


Abstract
This article is an updated report of a symposium held at the June 2000 annual meeting of the American Society for Pharmacology and Experimental Therapeutics in Boston. The symposium was sponsored by the ASPET Divisions for Drug Metabolism and Molecular Pharmacology. The report covers research from the authors' laboratories on the structure and regulation of UDP-glucuronosyltransferase (UGT) genes, glucuronidation of xenobiotics and endobiotics, the toxicological relevance of UGTs, the role of UGT polymorphisms in cancer susceptibility, and gene therapy for UGT deficiencies.
For most xenobiotics and many endobiotics, glucuronidation constitutes a major route of elimination and thereby may substantially modulate substrate concentrations and effects. In some cases, glucuronidation forms the biologically active molecule. Recent studies have revealed an extensive superfamily of UDP-glucuronosyltransferases (UGTs),2 previously termed glucuronyl transferases, which catalyze the conjugation of UDP-glucuronic acid with lipid-soluble substrates to form polar conjugates that are excreted in the urine and feces. These studies have provided fundamental insights into UGT gene structure and regulation, isozyme substrate selectivity, and interindividual variability. Whereas there remains much to learn about the potential biological relevance of UGTs, deficient glucuronidation can result in either elevated tissue concentrations and direct toxicity of substrates, as with the endobiotic bilirubin, or, alternatively, enhanced bioactivation of substrates to toxic reactive intermediates, as in the case of acetaminophen and benzo[a]pyrene. Interindividual UGT variability likely plays an important role in drug efficacy and xenobiotic toxicity, as well as in hormonal regulation and certain diseases, which in some cases may be amenable to therapeutic manipulations including gene therapy.
Structure and Tissue-Specific Regulation of UGT Genes (P.I.M., P.A.G., Y.I., A.J.H.)
The UGT content of cells and tissues is a major determinant of our response to those chemicals that are primarily eliminated by conjugation with glucuronic acid. There are marked interindividual differences in the content of UGTs in the liver and other organs including the gastrointestinal tract. For example, only one third of the population appears to express UGT1A1, UGT1A3, and UGT1A6 in their gastric epithelium (Strassburg et al., 1998). Studies on the mechanisms that regulate UGT genes, in a temporal and tissue-specific manner, should contribute significantly to understanding the basis for these differences. Such studies should also aid in the design of molecular probes to assess the capacity of individuals to metabolize specific drugs and toxins, before exposure to these agents.
The genes encoding UGTs that use UDP-glucuronic acid as sugar donor have been assigned to two families (Mackenzie et al., 1997). The UGT1 family constitutes a complex gene locus on human chromosome 2q37 and comprises 13 first exons that encode the unique N-terminal domains of the UGT1A proteins and exons 2 to 5 that encode the C-terminal domain, which is identical in all UGT1A family members (Owens and Ritter, 1992). The UGT2 family, in contrast, is encoded by separate genes clustered on chromosome 4q13 and consists of the UGT2A and UGT2B subfamilies. The UGT2B genes that have been analyzed to date, viz., rat UGT2B1 (Mackenzie and Rodbourn 1990) and UGT2B2 (Haque et al., 1991), and human UGT2B4 (Monaghan et al., 1997), UGT2B7 (Ishii et al., 2000), UGT2B15 (Turgeon et al., 2000), and UGT2B17 (Beaulieu et al., 1997), consist of six exons and have highly similar exon/intron boundaries. Accumulating evidence demonstrates that many of the 17 human UGTs characterized to date exhibit tissue-specific patterns of expression. For example, UGT1A3 is found in the liver, kidney, and prostate, and throughout the gastrointestinal tract, whereas UGT1A8 is mainly restricted to the colon (Mojarrabi and Mackenzie, 1998). The factors that govern this specificity of UGT expression remain largely unknown.
Studies on rodent UGT genes have demonstrated that the transcription factors hepatocyte nuclear factor 1 (HNF1) and CAAT-enhancer binding protein are important positive regulators of UGT expression in the liver (Hansen et al., 1997, 1998). These nuclear proteins, which are members of the homeodomain and basic region-leucine zipper groups of transcription factors, respectively, have been shown to be important in the liver-specific expression of other genes such as albumin (Lichtsteiner et al., 1987). Furthermore, mice in which the hepatic expression of these factors has been substantially reduced display deficiencies in hepatic UGT1A1 and UGT2B expression (Lee et al., 1997). With the recent isolation and characterization of several human UGT genes, evidence is accumulating that many of the factors that contribute to liver-specific expression of UGTs in rodents are also important in regulating UGTs in human liver. This is exemplified by studies on the human UGT2B7 gene (Ishii et al., 2000). Studies on this UGT gene were initiated since UGT2B7 glucuronidates many important therapeutic drugs, including nonsteroidal anti-inflammatory drugs, morphine, oxazepam, and zidovudine, and is found in both the liver and the gastrointestinal tract (Radominska-Pandya et al., 1999).
The proximal promoter of the UGT2B7 gene contains an A/T-rich region just upstream from the transcription start site. Cotransfection experiments in the human liver-derived cell line, HepG2, and gel electrophoretic mobility shift assays demonstrated that HNF1α bound to this region to enhance UGT2B7 promoter activity. The closely related transcription factor HNF1β had little effect on UGT2B7 promoter activity. Furthermore, mutations in the A/T-rich region abolished the capacity of HNF1α to enhance promoter activity. Further investigation is required to define other factors that regulate the UGT2B7 gene, either independently or via interactions with HNF1α. In this respect, it is interesting to note that the ubiquitous transcription factor, octamer transcription factor-1, acts synergistically with HNF1α to enhance UGT2B7 promoter activity in HepG2 cells. Octamer transcription factor-1 is another member of the homeodomain group of transcription factors. It acts as a positive regulator of UGT2B7 expression by tethering to HNF1α and presumably promoting assembly of the RNA polymerase complex, rather than by directly binding to DNA (Ishii et al., 2000).
A comparison of the proximal promoter sequences of several human UGT genes reveals the presence of a similar A/T-rich region in many of these genes (Fig. 1). The binding of HNF1α to this sequence has been confirmed for the UGT1A1 and UGT2B17 genes (Bernard et al., 1999; Gregory et al., 2000). It is interesting to note that this putative HNF1 binding site is present in a similar position in the proximal promoters of those UGT genes that are expressed in the liver, but it is not present in the same position in those genes expressed exclusively in extrahepatic tissues.

Alignment of UGT proximal promoter sequences.
The sequences, including those of UGT pseudogenes (denoted by P), were obtained from GenBank, accession numbers AF180372 (UGT1A1), M84126 (UGT1A2P), M84127 (UGT1A3), M84128 (UGT1A4), M84129 (UGT1A5), AF014112 (UGT1A6), U39570 (UGT1A7), U42604 (UGT1A8), U39550 (UGT1A10), U39551 (UGT1A11P), U39552 (UGT1A12P), AF179875 (UGT2B4), AF282881 (UGT2B7), AF179873 (UGT2B11), AF179881 (UGT2B15), AF179874 (UGT2B17), AF179876 (UGT2B24P), AF1798787 (UGT2B25P), AF179878 (UGT2B26P), AF179879 (UGT2B27P), and AF179880 (UGT2B28P). The putative HNF1 binding site is shown in italics. The sites that have been demonstrated experimentally to bind HNF1 protein are underlined. Note that UGT1A7, UGT1A8, and UGT1A10, which are not expressed in the liver, do not contain a consensus HNF1 binding site in a position equivalent to that of the other genes.
Although many UGTs are expressed in extrahepatic tissues, the mechanisms that regulate their tissue-specific expression have not been identified. The prostate contains UGT2B17, which is thought to play an important role in regulating intracellular androgen concentrations (Guillemette et al., 1997). The prostate-derived LNCap cell line contains large amounts of this enzyme and, hence, is a suitable model to investigate UGT2B17 regulation. The UGT2B17 gene promoter is substantially activated by exogenous HNF1α in transfected LNCaP cells. However, unlike HepG2 cells, LNCaP cells do not contain endogenous HNF1α but do contain the related transcription factor HNF1β. The latter appears to be a positive regulator of UGT2B17 expression in these cells and exerts its effects through a site that is distal to the HNF1α binding site utilized in HepG2 cells (Gregory et al., 2000). Factors specifying the expression of UGTs in other tissues remain to be identified.
Much work is required to define the mechanisms that contribute to the differential expression of UGT genes in the liver and other organs. Nevertheless, it is now evident that different factors and different combinations of factors regulate the same promoter in different cell types.
Glucuronidation of Xenobiotics and Endobiotics (J.K.R., F.K.K.)
Progress is continuing in cloning and establishing the substrate specificities of the individual UGT enzymes that catalyze this reaction. Their identification is essential for understanding why individuals vary in their rates of glucuronidation, altering the pharmacological and toxicological responses to different agents. Currently, there are ∼20 distinct UGTs known in both humans and rats, the best characterized animal species. These can be divided into two families, UGT1 and UGT2, on the basis of sequence homology. Studies in our laboratory have focused on the substrate specificity and regulation of UGT1 isoforms.
The UGT1A1 form appears to be the most abundant UGT1 isoform expressed in liver (Ritter et al., 1992; Ikushiro et al., 1995) and is the principal isoform involved in the glucuronidation of bilirubin (Bosma et al., 1994). UGT1A1 is also selectively active toward certain phenols [e.g., SN-38 (Iyer et al., 1998)] and 17α-ethinylestradiol (Ebner et al., 1993; Senafi et al., 1994). The available evidence points to high interindividual variability in UGT1A1 expression. In a study of UGT1A1 variation in human liver donor samples, marked variation (>50-fold) was observed in the microsomal UGT1A1 protein level (Ritter et al., 1999). Three samples with the highest UGT1A1 level were from patients who had received the anticonvulsant inducing agent, phenytoin, during their hospitalization. These findings are consistent with a report of elevated UGT1A1 mRNA in the liver of a phenytoin-exposed patient. They also agree with clinical evidence supporting the effectiveness of phenobarbital in inducing bilirubin elimination in patients with unconjugated hyperbilirubinemia.
Further evidence in support of environmental influences on UGT1A1 was provided by studies using primary human hepatocytes. After adaptation to culture, a common environment, UGT1A1 mRNA levels showed lower variability (<3- versus 50-fold). The three cultures from the phenytoin-exposed donors showed the most pronounced declines in UGT1A1 mRNA, likely due to the removal of inducing stimuli (phenytoin, diet). Induction studies showed that phenobarbital and oltipraz (prototypical inducing agents) resulted in elevated UGT1A1 mRNA, but exposure to 3-methylcholanthrene resulted in the most potent inducing effects (3- to 6-fold). These findings suggest that expression of UGT1A1 in human liver is under the control of multiple control mechanisms including the aryl hydrocarbon receptor. Exposure to polycyclic hydrocarbon-type inducers via cigarette smoking is another potential cause of UGT1A1 variation.
The possible role of the UGT1A1 promoter polymorphism (Bosma et al., 1995) in the observed variation was also investigated by genotyping the donors for the number of TA repeats in their UGT1A1 TATA boxes. A genetic influence on expression was supported by the observation that two individuals with the lowest UGT1A1 expression were homozygotes for the (TA)7TAA allele (Ritter et al., 1999). A third (TA)7TAA homozygote was one of the phenytoin-exposed patients. This patient exhibited lower UGT1A1 levels than did the two other phenytoin-exposed patients, who were both homozygous for the wild-type allele [(TA)6TAA]. These data provide support for both genetic and environmental factors in interindividual variation in hepatic UGT1A1 expression.
The Gunn rat provides a useful animal model to investigate the relative contribution of UGT1 isoforms in total glucuronidating activities. The frame-shift mutation associated with the loss of UGT1A1 activity and hyperbilirubinemia in Gunn rats also inactivates the other UGT1 family isoforms. The contributions of UGT1 isozymes were assessed using various in vitro (microsomal UGT assays) and in vivo approaches (pharmacokinetics and organ toxicity). Two examples are the analgesic and potential hepatotoxicant, acetaminophen (de Morais et al., 1992a), and the toxic environmental pollutant, benzo[a]pyrene (B[a]P) (Hu and Wells, 1992). Establishing the identities of UGT1 isoforms involved in the glucuronidation of specific substrates requires the use of cloned and expressed UGT cDNAs. Using human embryonic kidney cells expressing the major UGT1 isoforms found in rat liver [UGT1A1, UGT1A5, UGT1A6, and UGT1A7 (Ikushiro et al., 1995)], we investigated the selectivities of these isoforms in the glucuronidation of bilirubin, acetaminophen, and B[a]P metabolites. Only the UGT1A1 isozyme was active toward bilirubin. In contrast, two of the four rat liver isoforms tested were active toward acetaminophen (UGT1A6 and UGT1A7) (Kessler et al., 2002). In rats maintained on a standard laboratory diet, UGT1A6 and UGT1A7 are expressed at low levels in liver but are induced after exposure to certain inducers (Grove et al., 1997; Kessler and Ritter, 1997; Kobayashi et al., 1998). These data likely indicate an important role for UGT1A6 and UGT1A7 in protection against acetaminophen-induced hepatotoxicity. The activities of these isoforms resembles the reported activities of human UGT1A6 and UGT1A9 toward acetaminophen (Bock et al., 1993). Apparent differences in the affinity and/or capacity of UGT1A6 (high affinity, low capacity) and UGT1A9 (low affinity, high capacity) for acetaminophen likely indicate that they will contribute differently to protection against acetaminophen in overdose situations. However, the possibility that other UGT1 isoforms besides the phenol UGTs contribute to acetaminophen glucuronidation is suggested by the finding that human UGT1A1 is also significantly active (Court et al., 2001). These results highlight the species-specific nature of drug glucuronidation.
Glucuronidation also is known to modulate toxicities associated with B[a]P exposure (Hu and Wells, 1992; Hu and Wells, 1994]. The high activity of the rat UGT1A7 form toward B[a]P metabolites, including phenols, quinols, and dihydrodiols, has been demonstrated (Grove et al., 1997). Despite its inducibility by polycyclic hydrocarbons, UGT1A6 shows very low activity toward most B[a]P metabolites, a finding consistent with the preference of UGT1A6 for small phenolic compounds with simple ring substitutions. Interestingly, rat UGT1A1 was found to be significantly active. Qualitatively, UGT1A1 resembles the UGT1A7 form in its activity toward many different B[a]P metabolites including B[a]P-7,8-dihydrodiol. These findings are supported by the observation that the human bilirubin UGT is active toward B[a]P-7,8-dihydrodiol(Fang et al., 2002). Although the specific activities of UGT1A1 were generally 2- to 6-fold lower than that of UGT1A7, some activities were higher (e.g., toward B[a]P-4,5-dihydrodiol). The high natural abundance of this form in liver, together with the observation of UGT1A1 inducibility by polycyclic aromatic hydrocarbons, supports a role for UGT1A1 deficiency in the mechanism of increased sensitivity of Gunn rats to B[a]P-induced toxic effects. Variation in UGT1A1 has the potential to alter individual sensitivities to polycyclic aromatic hydrocarbons present in the diet and the environment.
Toxicological Relevance of UGTs (P.G.W., P.M.K.)
Toxicologic Implications of UGT Deficiencies. We have focused upon drugs and environmental chemicals for which toxicity depends upon the bioactivation of the xenobiotic or a stable metabolite by enzymes like the cytochromes P450 (P450s) or prostaglandin H synthases to highly toxic electrophilic and/or free radical reactive intermediates that damage cellular macromolecules (DNA, protein, lipid) and/or enhance oxidative stress (for reviews, see Wells and Winn, 1996; Wells et al., 1997). In such cases, glucuronidation and elimination of the xenobiotic and/or its stable metabolite serves as a toxicological gatekeeper, directing metabolism away from toxifying bioactivation. Furthermore, bioactivation often is a quantitatively minor pathway, often amounting to less than 10 to 15% of overall elimination, whereas glucuronidation usually is quantitatively major, constituting 40 to 75% or more of xenobiotic elimination. Hence, relatively minor deficiencies in glucuronidation theoretically can result in a substantial percentage increase in bioactivation, with toxicological consequences even at therapeutic drug concentrations or putatively safe concentrations of environmental chemicals.
Hepatic and Renal Toxicity. The widely used analgesic drug acetaminophen (paracetamol) at high doses is hepatotoxic and nephrotoxic, and eliminated primarily via UGT-catalyzed glucuronidation. Mutant Gunn and RHA rats, which lack all UGT1A enzymes (reviewed by Iyanagi et al., 1998), had reduced acetaminophen glucuronidation, enhanced P450-catalyzed bioactivation of acetaminophen and covalent binding to hepatocellular proteins, and enhanced hepatocellular centrilobular and renal cellular necrosis (de Morais and Wells, 1988, 1989; de Morais et al., 1992a). Interestingly, the homozygous UGT1A-deficient mutant rats exhibited measurable, albeit substantially reduced, acetaminophen glucuronidation, suggesting the likely contribution of a UGT2 isozyme. In people with a hereditary UGT1A1 deficiency (Gilbert's syndrome) (review: Tukey and Strassburg, 2000), similar studies using intravenous drug administration showed reduced glucuronidation of acetaminophen and, conversely, enhanced bioactivation determined by the formation of glutathione-derived acetaminophen metabolites, although measurable hepatotoxicity was not evident at the therapeutic dose employed (de Morais et al., 1992b). Among all the Gilbert's subjects and controls, a greater reduction in glucuronidation correlated highly with a greater enhancement in bioactivation. Conflicting data have been reported in other studies, with reduced acetaminophen glucuronidation apparent in some Gilbert's subjects and no effect in others (Ullrich et al., 1987; Esteban and Perez-Mateo, 1999). In our study, one subject with Gilbert's syndrome had normal acetaminophen glucuronidation, whereas one of the “normal” subjects without Gilbert's syndrome had deficient acetaminophen glucuronidation and enhanced bioactivation. These findings suggest that acetaminophen glucuronidation is more complex than previously thought, and may be explained by multiple UGT isozymes contributing to its glucuronidation, including UGT1A1 and 1A6. The extent to which each isozyme contributes may differ significantly among individuals, depending on exposure to inducing agents, and genetic and other factors. Interestingly, a recent study found that 8% of people with Gilbert's syndrome also possessed a homozygous deficiency in the UGT1A6 gene (Lampe et al., 1999).
Carcinogenesis. The environmental teratogens and/or carcinogens benzo[a]pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) are representative of a multitude of polycyclic aromatic hydrocarbons and nitrosamines found widely in the environment, particularly in tobacco smoke. Most benzo[a]pyrene and NNK metabolites are eliminated primarily via glucuronidation, catalyzed by several UGT1A and UGT2 isozymes (Grove et al., 2000; Tukey and Strassburg, 2000), which avoids the alternative P450- and/or prostaglandin H synthase-catalyzed bioactivation of the metabolites to toxic reactive intermediates that irreversibly damage DNA (for reviews, see Wells and Winn, 1996; Wells et al., 1997). Using UGT1A-deficient Gunn and RHA rats, the glucuronidation of benzo[a]pyrene metabolites was found to be reduced in vitro and in vivo, resulting in their enhanced bioactivation and covalent binding to both protein and DNA (Hu and Wells, 1992). UGT deficiency was the critical toxicologic determinant, since these animals compared with controls had similar activities of various P450 isozymes and glutathione S-transferase isozymes and, in the case of the RHA strain, the controls were congenic (Hu and Wells, 1992). To estimate carcinogenic risk in UGT-deficient rats, a skin fibroblast model was developed, with genotoxicity and potential carcinogenicity assessed by the formation of micronuclei (Vienneau et al., 1995; Kim and Wells, 1996a). Cultured fibroblasts from UGT-deficient Gunn and RHA rats incubated with either benzo[a]pyrene or NNK had increased oxidative DNA damage and micronucleus formation compared with UGT-normal controls, whereas no increase in micronuclei occurred with benzo[e]pyrene, a noncarcinogenic isomer. Benzo[a]pyrene- and NNK-initiated micronucleus formation was dependent upon P450- and/or peroxidase-catalyzed bioactivation (Kim and Wells, 1996a; Kim et al., 1997a).
Cellular Models for Human Risk Assessment. To facilitate human studies, we have evaluated lymphocytes, which can be obtained in relatively large quantities sufficient for determination of collective UGT activity. To determine whether lymphocytes accurately reflect hepatic activities, lymphocytes and hepatic microsomes were taken/prepared from the same RHA UGT-normal (+/+) and UGT-deficient (j/j) rats (Hu and Wells, 1994). To produce benzo[a]pyrene metabolites as substrates for glucuronidation or ultimate bioactivation, benzo[a]pyrene was preincubated with rat liver microsomes and NADPH, and the supernatant was immediately added to the lymphocyte incubations. Lymphocytes from UGT-deficient rats accurately reflected the decreased glucuronidation of benzo[a]pyrene metabolites and enhanced bioactivation, covalent binding, and cytotoxicity that were observed with hepatic microsomes from the same rats (Hu and Wells, 1994), and with related in vivo studies of benzo[a]pyrene glucuronidation, bioactivation, and covalent binding (Hu and Wells, 1992), and embryotoxicity (Wells et al., 1989). Lymphocytes may constitute a useful model for risk assessment.
In preliminary human studies, lymphocytes from 12 normal volunteers were tested as described in the rat lymphocyte studies above (Hu and Wells, 1993). All subjects had normal UGT activity for bilirubin (i.e., none had Gilbert's syndrome), but there was a 200-fold variability in UGT activities for benzo[a]pyrene metabolites, including two with no measurable activity. Decreasing UGT activity correlated with decreased UGT-dependent protection against benzo[a]pyrene covalent binding and, conversely, with increased cytotoxicity for benzo-[a]pyrene quinones and diols, but not monophenols. A similar protection against benzo[a]pyrene quinones, but not 3-OH-benzo[a]pyrene or benzo[a]pyrene-7,8-dihydrodiol, was shown in human lymphoblastoid cells transfected with rat UGT1A7 (Grove et al., 2000). Our lymphocyte studies suggest that substantial UGT deficiencies for potentially toxic benzo[a]pyrene metabolites are common in the normal population and indicate that these UGT deficiencies may constitute important determinants of toxicologic predisposition, particularly with respect to chemical carcinogenesis and teratogenesis.
Developmental Toxicity. Whereas there is little UGT activity in the rodent embryo, maternal UGT activity theoretically may play an important role in determining how much teratogen reaches the embryo. In preliminary studies, pregnant UGT-deficient Gunn rats were substantially more susceptible to benzo[a]pyrene-initiated embryonic death at a subcarcinogenic dose (25 mg/kg i.p.) that had no effect on UGT-normal Wistar controls (Wells et al., 1989). Similarly, the anticonvulsant drug phenytoin, a human teratogen, and its major metabolite, 5-(p-hydroxyphenyl)-5-phenylhydantoin, caused increased DNA oxidation and micronucleus formation in UGT-deficient cultured rat skin fibroblasts, and in vivo studies indicated that this enhanced genotoxicity was due to decreased N-glucuronidation of phenytoin and O-glucuronidation of 5-(p-hydroxyphenyl)-5-phenylhydantoin in the UGT-deficient rats (Kim et al., 1997b), resulting in increased hydroxyl radical formation (Kim and Wells, 1996b). Preliminary in vivo evidence suggests a similar enhancement in embryopathies in phenytoin-treated pregnant Gunn and RHA UGT-deficient rats treated with phenytoin (Kim and Wells, 1998). In the last trimester of pregnancy, increasing activities of some fetal UGTs may provide the offspring with a second layer of biochemical protection, particularly against functional (as distinct from structural) anomalies, although this has yet to be studied.
General Toxicologic Observations. Overall, several consistent observations emerged from the above studies. First, a relatively small percentage decrease in a quantitatively major pathway of elimination, such as glucuronidation, can produce a disproportionately large percentage increase in bioactivation. This effect is particularly evident if there are no alternative eliminating pathways, or if the alternative pathways are readily saturable, as is the case with sulfation. For example, even in the rat, which has at least twice the sulfating capacity of mice and humans for acetaminophen, there was no compensatory enhancement of acetaminophen sulfation in UGT-deficient rats (de Morais et al., 1992a). The toxicological consequences in humans may be more pronounced.
Second, consistent, progressively greater deficiencies in UGT activity in +/j and j/j RHA rats resulted in corresponding UGT gene dose-dependent decreases in the glucuronidation of both acetaminophen and benzo[a]pyrene in vitro and/or in vivo and, conversely, increasing xenobiotic bioactivation, covalent binding, and toxicity, all of which were reflected in the lymphocyte model. A consistent finding of particular clinical interest is that a heterozygous deficiency in UGTs was toxicologically relevant (de Morais et al., 1992a; Hu and Wells, 1992, 1994), with a risk equivalent to homozygotes in some systems (Kim and Wells, 1996b; Kim et al., 1997b).
Finally, despite our improving understanding of the pharmacogenomic basis for UGT deficiencies and the specific roles of UGT isozymes in xenobiotic glucuronidation (reviews: Tukey and Strassburg, 2000; Guillemette, 2003), the complexity of toxicological risk remains challenging. At a given xenobiotic concentration, individual toxicological susceptibility will depend upon both the levels of the relevant UGT proteins, which vary according to genetic and environmental determinants, and the overall balance among numerous associated biochemical pathways, including elimination via other conjugating pathways, membrane transport, bioactivation, reactive intermediate detoxification, and macromolecular repair, among others. Environmental modulation of the in vivo outcome can be particularly unpredictable, as exemplified by acetaminophen toxicity, which in rodents is reduced by the UGT inducer oltipraz due to enhanced glucuronidation (Davies and Schnell, 1991; Kessler et al., 2002), but conversely is enhanced by pretreatment with the UGT inducers phenobarbital and 3-methylcholanthrene, presumably due to their relatively greater induction of P450-catalyzed bioactivation.
Pharmacogenetics of UGT Enzymes: Implications for Cancer Susceptibility (C.G.)
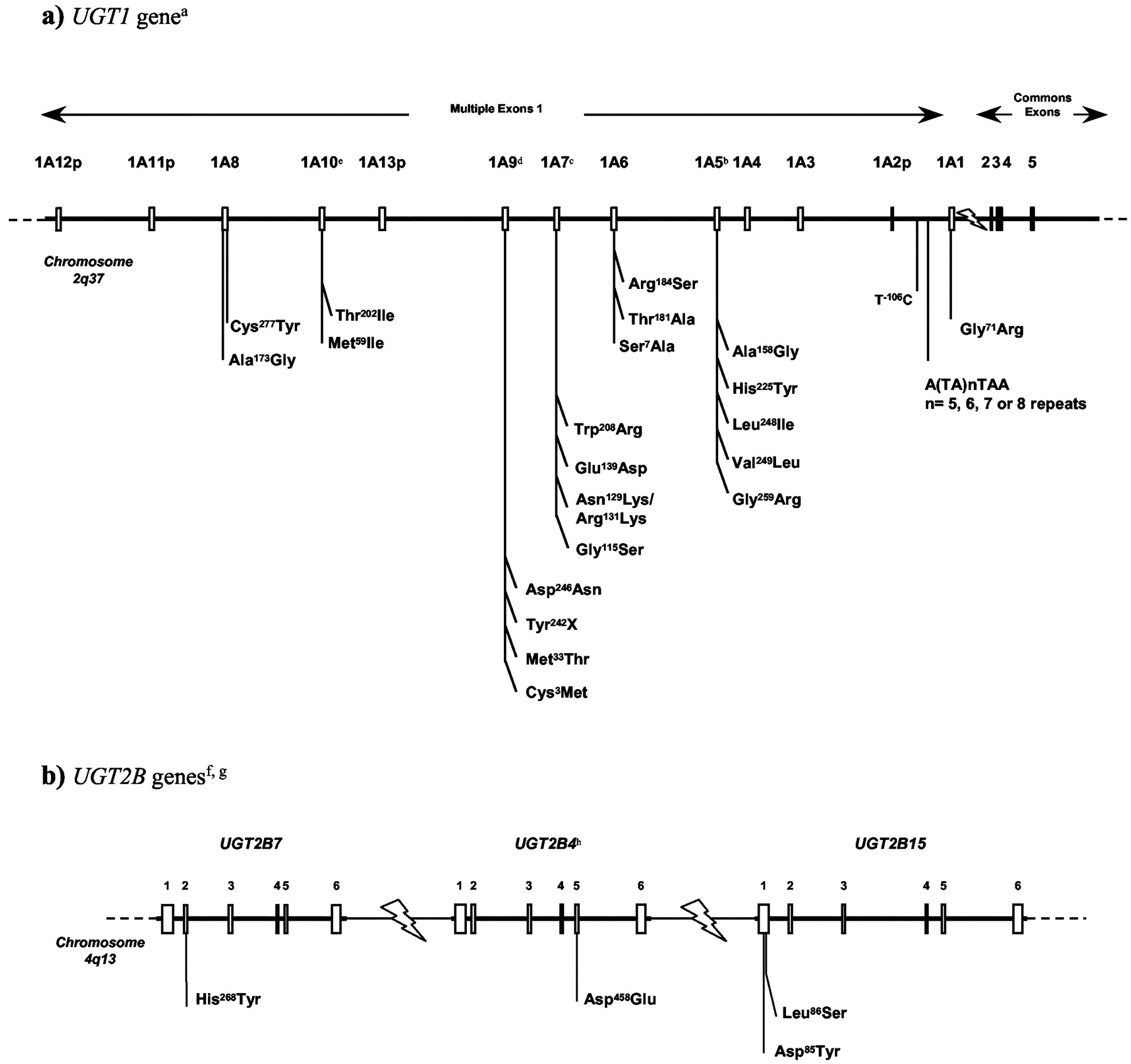
The molecular genetics of the UGT superfamily are well understood (Mackenzie et al., 1997; Gong et al., 2001), but the molecular mechanisms of large interindividual phenotypic variations remain to be elucidated. Breakthroughs in the identification of functional common genetic variations in UGT genes that may impact drug response and/or disease susceptibility are starting to emerge. Polymorphic variations that affect functional activity of the enzyme have been characterized in human UGT1A and UGT2B genes. Similar to other drug-metabolizing enzymes, evidence of differential prevalence among ethnic and racial groups have been observed in human populations (Fig. 2) (reviewed in Guillemette, 2003).

Common genetic polymorphisms in UGT1A and UGT2B genes.
A, common genetic polymorphisms at the UGT1A gene locus. The entire UGT1 family is derived from a single gene locus (UGT1), located on chromosome 2q37, which encodes nine functional proteins (UGT1A1, and UGT1A3 to UGT1A10) and four pseudogenes (UGT1A2p, and UGT1A11p to UGT1A13p) (Ritter et al., 1992; Gong et al., 2001). B, common genetic polymorphisms in functional UGT2B genes. The UGT2B family comprises several distinct genes and pseudogenes, which are not included here. The genomic organization of functional UGT2B genes is not entirely elucidated; therefore, the relative chromosomal localization used here was simplified for schematic purposes. Sequences that differ by less that 3% are considered alleles of the same gene (Mackenzie et al., 1997). To date, allelic variants have been reported for UGT1A1, UGT1A6, UGT1A7, UGT1A8, and UGT1A9, in addition to UGT2B4, UGT2B7, UGT2B15, and UGT2B28. The function and prevalence of some of these variants have also been described (Guillemette et al., 2000, 2001; Woolley et al., 2000; Girard et al., 2003; Beutler et al., 1998; Bosma et al., 1994; Ciotti et al., 1997; Coffman et al., 1998; Lampe et al., 1999, 2000; Guillemette et al., 2000; Hall et al., 1999; Riedy et al., 2000; Lévesque et al., 1997, 1999; Turgeon et al., 2000; Akaba et al., 1998; Maruo et al., 1999; Huang et al., 2002; Villeneuve et al., 2003).A Recent studies support a possible role of UGT in cancer risk (Guillemette et al., 2000, 2001; Zheng et al., 2001; Vogel et al., 2001; Strassburg et al., 2002; Gsur et al., 2002; MacLeod et al., 2000; Gestl et al., 2002), although additional studies are needed to confirm these findings.B. a The structure of the UGT1 gene presented here is based on the GenBank accession number AF297093 (Gong et al., 2001). b UGT1A5 alleles correspond to: UGT1A5*1 Ala158His225Leu248Val249Gly259; UGT1A5*2 Gly158His225Leu248Val249Gly259; UGT1A5*3 Ala158Tyr225Leu248Val249Gly259; UGT1A5*4 Ala158His225Ile248Leu249Arg259; UGT1A5*5 Gly158His225Ile248Leu249Arg259; UGT1A5*6 Ala158Tyr225Ile248Leu249Arg259; UGT1A5*7 Gly158Tyr225Ile248Leu249Arg259. c UGT1A7 alleles correspond to: UGT1A7*1 Gly115Asn129Arg131Glu139Trp208; UGT1A7*2 Gly115Lys129Lys131Glu139Trp208; UGT1A7*3 Gly115Lys129Lys131Glu139Arg208; UGT1A7*4 Gly115Asn129Arg131Glu139Arg208; UGT1A7*5 Ser115Asn129Arg131Glu139Arg208; UGT1A7*6 Gly115Asn129Arg131Glu139Trp208; UGT1A7*7 Gly115Lys129Lys131Asp139Trp208 UGT1A7*8 Gly115Lys129Lys131Asp139Arg208 and UGT1A7*9 Ser115Lys129Lys131Glu139Tr208. d UGT1A9 alleles correspond to: UGT1A9*1 Cys3Met33; UGT1A9*2 Tyr3Met 33; UGT1A9*3 Cys3Thr33 (Villeneuve et al., 2003); UGT1A9*4 Tyr242X (Y. Saito, unpublished data), and UGT1A9*5 Asp246Asn (Jinno et al., 2003a). e UGT1A10 alleles correspond to: UGT1A10*1 Met59Thr202; UGT1A10*2 Ile59Thr202; and UGT1A10*3 Met59Ile202 (Jinno et al., 2003b). f The relative positions of the UGT2B4, UGT2B7, and UGT2B15 genes on chromosome 4q13 are based on the data reported by Riedy et al. (2000). g Polymorphic expression of two truncated UGT2B28 variants (type II and type III) has been reported (Levesque et al., 2001). UGT2B28 type II differs from type I by a deletion of 308 bp in the cofactor binding domain, whereas UGT2B28 type III lacks 351 bp in the putative substrate binding domain. h An additional cDNA clone isolated from human liver corresponds to the UGT2B4 Phe109Leu, Phe396Leu allele but appears to be very rare since it was not found in two independent population studies.
The first evidence suggesting alterations in UGT genes as a genetic risk factor of cancer was recently obtained. In a first study, we hypothesized that constitutive alteration in UGTs involved in the inactivation of estradiol and its catechol-reactive metabolites may modify estrogen exposure and, consequently, estrogen-related cancer risk. We investigated UGT1A1 as a first candidate gene. UGT1A1 is a major UGT expressed in mammary gland and involved in the formation of estradiol-glucuronide (Senafi et al., 1994; Guillemette et al., 2000a). The most common genetic variant in the UGT1A1 gene is a dinucleotide repeat polymorphism in the atypical TATA-box region of the UGT1A1 promoter (Bosma et al., 1995). Using genetic epidemiological studies designed as population-based case-control studies, we first observed that the UGT1A1*28 (A(TA)7TAA) and the UGT1A1*34 (A(TA)8TAA) promoter alleles were associated with an increased risk of developing invasive breast cancer in premenopausal African-American women (OR = 2.1; 95% confidence interval, 1.0–4.2; p = 0.04) (Guillemette et al., 2000a). This finding is consistent with a role for estrogen-UGT in modulating the action of endogenous hormones in breast cancer risk in the African-American population. However, in a nested case-control study of white women within the Nurses' Health Study cohort, we were unable to detect a significant risk associated with the low transcriptional allele UGT1A1*28 (Guillemette et al., 2001). In the same population, an elevation of estradiol among women who are carriers of at least one UGT1A1*28 allele was observed and suggests a possible contribution of the glucuronidation pathway, and especially UGT1A1, in the maintenance of hormone homeostasis, although not sufficient to alter breast cancer risk in white women. We further studied the relationship between UGT1A1 polymorphisms and variation in breast density, a predictor of breast cancer. Premenopausal women homozygous for the UGT1A1*28 allele were found to have significantly lower breast density compared with those with the *1/*1 genotype (-43.1% difference; p = 0.04). In contrast, postmenopausal women with the UGT1A1*28/*28 genotype had greater breast density compared with those with the *1/*1 genotype (+32.0% difference; p = 0.05), which was even greater among current postmenopausal hormone users (+56.8% difference; p = 0.03). These results suggest that UGT1A1 genotype is a predictor of breast density within groups of different menopausal status and support that interindividual differences in estrogen glucuronidation influence local estrogen concentration and breast density (Haiman et al., 2003).
Recent studies support the idea that UGTs play a significant role in the detoxification of environmental carcinogens and therefore represent good candidates for low-penetrance susceptibility genes that possibly contribute to cancer risk by amplifying the effects of carcinogen exposure. Among these, UGT1A7 is an important extrahepatic UGT and represents one of several UGTs that were shown to be active on chemical carcinogens. In a recent study, we identified three common UGT1A7 allelic variant isozymes that encode novel UGT1A7 proteins differing in primary structure at three amino acid positions (Fig. 2) (Guillemette et al., 2000b; Woolley et al., 2000). Prevalence studies confirmed the presence of all four UGT1A7 alleles in the African-American and Asian populations, although at various frequencies. Functional studies revealed significant differences in catalytic activity of the UGT1A7 alleles toward benzo(a)pyrene metabolites, known tobacco-carcinogens, as well as for a number of additional substrates (Guillemette et al., 2000b). UGT1A7 was shown to be expressed in normal orolaryngeal tissue specimens including tongue, tonsil, floor of mouth, and larynx, which are target tissues of carcinogenic environmental molecules (Zheng et al., 2001). The UGT1A7 allele, associated with low activity, was subsequently associated with an increased risk of orolaryngeal cancer (Zheng et al., 2001). Genotypes containing different combinations of the two lowest activity alleles, UGT1A7*3 (129Lys131Lys208Arg) and UGT1A7*4 (129Arg131Asn208Arg), were strongly linked to an increased risk for orolaryngeal cancer in white (OR = 2.8; 95% IC 1.1–7.6) and in African-American people (OR = 6.2; 95% IC 1.2–31) compared with the wild-type genotype (homozygous for the UG1A7*1 allele [129Arg131Asn208Trp]). Upon stratification by cancer site, predicted low-activity UGT1A7 genotypes were strongly linked to increased risk for both oral cavity (OR = 4.2; 95% IC 1.7–0) and laryngeal cancers (OR = 3.7; 95% IC 0.99–14). In addition, subjects with low-activity genotypes who were light or heavy smokers had a significantly increased risk compared with the wild-type genotype (OR = 3.7; 95% IC 1.1–12 and OR = 6.1; 95% IC 1.5–25, respectively) (Zheng et al., 2001). These results revealed for the first time that genetic variations in the UGT1A7 gene that reduce the carcinogen-detoxifying activity increase the risk of developing a smoking-related orolaryngeal cancer. The association of UGT1A7 alleles with risk of hepatic and colorectal cancers was further demonstrated (Vogel et al., 2001; Zheng et al., 2001; Strassburg et al., 2002). Recently, we conducted a case-control study, 400 cases and 400 controls matched for age, sex, and race, to assess the relation between characteristics of meat consumption, heterocyclic amine (HCA) exposure, the UGT1A7 genotype, and colon cancer. No main effect of the UGT1A7 genotype was observed on colon cancer risk. On the other hand, the association between dietary HCA exposure and colon cancer was modified in individuals with the low-activity UGT1A7 genotypes (C. Guillemette, unpublished observations). These data suggest that the relation between dietary sources of HCA and colon cancer may be modulated by the UGT1A7 detoxification pathway. These results also point to HCA exposure as an important etiologic factor in colon cancer. Altogether, these findings warrant additional epidemiological studies to confirm the role of low UGT1A7 conjugator genotypes in risk for cancers. Besides, our group recently discovered two additional UGT1A7 single nucleotide polymorphisms, found exclusively in African-American subjects, which generate five additional alleles (UGT1A7*5 to *9) when combined with the four known single nucleotide polymorphisms present in UGT1A7*2, *3, and *4. Upon functional analysis, several of these UGT1A7 variant isozymes exhibited much lower glucuronidation activities compared with UGT1A7*1; their role in cancer remains unverified at the present time (Girard et al., 2003).
In conclusion, much research in this area is needed, although promising leads have emerged regarding the role of UGT genetic polymorphisms in cancer etiology and on their possible implication in modulating the degree of exposure to several carcinogenic compounds.
Gene Therapy for UGT Deficiencies (J.R.C., N.R.C.)
Genetic lesions of UGT1A1 can result in three grades of hyperbilirubinemia in humans. Insertions, deletions, or mutations of any of the five exons encoding UGT1A1 that cause near-complete loss of the enzyme activity result in the potentially lethal disorder, Crigler-Najjar syndrome type 1 (CN-1). Mutations causing lesser degrees of reduction of UGT1A1 activity cause a milder form of the disease, termed Crigler-Najjar syndrome type 2. The third grade of hyperbilirubinemia is an even milder form, termed Gilbert's syndrome, which results from the insertion of TA dinucleotides within the TATAA element of the UGT1A1 promoter or base substitutions in the UGT1A1 coding region (Roy Chowdhury et al., 2001). Of these three disorders, CN-1 is associated with levels of unconjugated hyperbilirubinemia that are severe enough to cause bilirubin encephalopathy. Before the routine use of phototherapy, CN-1 was generally lethal during infancy. Although phototherapy permits survival beyond adolescence, it becomes progressively less effective around puberty, so that the risk of kernicterus persists life-long. Currently, liver transplantation is the only definitive therapy for CN-1. However, discovery of the molecular bases of inherited jaundice, and advances in the techniques of hepatocyte transplantation and nucleic acid transfer to the liver have brought gene therapy for Crigler-Najjar syndrome close to reality. Gene therapy methods can be classified into approaches based on isolated hepatocyte, and methods using gene delivery in vivo.
Hepatocyte-Based Gene Therapy. In the simplest form, normal genes may be introduced into the liver of patients by transplantation of allogeneic normal human hepatocytes. Hepatocytes introduced into the portal circulation by infusion into the portal vein or injection into the splenic pulp integrate into normal liver chords of structurally normal liver with remarkable rapidity and function on a long-term basis (Gupta et al., 1991). Immunosuppression is required for prevention of allograft rejection. This method should be particularly useful for diseases, such as CN-1, in which the liver architecture is normal. Because the host liver remains intact, the metabolic cost of graft rejection is limited. A 10-year-old girl with CN-1 was the first to receive liver transplantation (Fox et al., 1998). Introduction of 7.5 × 109 normal isolated hepatocytes into the liver through a percutaneously placed portal vein catheter reduced serum bilirubin to approximately half the pretransplant level for over 2 years. This study demonstrated the safety and efficacy of hepatocyte transplantation, but despite the replacement of approximately 5% of the hepatic UGT1A1 activity, the need for phototherapy was not obviated (Roy Chowdhury et al., 1998). It appears that complete cure of metabolic liver diseases will require repeated hepatocyte transplantation or preferential proliferation of the transplanted hepatocytes over the host cells.
To solve many of the lingering problems associated with hepatocyte transplantation, extensive studies are ongoing in Gunn rats that are both a molecular and pathophysiological model of CN-1. Such preferential proliferation requires a strong mitotic stimulus to the liver, to which only the engrafted cells, but not the host hepatocytes, can respond. This was achieved by the combination of preparative irradiation of the liver and partial hepatectomy, prior to hepatocyte transplantation (Guha et al., 1999, 2002). Controlled irradiation of the liver prevents proliferation of the host Gunn rat hepatocytes. Consequently, transplanted congeneic normal hepatocytes progressively repopulate the liver almost completely by 12 weeks, fully normalizing serum bilirubin levels.
Another approach involves isolating hepatocytes from a resected liver segment, transducing the primary hepatocytes with a therapeutic gene in culture and subsequently transplanting the cells back into the donor (Roy Chowdhury et al., 1991). Because the cells are autologous, immune suppression is not needed. However, the number of cells that can be harvested, transduced, and engrafted after transplantation is limited, which severely restricts the efficiency of ex vivo gene therapy. Some of these limitations may be overcome by conditionally immortalizing the hepatocytes, so that the transduced cells can be expanded in culture before transplantation (Tada et al., 1998).
Gene Transfer in Vivo. The conventional in vivo gene therapy methods consist of replacing a missing functional gene by transferring nucleic acids to the target cells using viral or nonviral vectors. A radically new approach involves repairing genetic mutations in situ. These approaches, as related to the treatment of UGT1A1 deficiency, are briefly described below.
Nonviral vectors. Physical methods, such as ballistics or direct injection into the liver parenchyma, have resulted in a limited extent of gene transfer. Naked DNA can be transfected into the liver by rapid intravenous administration of plasmids at high volumes, causing a volume overload and hepatic congestion. Obviously this approach is not translatable to clinical application. DNA can be complexed electrostatically with polycations, which can be lactosylated or complexed with galactose-terminated peptides for hepatocyte-targeted delivery in vivo. The ligand-DNA complex is internalized by receptor-mediated endocytosis via hepatocyte-specific asialoglycoprotein receptors. Although the majority of the ligand is translocated to the lysosome, a small fraction reaches the nucleus, where it is expressed transiently. The transgene expression can be prolonged to several weeks by performing partial hepatectomy or transient pharmacological disruption of the microtubules (Roy Chowdhury et al., 1996) or by using polycations, such as polyethyleneimine, that destabilize the endosomal vesicles. An important new advance in plasmid-directed gene transfer has been the use of a Tc1 Mariner-type transposon system, termed the Sleeping Beauty. Expression of the Sleeping Beauty transposase results in the transposition of stretches of DNA, flanked by inverted/direct repeats of the transposon sequences, into host cellular chromosomes, leading to long-term transgene expression (Izsvák et al., 2000).
Viral vectors. Recombinant viruses capable of transferring genes into cells in vitro or in vivo are commonly used for gene therapy. These vectors can be classified into those that remain episomally and those that integrate into the host genome. Recombinant adenoviruses are prototypes of episomal vectors. Adenoviral vectors transfer genes into nondividing hepatocytes in vivo with great efficiency, although the longevity of the episomal vector is limited. Moreover, adenoviral proteins are strongly immunogenic, and host humoral and cellular immune response precludes repeated gene transfer. Specific tolerization of the host by administration in neonatal rats (Takahashi et al., 1996), intrathymic injection of adenoviral proteins (Ilan et al., 1996), or oral administration of small doses of adenoviral proteins (Ilan et al., 1997) have resulted in specific host tolerance to the viral proteins and have permitted repeated administration of the recombinant virus and long-term amelioration of jaundice in Gunn rats. Recently, immunomodulatory genes, such as CTLA4Ig, have been incorporated into adenoviral vectors (Thummala et al., 2002). Coexpression of such genes, along with the transgene, makes the virus nonimmunogenic, prolongs the transgene expression, and permits repeated administration.
Retroviral vectors integrate into the host genome. Although recombinant oncoretroviruses have been used extensively for gene therapy (Roy Chowdhury et al., 1991; Tada et al., 1998), these vectors require host cell mitosis for integration, which is infrequent in the quiescent liver. Vectors based on immunodeficiency-type retroviruses (lentiviruses) that can integrate into nondividing cells are being developed for the treatment of UGT1A1 deficiency. Recombinant adenoassociated viruses (rAAVs) are also being tested in Gunn rats, although, so far, it has been possible to transduce only up to 5% of rat hepatocytes by intraportal infusion. Recent studies indicate that rAAVs do not integrate to a significant extent in immunocompetent animals, suggesting that, in contrast to previous expectations, the transgene expression may not be permanent (Nakai et al., 2001). Moreover, because rAAV vectors evoke a host antibody response, repeated administration may be problematic. Therefore, the search for newer vectors continues. Recombinant simian virus 40 is promising as an integrating vector because this T-antigen-deleted virus does not evoke host immune response. Recombinant simian virus 40 integrates into the host genome progressively over several days. After infusion of a recombinant SV40 expressing human UGT1A1 into the portal vein of the Gunn rat, serum bilirubin was reduced by up to 60% and remained at that level throughout an 18-month period of observation, suggesting a permanent therapeutic effect. There was no detectable antibody response to the recombinant virus, and gene transfer was repeatable upon injection of a recombinant SV40 vector expressing a different transgene (Sauter et al., 2000). These viruses can be grown and concentrated to infectious titers of 1011 to 1012. Thus, the recombinant SV40 appears to have a great potential in liver-directed gene therapy, its major limitation being a relatively small DNA packaging space (4–4.5 kilobases) (Strayer et al., 2002).
Gene repair therapy. Site-directed gene repair in vivo is a novel form of gene therapy, which relies on the cellular DNA repair mechanisms to correct point mutations or short deletions (Kren et al., 1999). The method utilizes synthetic RNA-DNA chimera, which align to a target sequence in the genome with high specificity and efficiency. The nucleotide sequence in the chimera is complementary to the target genomic sequence, except for a single mismatch. After alignment, the mismatch between the DNA limb of the chimeric molecule and the complementary strand of the genomic DNA triggers the cell's mismatch repair enzymes, resulting in correction of the mutation. For correction of the single guanosine base deletion in the UGT1A1 exon 4 of Gunn rats, the RNA-DNA chimera was constructed to contain the wild-type sequence (Kren et al., 1999). The chimeric molecules were complexed with lactosylated polyethyleneimine or with galactosylated liposomes. After intravenous infusion of the carrier-chimera complex, the genetic lesions were corrected in 1 to 10% of the alleles, resulting in the appearance of the enzymatically active full-length UGT1A1 in the liver of the recipient Gunn rats. Function of the enzyme was demonstrated by the appearance of bilirubin glucuronides in bile and reduction of plasma bilirubin levels to 35 to 40% of pretreatment levels. Long-term follow-up indicates permanent correction of the genetic lesion. Figure 3 shows immunohistochemical staining of liver sections from Gunn rats treated with recombinant adenoviral or SV40 vectors, or with synthetic DNA-RNA chimera for gene repair. In summary, both nonviral and viral vectors are being rapidly improved for overcoming the challenges of liver-directed gene therapy for CN-1. Hepatocyte transplantation and gene therapy are being developed in parallel to complement each other.
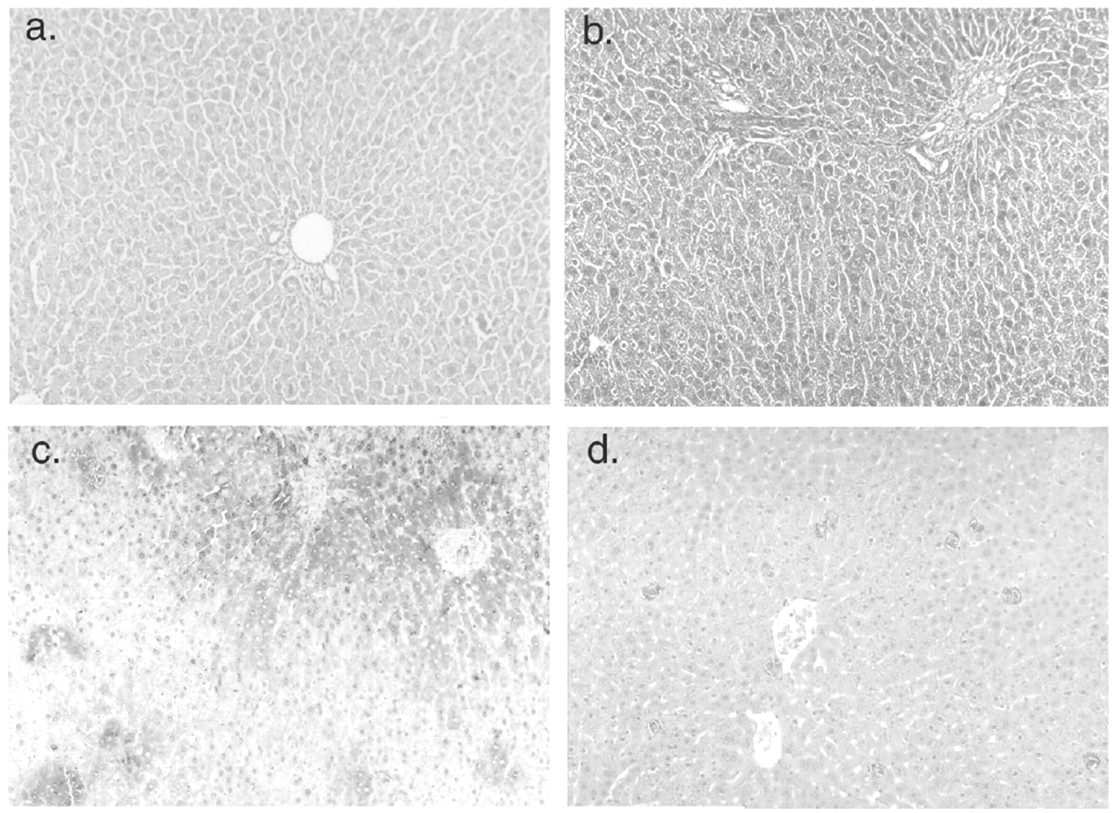
Immunohistochemical staining of sections of Gunn rat liver with anti-UGT1A1 antibodies.
For panels a–c, immunohistochemical staining was performed using a human UGT1A-specific monoclonal antibody. Panel a, control (untreated) Gunn rat; panel b, liver from a Gunn rat 1 week after the injection of a first-generation adenovirus expressing human UGT1A1 (109 pfu). Panel c, liver from a Gunn rat 1 year after the injection of 108 pfu of a recombinant SV40 expressing human UGT1A1. Panel d, gene repair therapy in Gunn rat. Liver biopsy was performed 1 year after five injections of an RNA-DNA chimera designed to insert the missing guanosine residue in Gunn rat UGT1A1 (exon 4). Immunohistochemical staining was performed using a rabbit antiserum specific for rat UGT1A1. Please note that the staining of the positive cells is light, probably indicating that only one allele per cell is corrected.
It is apparent that significant progress continues to be made in our understanding of the UGT gene family and their roles in physiology and pathophysiology. Molecular factors involved in the control of expression of the UGTs are being identified, providing clues to how these genes are regulated from tissue to tissue, during development, and in response to certain chemical exposures. The substrate specificities and activities of the different UGT family members in humans are beginning to be understood, and information on their rodent counterparts is now coming to light which will be valuable for interpretation of toxicity studies in rodents. Identification of genetic differences among individuals in the sequences of UGT genes and their impact on risks of toxicity from exposure to drugs and environmental chemicals is a particularly active area of investigation and will be useful for predictive risk assessment. The research comes full circle with current efforts toward development of an effective gene therapy for treatment of Crigler-Najjar syndrome, providing a greatly needed alternative to liver transplantation. It should be clear that advances from both basic and clinical research involving animals and humans have provided critical insights into the role of glucuronidation and UGTs in health, drug therapy, and disease.
Footnotes
-
↵2 Abbreviations used are: UGT, UDP-glucuronosyltransferase; B[a]P, benzo-[a]pyrene; CN-1, Crigler-Najjar syndrome type 1; HCA, heterocyclic amine; HNF1, hepatocyte nuclear factor 1; OR, odds ratio; SN-38, 7-ethyl-10-hydroxycamptothecin; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; P450, cytochrome P450; rAAV, recombinant adenoassociated virus.
-
This work was supported in part by grants from the Canadian Institutes of Health Research (P.G.W., C.G.), the National Health and Medical Research Council of Australia and the Anti-Cancer Foundation of South Australia (P.I.M., P.A.G., A.J.H., Y.I.), the Canada Research Chair Program and the Fonds de la Recherche en Sante du Quebec (C.G.), and United States Public Health Service Grants R01-DK46057, R01-DK34357 and P30DK41296 (J.R.C., N.R.C.), and R01ES07762 (J.K.R.).
-
↵1 These authors contributed equally as symposium speakers.
- Received October 10, 2003.
- Accepted December 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics
References




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Structure and Tissue-Specific Regulation of UGT Genes (P.I.M., P.A.G., Y.I., A.J.H.)
- Glucuronidation of Xenobiotics and Endobiotics (J.K.R., F.K.K.)
- Toxicological Relevance of UGTs (P.G.W., P.M.K.)
- Pharmacogenetics of UGT Enzymes: Implications for Cancer Susceptibility (C.G.)
- Gene Therapy for UGT Deficiencies (J.R.C., N.R.C.)
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters