3A4, CYP3A5, CYP3A43, and PXR hepatic mRNA expression correlate, indicating common regulatory features, and that the contribution of CYP3A5 to hepatic drug metabolism in Caucasians is insignificant.
Interindividual variation in the activity and levels of expression of CYP3A is considerable. This variation is manifested by differences greater than 10-fold in the in vivo metabolism of drugs that are substrates for CYP3As (Kleinbloesem et al., 1984; Lown et al., 1995; Wilkinson, 1996; Thummel and Wilkinson, 1998; Guengerich, 1999; Kinirons et al., 1999; Schmidt et al., 2001) and a 31-fold variation in CYP3A4 activity in vitro as assessed in our liver bank (Westlind et al., 1999). Furthermore, such differences can be increased significantly by induction and inhibition of CYP3A4. Indeed, a 400-fold variation of the area under the curve is seen between subjects receiving rifampicin as an inducer and itraconazole as an inhibitor of CYP3A4 (Backman et al., 1998). Such variation can cause clinically important differences in drug toxicity and response. Aside from the effects of drugs as inducers and inhibitors, genetic, pathological, hormonal, and dietary factors also may contribute to variability in CYP3A4 activity. Of these, it appears that genetic factors are of great importance for the interindividual variability in constitutive expression and activity of the CYP3A subfamily, accounting for 70 to 90% of the variation, although the underlying genetic factors are essentially unknown (Ozdemir et al., 2000). In the search for such factors, the functional polymorphism of the CYP3A4 gene has been investigated, and 19 different polymorphic CYP3A4 alleles carrying missense mutations have been found (see http://www.imm.ki.se/CYPalleles/cyp3a4.htm). However, these are very rare, and among them, only a few have been found to carry mutations that influence function. Thus, additional factors that control the genetically determined differences in CYP3A4 expression remain to be identified.
It has been suggested that variable expression of other CYP3A enzymes might contribute to the overall differences in CYP3A enzyme activity. Thus, we and others (Domanski et al., 2001; Gellner et al., 2001; Westlind et al., 2001) identified the CYP3A43 gene. However, CYP3A43 is expressed at very low levels, and the enzyme is likely to be inactive (Westlind et al., 2001). In addition, CYP3A5 has been found to be expressed at high levels in 10 to 30% of human livers (Aoyama et al., 1989; Wrighton et al., 1990; Schuetz et al., 1994; Kuehl et al., 2001). Kuehl et al. (2001) suggested that CYP3A5 contributes substantially to the CYP3A-dependent drug metabolism due to very high hepatic expression in about 30% of Caucasian livers. They identified a single nucleotide polymorphism in intron 3 creating a cryptic splice site and three different splice variants of CYP3A5 (CYP3A5*3) explaining the lack of expression in the majority of Caucasian livers.
Another explanation for the variable CYP3A expression might be variation in the expression of human pregnane X receptor (PXR2; NR1I2), a key mediator of the xenobiotic induction of CYP3A genes. Inducible regulatory control of the expression of PXR itself may constitute a factor of importance for the variation in CYP3A induction.
To elucidate the background behind the interindividual variability in CYP3A expression and activity, we evaluated the relative expression of PXR, CYP3A4, CYP3A5, and CYP3A43 mRNA in a panel of human livers characterized with respect to CYP3A-dependent catalytic activity and put emphasis on the quantification of the CYP3A4 and CYP3A5 enzymes using enzyme-specific antibodies raised against the C termini and carrier protein-coupled peptides as standards. Our results indicate that the contribution of CYP3A5 to hepatic CYP3A activity in Caucasians is insignificant and that all the CYP3A gene products investigated correlate in expression, indicating common regulatory factors of importance for their expression and for the interindividual differences in hepatic content of CYP3A4.
Materials and Methods
Human Liver Material. Liver samples from 46 individuals were collected from three different sources. Seventeen samples (HL110–HL129) were obtained from Sahlgrenska Hospital (Gothenburg, Sweden), originating from patients undergoing liver resection, in most cases due to malignant tumors (usually metastatic colon cancer). The liver samples were collected from parts of the liver without pathological changes. Fifteen liver samples (HL19–HL55) were obtained from Huddinge Hospital (Stockholm, Sweden), from organ donors who met accidental deaths. Procedures for isolation of liver pieces have previously been described by Ekstrom et al. (1989). Fourteen liver samples were obtained from the International Institute for the Advancement of Medicine (IIAM; Exton, PA) and chosen to be either high (≥2900 pmol/mg/min) or low (≤600 pmol/mg/min) with respect to activity measured as 6β-hydroxylation of testosterone. Eight of these exhibited low activity and six exhibited high activity. All livers except one (HL46) were documented to be of Caucasian origin.
Human Liver Microsomes. Human liver microsomes were prepared by subcellular fractionation as described elsewhere (Westlind et al., 1999). The microsomes were suspended in 100 mM potassium phosphate buffer (pH 7.4) containing 20% glycerol, snap-frozen in liquid nitrogen, and stored at -70°C. Total protein content was determined by the method of Lowry et al. (1951).
Testosterone 6β-Hydroxylation Assay. Measurement of CYP3A activity based on 6β-hydroxylation of testosterone was carried out as previously described (Westlind et al., 1999).
Genomic DNA Preparation. Genomic DNA was prepared according to the manufacturer's protocol using a QIAamp tissue kit (QIAGEN GmbH, Hilden, Germany). Final DNA concentration was determined with the use of a PicoGreen dye-based fluorescence assay (Molecular Probes, Leiden, The Netherlands).
Isolation of Total RNA from Human Liver Tissue. Total RNA was prepared from 25 to 50 mg of liver tissue using the QuickPrepTotal RNA Extraction Kit (Amersham Biosciences, Freiburg, Germany). RNA integrity was examined and ensured by 1.2% agarose-formaldehyde gel electrophoresis. The concentration of purified RNA was determined spectrophotometrically, as was the purity, by the 260/280 absorbance ratio. To eliminate any contamination of chromosomal DNA, the isolated total RNA was treated with DNase using RQ1 RNase-Free DNase (Promega, Madison, WI). The DNase-treated RNA was then subject to phenol extraction to eliminate carryover of magnesium that can inhibit subsequent cDNA synthesis. Subsequently, RNA was dissolved in diethyl pyrocarbonate-H2O together with the ribonuclease inhibitor RNasin.
First Strand cDNA Synthesis. cDNA was prepared from 2 μg of DNase-treated total RNA using the SuperScript Preamplification System for First Strand cDNA Synthesis (Invitrogen, Stockholm, Sweden) and random hexamer primers.
Real-Time Quantitative PCR Detection.CYP3A4, CYP3A5, CYP3A43, and PXR mRNA expression was quantified in 46 livers. Quantitative real-time PCR assay of transcripts was carried out with the use of gene-specific double fluorescent labeled probes in a 7700 Sequence Detector (Applied Biosystems, Foster City, CA). The probes were labeled with VIC as the 5′-fluorescent reporter and 5-carboxytetramethylrhodamine at the 3′-end as the quencher. The sets of primers and probes for CYP3A4, CYP3A5, CYP3A43, and PXR (Table 1) were designed to span exon junctions to prevent detection of any possible contamination of genomic DNA. The specificity of the CYP3A primers and probes was verified by the lack of amplification from plasmids containing other CYP3A cDNAs than the intended target. The optimized concentrations of primers and probes were as follows: CYP3A4 forward primer (F) 900 nM, reverse primer (R) 900 nM, and probe (P) 250 nM; CYP3A5 (F) 400 nM, (R) 400 nM, and (P) 100 nM; CYP3A43 (F) 300 nM, (R) 900 nM, and (P) 100 nM; PXR (F) 300 nM, (R) 300 nM, and (P) 175 nM; and huPO (F) 200 nM, (R) 200 nM, and (P) 100 nM. The primers and probes used for CYP3A5 mRNA quantification do not distinguish between the properly spliced mRNA resulting from the CYP3A5*1 genotype and the aberrantly spliced mRNA, which is a consequence of the CYP3A5*3 genotype. However, it was recently shown that measurement of total CYP3A5 mRNA is an acceptable substitution for CYP3A5*1 mRNA (wild-type mRNA) since CYP3A5*1 mRNA levels highly correlate with total CYP3A5 mRNA levels (Lin et al., 2002). Primers and probes for CYP3A4 do not distinguish between CYP3A4*1 and known mutant alleles. Primers and probes for the human PXR were designed to exclude amplification of the PXR variant mRNA that contains an in-frame deletion of 111 nucleotides (Dotzlaw et al., 1999; Fukuen et al., 2002).
Sequences and positions of primers and probes used for real-time quantitative PCR.
Standard curves were constructed by serial 10-fold dilutions, ranging from 1.0 fg to 10 pg, of a plasmid containing the cDNA of interest as template. For normalization of the mRNA data, the endogenous control huPO (human acidic ribosomal phosphoprotein), validated for stable expression in liver, was used. Since equal amplification efficiency in the product formation of CYP3A4, CYP3A5, CYP3A43, and PXR targets was achieved, both when using plasmids and diluted liver samples as standard curves, plasmids were selected as standards for determining the arbitrary units for the respective target. The PCR reaction mixture contained 1 μl of cDNA, 1× TaqMan Universal PCR Master Mix (Applied Biosystems) and optimized concentrations of primers and probes (see above). After two initial steps of 50°C for 2 min and 95°C for 1 min, the PCR reaction ran 40 cycles of 95°C for 15 s and 60°C for 1 min. Each sample was analyzed twice in triplicate, and data were analyzed using the Sequence Detector V1.6 program (Applied Biosystems).
Western Blot Analysis. Quantification of CYP3A4 and CYP3A5 protein in human liver microsomes was performed by immunoblotting analysis using antibodies raised against peptides representing the C terminus of each enzyme. The procedures for antibody production and characterization are described elsewhere (Edwards et al., 1998). Conjugates consisting of the peptides linked to the carrier protein lysozyme were used as standards. The specificities of the antibodies were tested using heterologously expressed CYP3A4, CYP3A5, and CYP3A7 (BD Gentest, Woburn, MA). The antibody raised against the C terminus of CYP3A4 showed reactivity with CYP3A4 and CYP3A7 because the pentapeptide used in antibody development represents the C terminus of both CYP3A4 and CYP3A7, whereas the antibody raised against the C terminus of CYP3A5 did not react with any of the other CYP3A isoforms.
The level of CYP3A4 and CYP3A5 was determined in two separate human liver microsomal samples that contained relatively high levels of CYP3A4 and CYP3A5. Quantification was achieved using lysozyme-peptide conjugates as standards and antisera depleted of antibodies reacting with the linker group. The amount of peptide antigen in each conjugate was determined by SDS-polyacrylamide gel electrophoresis, using 15% gels and Coomassie blue staining to identify bands corresponding to unconjugated lysozyme and lysozyme conjugated to one, two, three, or more molecules of peptide. The relative intensity of bands was measured by densitometry and the average number of peptides conjugated per mole of lysozyme was calculated.
Subsequently, the two microsomal samples were used to produce a concentration curve for quantification of CYP3A4 and CYP3A5 in the liver bank samples. These samples were then separated, together with 10 μg of microsomal samples with unknown amounts of CYP3A4 and CYP3A5 by 8.5% SDS-polyacrylamide gel electrophoresis using the Mini-PROTEAN II electrophoresis cell (Bio-Rad, Sundbyberg, Sweden). After transferring of the proteins to Hybond-C nitrocellulose membranes (Amersham Biosciences), the membranes were incubated with a 1:4000 dilution of the primary antibody and a 1:1000 dilution of horseradish peroxidase-conjugated protein A as secondary antibody (Bio-Rad). The signals were visualized by the SuperSignal West Pico Chemiluminescence method (Pierce Chemical, Rockford, IL) and scanned using LAS-1000 (Fujifilm, Dusseldorf, Germany). The relative intensity of each band was determined by Image Gauge V3.6, and the CYP3A4 and CYP3A5 content for each microsomal sample was determined by the linear slope produced by the microsomal sample used as a standard.
The effect of quenching of the signal by the amount of protein loaded for Western blotting was investigated by loading recombinantly expressed CYP3A5 together with different amounts of liver microsomes lacking CYP3A5. When higher amounts than 20 μg of total protein were used, the linearity was impaired. Thus, no more than 20 μg of total protein of any standard or sample was used in any Western blot analysis. All liver microsomal samples were assessed for CYP3A7 protein using an enzyme-specific antibody raised against a peptide of 11 amino acids corresponding to a CYP3A7-specific sequence.
Identification of CYP3A5*1 and CYP3A5*3 by Allele-Specific PCR. For detection of CYP3A5*1 and CYP3A5*3 (either allele A, B,or C), an allele-specific PCR was developed. CYP3A5 gene-specific primers, 5′-TTATAAGGTGGTCTCAGCCAAT-3′ (CYP3A5-3 F1) and 5′-CAGGGAGTTGACCTTCATACGTT-3′ (CYP3A5-3 R1), specifically amplified a fragment of the region of interest. Genomic DNA (50 ng) was used as template for a first PCR starting with 60 s at 95°C, followed by 35 cycles of 15 s at 95°C, 20 s at 58°C, 60 s at 72°C, and a final extension of 7 min at 72°C. This PCR product was diluted 10 times and served as template for the subsequent allele-specific PCR reaction separating CYP3A5*1 and CYP3A5*3 (A, B, or C) alleles; the A>G polymorphism at 6986 (NG_000004.1), where 6986G causes splice variants, was assessed using 5′-CATGACTTAGTAGACAGATGA-3′ (3A5-3 F5) together with either 5′-CAGGGAAGAGATAC-3′ (3A5-3 R MUT) for identification of the CYP3A5*3 allele or 5′-CAGGGAAGAGATAT-3′ (3A5-3 R WT) for identification of the CYP3A5*1 allele. The PCR condition for the nested allele-specific PCR was identical to the first PCR with the exceptions that it was only run for 14 to 15 cycles and with an annealing temperature of 48°C. The CYP3A5*3 A, B, and C allelic variants will be collectively referred to here as the CYP3A5*3 allele.
Sequencing for Detection of the CYP3A5*6 Allelic Variant. Genomic DNA (∼50 ng) was used for gene-specific amplification of the whole CYP3A5 exon 7 and parts of intron 6 and intron 7 using primer 5′-GGTGTTGACAGCTAAAGTGTG-3′ (3A5-6 R3) and primer 5′-TAGATGGAAGATGATTCAGCAGA-3′ (CYP3A5-6 F5). The amplified product of 525 base pairs was purified (Wizard, Promega) before sequencing in both directions, using the forward primer 5′-TCTGAAAGTCTGTGGCTCTTTATG-3′ (CYP3A5-6 F4) and the reverse primer 5′-TGGAATTGTACCTTTTAAGTGGA-3′ (3A5-6 R SEQ), respectively. Approximately 50 ng of purified product was used in a cycle sequencing reaction using the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction kit (PerkinElmer, Boston, MA), and the precipitated product was analyzed on an ABI PRISM 377 DNA sequencer (PerkinElmer). The resulting sequences were analyzed for CYP3A5*6 using CHROMAS 2.13 (Technelysium Pty. Ltd., Helensvale, QLD, Australia).
Statistical Analysis. Spearman rank (rs) nonparametric correlation analysis was performed using GraphPad InStat version 3.0 for Windows 95 (GraphPad Software, San Diego, CA). The significance of differences in median protein, mRNA, or activity levels between different groups was assessed using the two-tailed nonparametric Mann-Whitney U test. Linear regression was performed using Excel 97 (Microsoft, Kista, Sweden).
Results
CYP3A4 and CYP3A5 Protein Expression in Relation to CYP3A5 Genotype. Liver samples from 46 individuals of Caucasian origin were genotyped for CYP3A5*1 and CYP3A5*3 by allele-specific PCR. Four individuals heterozygous for the wild-type CYP3A5*1 allele (allele frequency 4.3%) were identified, whereas the remaining 42 individuals were homozygous for the CYP3A5*3 (A, B, or C) allele. The presence of the CYP3A5*6 allele among these liver samples was also investigated; however, none carried this allele as determined by sequence analysis (see Materials and Methods).
The level of CYP3A4 and CYP3A5 protein was quantified using antisera raised against the C terminus of each enzyme and carrier protein-coupled peptides as standards. Furthermore, the extent of quenching was taken into account as described under Materials and Methods. The CYP3A5 protein was expressed at quantifiable levels in 5 (10.9%) of the 46 livers (Fig. 1). The four individuals heterozygous for the CYP3A5*1 allele expressed CYP3A5 at levels from 17.9 to 48.4 pmol/mg (Table 2), whereas one individual homozygous for the CYP3A5*3 allele expressed a smaller amount of CYP3A5, 6.6 pmol/mg. The remaining livers had less than 6 pmol/mg CYP3A5, which constituted the limit of detection under the conditions used. For some of the liver microsomal samples, there was a faint band indicating trace amounts of CYP3A5 protein.

Immunoblotting analysis of CYP3A5 and CYP3A4 protein levels in 10 μg of human liver microsomes with a, HL110; b, HL118; c, HL120; d, IIAM1101931; e, HL55; f, HL41; g, HL52; h, HL53; and i, HL124.
High CYP3A5 expression phenotypes are displayed with corresponding CYP3A5 genotypes.
List of human liver samples
CYP3A4 protein was detected when present at a level higher than 15 pmol/mg, which was the limit of detection, in 42 of the samples of liver microsomes. The median CYP3A4 protein amount among these 42 samples was 155.8 pmol/mg (15.2– 600.7); thus, the variability of CYP3A4 protein expression among quantified samples was >40-fold. The livers from IIAM had been selected for having high or low testosterone hydroxylase activity, and exclusion of those revealed a median CYP3A4 protein expression (n = 32) of 145.5 pmol/mg (15.2– 491.4) and a 32-fold variability. Based on these quantifications, it can be calculated that individuals heterozygous for the CYP3A5*1 allele expressed CYP3A5 at a level 15 to 37% of that of CYP3A4, which accounts for 13 to 27% of total CYP3A protein in these four livers (Table 2, Fig. 2). Overall, the combined contribution of CYP3A5 protein to total pool CYP3A protein in the liver bank was only 2% on the basis of the limit of detection. None of the livers expressed significant amounts of CYP3A7 (data not shown).

Contribution of CYP3A4 and CYP3A5 protein to total CYP3A content in human liver microsomes.
CYP3A5*1/*3 genotypes are displayed with the remainder of samples being CYP3A5*3 homozygous.
There was no correlation between CYP3A5 and CYP3A4 protein expression in livers displaying a CYP3A5*1/*3 genotype, and there was no statistically significant difference in median levels of total CYP3A protein in those individuals displaying CYP3A5*1/*3 genotype (200.4 pmol/mg) compared with those displaying CYP3A5*3/*3 genotype (155.8 pmol/mg) among liver samples with protein expression above the level of detection. Also, no statistically significant difference in median testosterone 6β-hydroxylation was achieved between the same groups. This was true whether the selected livers were included in the analysis or not.
Quantification and Correlation of CYP3A and PXR mRNA Expression and Testosterone 6β-Hydroxylation. The level of expression of CYP3A4, CYP3A5, CYP3A43, and PXR mRNA was determined by quantitative real-time PCR. Of total CYP3A transcripts measured in all livers (n = 46), CYP3A4 accounted for 96%, CYP3A5 for 3.9%, and CYP3A43 for 0.1%. The variability in CYP3A4 mRNA was very high, 860-fold among the randomly collected liver samples (n = 32) and 2900-fold among the selected liver samples, even when an extreme outlier with respect to mRNA expression of CYP3A4 and PXR (923,857 and 8,755 arbitrary units, respectively) was excluded (n = 13). The variation in CYP3A5 mRNA expression was considerably lower, approximately 50-fold in both groups. This can be compared with the variability in the mRNA of the low-expressed CYP3A43, which was 400-fold in both sets of human liver samples. PXR transcripts exhibited, as did CYP3A4, a significant difference in the variability between the randomly collected and selected livers, 18-fold and 150-fold, respectively. This was also true for 6β-hydroxylation of testosterone, which varied by a factor of 16 and 80, respectively.
For CYP3A5*1/*3 genotypes, the CYP3A5 transcripts constituted on average 10% of the total CYP3A transcripts measured. The median level of hepatic CYP3A5 mRNA was 3.7-fold higher in CYP3A5*1/*3 individuals than in individuals with a CYP3A5*3/*3 genotype (Fig. 3). The median level of CYP3A4 mRNA was 12-fold higher than that of CYP3A5 mRNA in CYP3A5*1/*3 individuals.

Statistically significant difference (p < 0.001) between median CYP3A5 mRNA levels in human livers with CYP3A5*3/*3 and CYP3A5*1/*3 genotype.
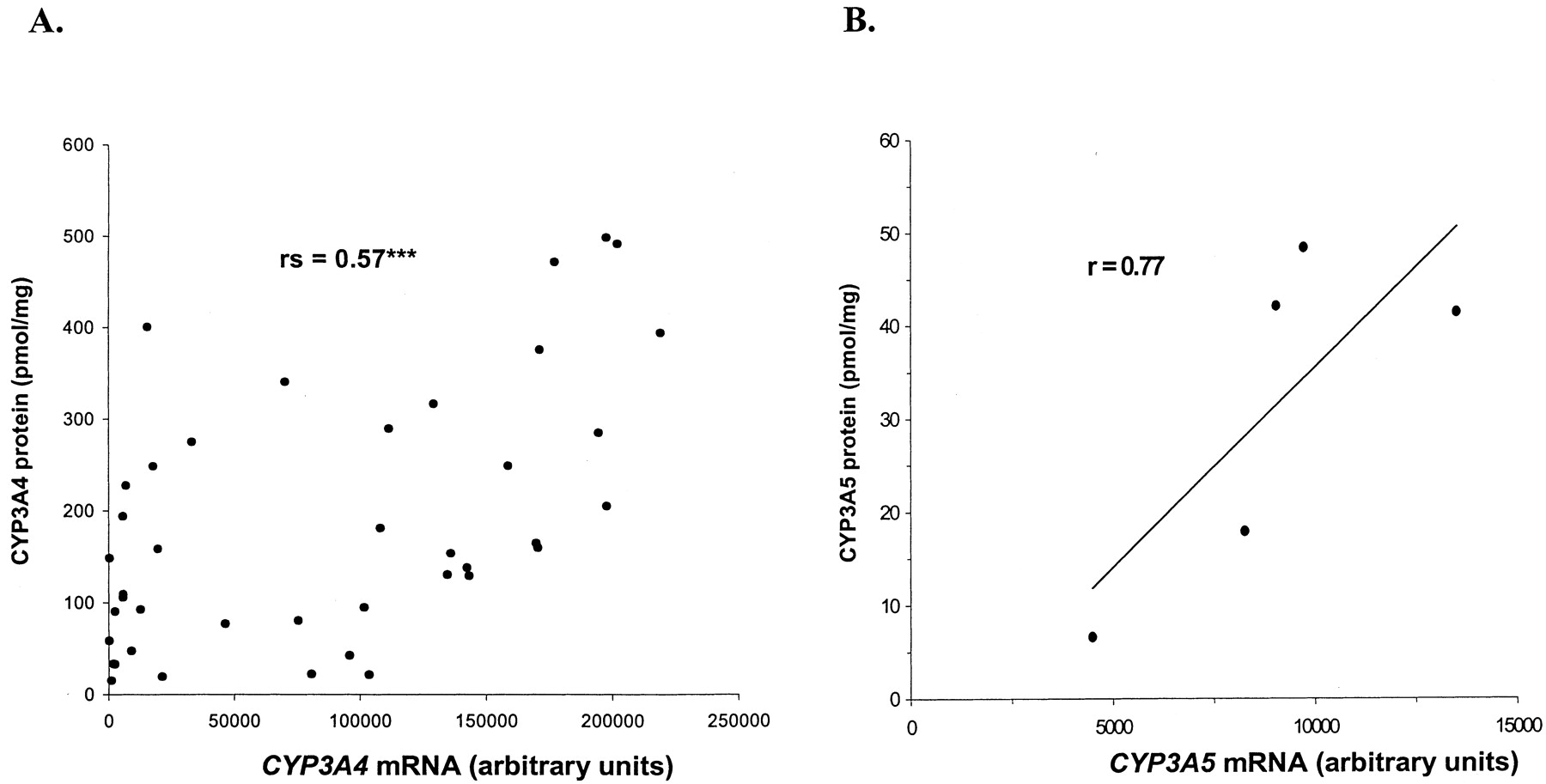
A significant correlation (Table 3) was found when CYP3A4, CYP3A5, CYP3A43, and PXR mRNA levels were related to each other. The extreme outlier with respect to expression of CYP3A4 and PXR mRNA was excluded in the correlation analyses of these transcripts. CYP3A4 mRNA expression (n = 41) correlated significantly with CYP3A4 protein expression, rs = 0.57 (0.31– 0.75, 95% CI), p < 0.0001. However, we found no correlation between CYP3A5 mRNA and CYP3A5 protein levels in those livers where protein could be detected (n = 5), although a linear regression coefficient of r = 0.77 was achieved (Fig. 4). The CYP3A4 protein expression (n = 42) was correlated significantly with testosterone 6β-hydroxylation activity, rs = 0.78 (0.62– 0.88, 95% CI), p < 0.0001 (Fig. 5). 6β-Hydroxylation of testosterone mainly reflects CYP3A4 activity, since CYP3A5 is less capable of catalyzing this reaction (Williams et al., 2002). Also CYP3A4 mRNA (n = 45) correlated with testosterone 6β-hydroxylation, rs = 0.63 (0.40 – 0.78, 95% CI), p < 0.0001, whereas there was only a very weak correlation between PXR mRNA (n = 45) and testosterone 6β-hydroxylation activity, rs = 0.33 (0.03– 0.57, 95% CI), p < 0.05.
Covariation of mRNA expression of CYP3As and PXR with exclusion of an outlier with respect to CYP3A4 and PXR mRNA

A, Spearman rank correlation for CYP3A4 protein and mRNA (***, p < 0.001) and B, linear regression for CYP3A5 protein and mRNA, with exclusion of livers with CYP3A protein expression beneath the level of detection and the outlier in CYP3A4 mRNA.

Spearman rank correlation of CYP3A4 protein with testosterone 6β-hydroxylation (***, p < 0.001).
No significant gender differences in the expression of either mRNA or protein and no activity was found when the female outlier in CYP3A4 and PXR mRNA levels was excluded.
Discussion
This study was designed to quantitatively characterize the hepatic expression and interindividual variation of members of the CYP3A subfamily, and the regulating factor PXR to find clues concerning the mechanisms of expression. Moreover, a major aim was to determine the relative level of expression of CYP3A5 and its contribution to CYP3A activity and variability using novel tools as C terminus-specific antibodies and standards consisting of carrier protein-coupled peptides. The use of peptide conjugates as standards for quantification of protein expression instead of recombinant cytochromes P450 eliminates the risk of underestimating the amount of apoprotein in spectrophotometrically determined cytochromes P450 produced by heterologous expression systems. Also, protein modifications specific for different expression systems can interfere with the affinity of different antibodies (Hustert et al., 2001). Furthermore, to achieve an accurate quantification of protein expression using immunoblotting, the effect of quenching on linearity needs to be considered since loading high amounts of protein results in impaired linearity.
The results confirmed that a high expression of CYP3A5 mRNA and protein cosegregates with the CYP3A5*1 genotype. However, our results of a relative contribution of 13 to 27% of CYP3A5 to total CYP3A protein in individuals carrying at least one CYP3A5*1 allele is considerably lower than that reported by Kuehl et al. (2001). Indeed, in the whole bank of 46 livers, CYP3A5 only accounted for, in total, 2% of the amount of CYP3A protein determined. It appears that Kuehl et al. (2001) might have overestimated the levels of CYP3A5 protein (60 –205 pmol/mg for CYP3A5*1/*3 genotypes and accounting for over 50% of total CYP3A protein in one third of Caucasian livers), possibly due to problems with the methods of quantification. In a recent publication from the same group (Lin et al., 2002), the total contribution of CYP3A5 to CYP3A protein comprised 23%. In our hands, we found CYP3A5 and CYP3A4 protein levels of 6.6 to 48.3 and 15.2 to 600.7 pmol/mg, respectively. These values agree relatively well with previous data published by other authors (Wrighton et al., 1990; Shimada et al., 1994; Paine et al., 1997; Wandel et al., 1998; Tateishi et al., 1999; Snawder and Lipscomb, 2000; Hirota et al., 2001). The interindividual variability seen in CYP3A4 and CYP3A5 mRNA expression levels was much higher than that observed for the amount of protein and testosterone hydroxylase activity. Indeed, we found a descending magnitude of variability at each level of expression studied, mRNA > protein > activity.
With respect to the amount of mRNA transcripts, we found that CYP3A5 mRNA accounted for 3.9% of the total CYP3A mRNAs. This is similar to data presented by Koch et al. (2002), who found that CYP3A5 contributed on average 2% of the CYP3A mRNA pool (Koch et al., 2002).
It has been suggested that 60 to 90% of the variation in CYP3A4 activity is under genetic control (Ozdemir et al., 2000) and that unidentified genetic variants of CYP3A4 or genes elsewhere in the genome would contribute to the variability in CYP3A4 expression. Accordingly, it has been suggested that the polymorphism of the CYP3A5*1 allele, responsible for the expression of CYP3A5 protein, could contribute considerably to total CYP3A activity and variability. However, our data, based on the quantification of CYP3A5 using C-terminal peptide-specific antibodies, suggest that this is not the case. Indeed, a recent in vivo study failed to show any difference in midazolam and 1-hydroxymidazolam pharmacokinetics between homozygous CYP3A5*3/*3 individuals and heterozygous CYP3A5*1/*3 individuals (Shih and Huang, 2002). Thus, we conclude that polymorphic expression of CYP3A5 is not likely to have substantial impact on the quantitative overall hepatic CYP3A metabolism and is not a major determinant for the variability in CYP3A catalytic activity in a Caucasian population, although high expression of CYP3A5 could influence the metabolic profile in individuals carrying the CYP3A5*1 allele.
We found a significant coregulation of the CYP3A transcripts and of PXR mRNA. The basis behind this is unknown, but a coregulation of the CYP3A transcripts is in accordance with results presented by Lin et al. (2002). It might be suggested that a common factor governs the expression of all genes in the CYP3A locus. PXR, having bile acids as endogenous ligands (Schuetz et al., 2001; Staudinger et al., 2001; Xie et al., 2001; Kliewer and Willson 2002), is a key factor in the regulation of CYP3A expression. However, experiments in PXR-null mice show that the presence of PXR does not alter the constitutive expression of murine CYP3A proteins (Xie et al., 2000). This indicates that other transcription factors are of great importance in this respect and that variability in such a factor could possibly explain the variability seen in expression and activity. Other results have been indicative of a compensatory elevation in the basal CYP3A11 expression in PXR-null mice, suggesting that the absence of PXR receptor binding to the PXR motif would enable other factors with higher basal transcriptional activities to exert higher transcriptional impact (Staudinger et al., 2001).
It is likely that not all variability seen within the liver bank examined is due to variation in constitutive control of expression. Three of the livers, with higher than average levels of testosterone activity, originated from individuals who were reported to have taken glucocorticoids and/or phenytoin during their hospital stay. One of these exhibited very high expression levels and was considered an outlier above, in particular, with respect to the level of CYP3A4 and PXR mRNA expression.
In conclusion we find a high extent of coregulation of the various transcripts in the CYP3A locus, implicating variation in expression of common regulatory factors as a basis for the important interindividual variability seen among the members of this locus, and our data indicate that CYP3A5 does not contribute to such variability due to its very low abundance in Caucasian livers.
Acknowledgments
We are indebted to Brith Leidvik for initial work with the real-time PCR optimizations. We also thank Prof. Rune Toftgård at the Department of Bioscience, Center for Nutrition and Toxicology, Karolinska Institutet, for access to the 7700 Sequence Detector (Applied Biosystems).
Footnotes
-
↵ 1 These two authors contributed equally to this work
-
↵ 2 Abbreviations used are: PXR, pregnane X receptor; IIAM, International Institute for the Advancement of Medicine; PCR, polymerase chain reaction; huPO, human acidic ribosomal phosphoprotein; rs, Spearman rank correlation; CI, confidence interval.
-
This work was supported by grants from The Swedish Research Council and from AstraZeneca AB.
- Received January 2, 2003.
- Accepted March 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}