


Abstract
Cytochrome P450 (P450) and flavin-containing monooxygenase (FMO) enzymes are major catalysts involved in the metabolism of xenobiotics. The sulfoxidation of the thioether pesticides, phorate, disulfoton, sulprofos, and methiocarb, was investigated. Using pooled human liver microsomes (HLMs), thioether compounds displayed similar affinities; however, phorate and disulfoton displayed higher intrinsic clearance rates than either sulprofos or methiocarb. The sulfoxidation of thioethers by HLMs was found to be predominantly P450-driven (85-90%) compared with FMO (10-15%). Among 16 cDNA-expressed human P450 isoforms and 3 human FMO isoforms examined, the following isoforms and their polymorphisms had the highest rates for sulfoxidation, as follows: phorate, CYP1A2, 3A4, 2B6, 2C9*1, 2C18, 2C19, 2D6*1, and FMO1; disulfoton, CYP1A2, 3A4, 2B6, 2C9*1, 2C9*2, 2C18, 2C19, 2D6*1, and FMO1; sulprofos, CYP1A1, 1A2, 3A4, 2C9*1, 2C9*2, 2C9*3, 2C18, 2C19, 2D6*1, and FMO1; methiocarb, CYP1A1, 1A2, 3A4, 2B6, 2C9*1, 2C19, 2D6*1, and FMO1. Among these isoforms, members of the CYP2C subfamily often had the highest affinities and clearance rates. Moreover, sulfaphenazole, a CYP2C9 competitive inhibitor, inhibited disulfoton sulfoxidation by CYP2C9 (IC50 0.84 μM) as well as in HLMs. Ticlopidine, a CYP2C19 mechanism-based inhibitor, inhibited disulfoton sulfoxidation by CYP2C19 (IC50 after coincubation, 43.5 μM; IC50 after preincubation, 4.3 μM) and also in HLMs. Our results indicate that current models of the substrate binding site of the CYP2C subfamily would not effectively predict thioether pesticide metabolism. Thus, the substrate specificity of CYP2Cs is more extensive than is currently believed, and some reevaluation of structure-activity relationships may be required.
The oxidative metabolism of xenobiotics, including pesticides, is primarily carried out by cytochrome P450s (P450s)1 and to a lesser extent by the flavin-containing monooxygenases (FMOs). In the adult human liver, members of the 3A and 2C subfamilies are the predominant P450 isoforms involved, while FMO3 is the primary FMO isoform involved (Guengerich, 1995; Phillips et al., 1995). P450 isoforms of the 3A and 2C subfamilies were found to account for 30% and 20%, respectively, of the total P450 content of the human liver (Shimada et al., 1994). In humans, the CYP 2C subfamily includes at least four functional isoforms: 2C8, 2C9, 2C18, and 2C19, of which 2C8, 2C9, and 2C19 are found primarily in the liver, with 2C18 being primarily expressed in the skin (Ged et al., 1988; Romkes et al., 1991; Minoletti et al., 1999). Of the 2C liver isoforms, 2C9 and 2C19, which are 92% identical in their amino acid structure (Klose et al., 1998), appear to play the greatest role in metabolism of pharmaceuticals (Miners et al., 2000). Polymorphisms of CYP 2C9, 2C18, and 2C19 have been reported; in the current studies we have used the 2C9 isoforms 2C9*1(Arg144), 2C9*2(Cys144) and 2C9*3(Leu359).
The mammalian FMOs, like the P450s, although not as catalytically or structurally diverse as the P450 superfamily, are important phase I enzymes that are responsible for the conversion of lipophilic xenobiotics to more hydrophilic metabolites by the addition of oxygen through an NADPH-dependent pathway. Substrates for FMOs include several nitrogen-, sulfur-, selenium-, and phosphorus-containing compounds (Ziegler, 1991; Hodgson and Goldstein, 2001). Currently, five distinct mammalian FMO isoforms have been identified and categorized through amino acid or cDNA sequencing as FMOs 1, 2, 3, 4, and 5 (Lawton et al., 1994). Isoforms of human FMO1, 3, and 5 have been sequenced and expressed as functional proteins in heterologous expression systems (Overby et al., 1995; Phillips et al., 1995).
P450s and FMOs have been shown to be important in the activation and detoxification of a variety of pesticides including thioether-containing organophosphorus and carbamate compounds (Hajjar and Hodgson, 1980; Smyser et al., 1985, Tynes and Hodgson, 1985). In this study, we examined the sulfoxidation of three organophosphorus thioether pesticides, phorate (O,O-diethyl S-[(ethylthio)methyl] phosphorodithioate), disulfoton (O,O-diethyl S-[2-(ethylthio)ethyl] phosphorodithioate), and sulprofos (O-ethyl O-[4-(methylthio)phenyl] S-propyl phosphorodithioate), and a carbamate thioether pesticide, methiocarb (3,5-dimethyl-4-(methylthio)phenyl methylcarbamate).
The metabolism of phorate includes a series of complex oxidation reactions. The reaction is an activation process with products that are generally more toxic than the parent compound (Hodgson et al., 1998). Mouse FMO1 only forms one product, phorate sulfoxide, whereas P450 forms produce phorate sulfoxide, phorate sulfone, and other products (Hodgson et al., 1998). Preliminary results in this laboratory demonstrated the oxidation of phorate by the human P450 isoforms 1A2, 2C8, 2C9, 2C18, 2C19, 2E1, and 3A4 (Hodgson et al., 1998). Methiocarb also undergoes a series of oxidation reactions in rat liver, including the formation of methiocarb sulfoxide and additional products, by both the P450 and FMO pathways (Buronfosse et al., 1995). However, the P450 and FMO isoforms involved in methiocarb metabolism in humans have not been identified. Similarly, no previous studies have identified and characterized the human P450 or FMO isoforms involved in the sulfoxidation of disulfoton or sulprofos.
Our results indicate that members of the CYP2C subfamily are among the most important isozymes involved in the sulfoxidation of thioether compounds. Considerable interest has recently been focused on CYP2C subfamily members not only because of their involvement in the oxidation of a number of clinically used drugs but also due to the discovery of numerous polymorphic forms and substrate recognition sites within these isoforms. Molecular modeling techniques have also been employed to derive predictive models for CYP2C substrates, particularly CYP2C9 (Mancy et al., 1995; Mancy et al., 1996; Poli-Scaife et al., 1997; Ha-Duong et al., 2001b; Melet et al., 2003). The thioether compounds used in the present study were not previously known as CYP2C substrates and do not fit well within the previous predictive models for CYP2C isoform substrates. To further confirm that these thioether compounds are metabolized at the same catalytic site as previously predicted substrates, we used sulfaphenazole, a selective competitive inhibitor of human liver CYP2C9, and ticlopidine, a selective mechanism-based inhibitor of human liver CYP2C19.
Materials and Methods
Chemicals. Phorate, phorate sulfoxide, disulfoton, disulfoton sulfoxide, sulprofos, sulprofos sulfoxide, methiocarb, and methiocarb sulfoxide were purchased from Chem Service, Inc. (West Chester, PA). HPLC-grade acetonitrile and water were purchased from Fisher Scientific (Pittsburgh, PA). 1-Benzylimidizole was purchased from Aldrich Chemical Co. (Milwaukee, WI). All other chemicals, if not specified, were purchased from Sigma-Aldrich (St. Louis, MO).
Human Liver Microsomes, Human P450 Isoforms, and Human FMO isoforms. Pooled human liver microsomes (HLMs) (pooled from 21 donors), human P450 isoforms [CYP1A1, 1A2, 2B6, 3A4, 3A5, 3A7, 4A11, 2B6, 2C8, 2C9*1 (Arg144), 2C9*2 (Cys144), 2C9*3 (Leu359), 2C18, 2C19, 2D6*1(Val374), and 2E1], and FMO isoforms (FMO1, FMO3, and FMO5) expressed in baculovirus-infected insect cells (Sf9) (BTI-TN-5BI-4) were purchased from BD Gentest (Woburn, MA).
In Vitro Metabolism. Metabolic activity assays designed to screen human P450 and FMO isoforms for metabolic activity toward each substrate were performed by incubation with the appropriate substrate (final concentration 200 μM) with an NADPH regenerating system (0.25 mM NADP, 2.5 mM glucose 6-phosphate, and 2 U/ml glucose-6-phosphate dehydrogenase) in specific buffers recommended by the enzyme supplier (BD Gentest). After preincubation at 37°C for 5 min, the reactions were initiated by the addition of ice-cold P450 isoforms (final concentration 50 pmol/ml) or FMO isoforms (final concentration 104-300 pmol/ml) with gentle mixing and incubated for 30 min at 37°C. Reactions were terminated by the addition of an equal volume of acetonitrile and vortexing. The control reactions were performed under identical conditions with an Sf9 insect cell control. For the P450 and FMO isoforms 1A1, 1A2, 2E1, 2C8, 2D6*1, 3A5, 3A7, 2B6, 2C18, 3A4, 2C19, FMO1, FMO3, andFMO5, and Sf9 cell control, a 100 mM potassium phosphate buffer with 5 mM MgCl2 (pH 7.4) was used. For the P450 isoforms 2C9*1 (Arg144), 2C9*2 (Cys144), 2C9*3 (Leu359), 4A11, and 2A6, a buffer consisting of 100 mM Tris-HCl with 5 mM MgCl2 (pH 7.5) was used.
After screening assays were completed for each substrate, isoforms determined to have the greatest impact on metabolism were selected for further metabolic characterization. For phorate, enzyme kinetic assays were conducted using HLMs and human P450 and FMO isoforms including 1A2, 3A4, 2B6, 2C9*1, 2C18, 2C19, and FMO1. Each isoform was incubated with serial dilutions of phorate (final concentration 6.25-200 μM) together with appropriate buffer and cofactors as described below. For disulfoton and sulprofos, enzyme kinetic assays were conducted using HLMs, CYP1A2, 3A4, 2B6, 2C9*1, 2C9*2, 2C19, and 2D6*1, and FMO1 using serial dilutions of substrate (final concentration 1.56-200 μM). Additional kinetic assays with sulprofos were performed for 1A1, 3A5, 2C9*3, and 2C18. The enzyme kinetic assays for methiocarb with HLMs, and human P450 and FMO isoforms 1A1, 1A2, 3A4, 2B6, 2C9*1, 2C18, 2C19, 2D6*1, and FMO1 were performed with serial dilutions of methiocarb (final concentration 0.35-100 μM). The initial combination of substrate, NADPH regenerating system, and 100 mM potassium phosphate buffer with 5 mM MgCl2 (pH 7.4) was incubated at 37°C for 5 min. The reactions were initiated by the addition of ice-cold HLMs (protein concentration 1 mg/ml), P450 isoforms (final concentration 50 pmol/ml), or FMO1 (62 pmol/ml) with gentle mixing followed by incubation for 10 min (phorate, sulprofos, and methiocarb assays) or 5 min (disulfoton assay) at 37°C. Reactions were terminated by the addition of an equal volume of acetonitrile and vortexing. The control reactions were performed under identical conditions using Sf9 cells.
Inhibition of P450 or FMO in Human Liver Microsomes. P450 and FMO inhibition assays with human liver microsomes were performed as described by Grothusen et al. (1996) with slight modifications. To investigate the role of FMO in the reaction, assays were conducted in the presence of a P450 inhibitor, 1-benzylimidazole. A 5-min preincubation at 37°C consisted of ice-cold HLMs (protein concentration 1 mg/ml) added to 100 mM potassium phosphate buffer with 5 mM MgCl2 (pH 7.4), an NADPH regenerating system, and 1-benzylimidazole (1 mM final concentration). Following the preincubation step, the reaction was initiated by the addition of substrate (200 μM) with gentle mixing, followed by 10-min (disulfoton and sulprofos) or 30-min (phorate and methiocarb) incubations at 37°C.
Reactions investigating the role of P450 utilized human liver microsomes that had been heat treated at 50°C for 1 min to inhibit FMO activity and then immediately placed on ice. In these assays, substrate (final concentration 200 μM) was added to an NADPH regenerating system (0.25 mM NADP+, 2.5 mM glucose 6-phosphate, and 2 U/ml glucose-6-phosphate dehydrogenase) in 100 mM potassium phosphate buffer with 5 mM MgCl2 (pH 7.4). After preincubation at 37°C for 5 min, the reactions were initiated by the addition of ice-cold heat-treated human liver microsomes (protein concentration 1 mg/ml) with gentle mixing and incubated for 10 min (disulfoton and sulprofos) or 30 min (phorate and methiocarb) at 37°C.
To demonstrate whether thioether compounds are being metabolized by the same catalytic site as other CYP2C9 substrates, coincubations of CYP2C9 were conducted with disulfoton (37 μM) and sulfaphenazole (0.01-10 μM), a competitive inhibitor of CYP2C9. The reaction mixtures consisted of substrate, NADPH regenerating system, the inhibitor, and buffer as described above and were initiated by the addition of ice-cold CYP2C9*1 (final concentration 50 pmol/ml), followed by gentle mixing and incubation for 5 min at 37°C.
Ticlopidine (0.5-50 μM), a mechanism-based inhibitor of CYP2C19, was co- or preincubated with CYP2C19 (final concentration 50 pmol/ml) and disulfoton (37 μM). For the coincubation study, the reactions were initiated by the addition of ice-cold CYP2C19 to the buffer containing ticlopidine, disulfoton, and the NADPH regenerating system with gentle mixing and incubation for 5 min at 37°C. In the preincubation study, ice-cold CYP2C19 was added to the buffer and regenerating system containing ticlopidine at 37°C, 5 min before the addition of disulfoton (37 μM). The reaction was terminated after an additional 5 min and the supernatant was analyzed for sulfoxidation products by HPLC.
The inhibition of HLM sulfoxidation of disulfoton was also investigated using sulfaphenazole and ticlopidine. HLMs were preincubated for either 5 or 10 min with either sulfaphenazole (10 μM) or ticlopidine (20 μM) or both (10 μM and 20 μM, respectively) in the reaction described above for thioether metabolism by HLMs. The reaction was started by the addition of disulfoton, incubated for 5 min, and stopped by the addition of acetonitrile.
All assays were conducted in duplicate or triplicate. All reactions were terminated by the addition of equal volumes of acetonitrile and vortexing. After 10 min of centrifugation at 15,000 rpm in a microcentrifuge, the supernatants were analyzed for sulfoxidation concentrations by HPLC. The protein concentrations and incubation times used in the assays were found to be in the linear range in preliminary experiments. No metabolites were detected when incubations were carried out in the absence of an NADPH generating system.
Analysis of Metabolites by HPLC. Metabolites were analyzed using a Shimadzu HPLC system (Shimadzu, Kyoto, Japan). This Shimadzu HPLC system consisted of one pump (LC-10ATVP), an autoinjector (SIL-10ADVP), a UV-VIS detector (SPD-10AVVP), a system controller (SCL-10AVP), a four-position solvent selection valve (FCV-10ALVP), and a degasser (DGU-14A). Chromatography software was CLASS-VP version 4.3.
Phorate and its metabolite, phorate sulfoxide, were separated with a mobile phase consisting of A (20% acetonitrile, 80% water) and B (80% acetonitrile, 20% water). A gradient system was set up in the following manner: 0 to 3 min (0% B), 3 to 10 min (gradient to 100% B), 10 to 15 min (100% B), 15 to 18 min (gradient to 0% B), and 18 to 20 min (0% B). The flow rate was 1.0 ml/min. Metabolites were separated with a C18 column (Luna 5 μm, 150 × 3.00 mm; Phenomenex, Torrance, CA) at a wavelength of 210 nm. The injection volume was 25 μl. Under these conditions, the retention times for phorate and phorate sulfoxide were 12.0 min and 8.6 min, respectively. The limit of detection for phorate sulfoxide was 0.2 μM. The concentrations of sulfoxide metabolites for phorate and each of the other thioether pesticides were obtained through extrapolation of peak area from a standard curve prepared in equal volumes of acetonitrile and water.
Disulfoton and its metabolite, disulfoton sulfoxide, were separated with a mobile phase consisting of A (100% water) and B (100% acetonitrile). A gradient system was set up in the following manner: 0 to 1 min (35% B), 1 to 15 min (70% B), 15 to 18 min (70% B), 18 to 19 min (35% B), and 19 to 20 min (35% B). The flow rate was 0.75 ml/min. Metabolites were separated with a C18 column (Luna 5 μm, 150 × 3.00 mm; Phenomenex) at a wavelength of 210 nm. The injection volume was 25 μl. Under these conditions, the retention times for disulfoton and disulfoton sulfoxide were 16.0 min and 5.8 min, respectively. The limit of detection for disulfoton sulfoxide was 0.2 μM. The concentration of metabolite was obtained through extrapolation of peak area from a standard curve.
Sulprofos and its metabolite, sulprofos sulfoxide, were separated using a mobile phase consisting of A (100% water) and B (100% acetonitrile). A gradient system was set up in the following manner: 0 to 1 min (55% B), 1 to 10 min (80% B), 10 to 20 min (80% B), 20 to 21 min (55% B), and 21 to 25 min (55% B). The flow rate was 0.65 ml/min. Metabolites were separated with a Synergi column (Synergi 4 μm, 150 × 4.6 mm, MAX-RP, 80A; Phenomenex) at a wavelength of 260 nm. The injection volume was 50 μl. Under these conditions, the retention times for sulprofos and sulprofos sulfoxide were 19.3 min and 9.0 min, respectively. The limit of detection for disulfoton sulfoxide was 0.1 μM. The concentration of metabolite was obtained through extrapolation of peak area from a standard curve.
Metabolites of methiocarb were separated with a mobile phase consisting of A (20% acetonitrile, 80% water) and B (80% acetonitrile, 20% water). A gradient system was set up in the following manner: 0 to 3 min (0% B), 3 to 10 min (100% B), 10 to 25 min (100% B), 25 to 27 min (0% B), and 27 to 30 min (0% B). The flow rate was 0.5 ml/min. Metabolites were separated with a Prodigy column [Prodigy 3 μm, 150 × 4.0 mm, ODS (3), 100A; Phenomenex] at a wavelength of 265 nm. The injection volume was 25 μl. Under these conditions, the retention times for methiocarb and methiocarb sulfoxide were 16.7 min and 10.8 min, respectively. The limit of detection for methiocarb sulfoxide was 1.0 μM. The concentrations of metabolites were obtained by extrapolation of peak area from a standard curve.
Data Analysis and Statistics. The apparent Km and Vmax parameters were calculated using a nonlinear regression analysis program (SigmaPlot Enzyme Kinetic Module; SPSS Inc., Chicago, IL). These Km and Vmax values were then used to calculate the intrinsic clearance value (Km/Vmax) for each isoform. The percentage of total normalized rates (%TNR) was determined as described by Rodrigues (1999a). This is done by multiplying the intrinsic clearance value of each isoform by the nominal specific content (nmol of P450/mg protein) of the corresponding P450 form in native human liver microsomes to derive the normalized rate for each isoform. The nominal specific content utilized in these determinations was derived from a pool of liver microsomes (n = 12) phenotyped by BD Gentest (Rodrigues, 1999b). Since subsequent data indicated that CYP2B6 levels may have been high in this population, we used a value of 0.0207 nmol/mg protein derived from a different pool of 12 individuals phenotyped by BD Gentest. The normalized rate values obtained were then summed and the %TNRs were determined for each isoform.
Significant differences between data sets were determined by one-way analysis of variance, and multiple comparisons were performed with the Tukey-Kramer honestly significant different test by using the JMP 4.0.2 SAS program (SAS, 1989).
Results
Figure 1 shows structures of the compounds examined for sulfoxidation and the metabolites monitored in this study. Table 1 indicates the kinetic parameters for the sulfoxidation of thioethers by pooled HLMs. All the compounds displayed similar Km values for sulfoxidation; however, the intrinsic clearance rates [CLint(Vmax/Km)] for phorate (136 μl/mg protein/min) and disulfoton (130 μl/mg protein/min) were highest, followed by methiocarb (59 μl/mg protein/min) and sulprofos (20 μl/mg protein/min).
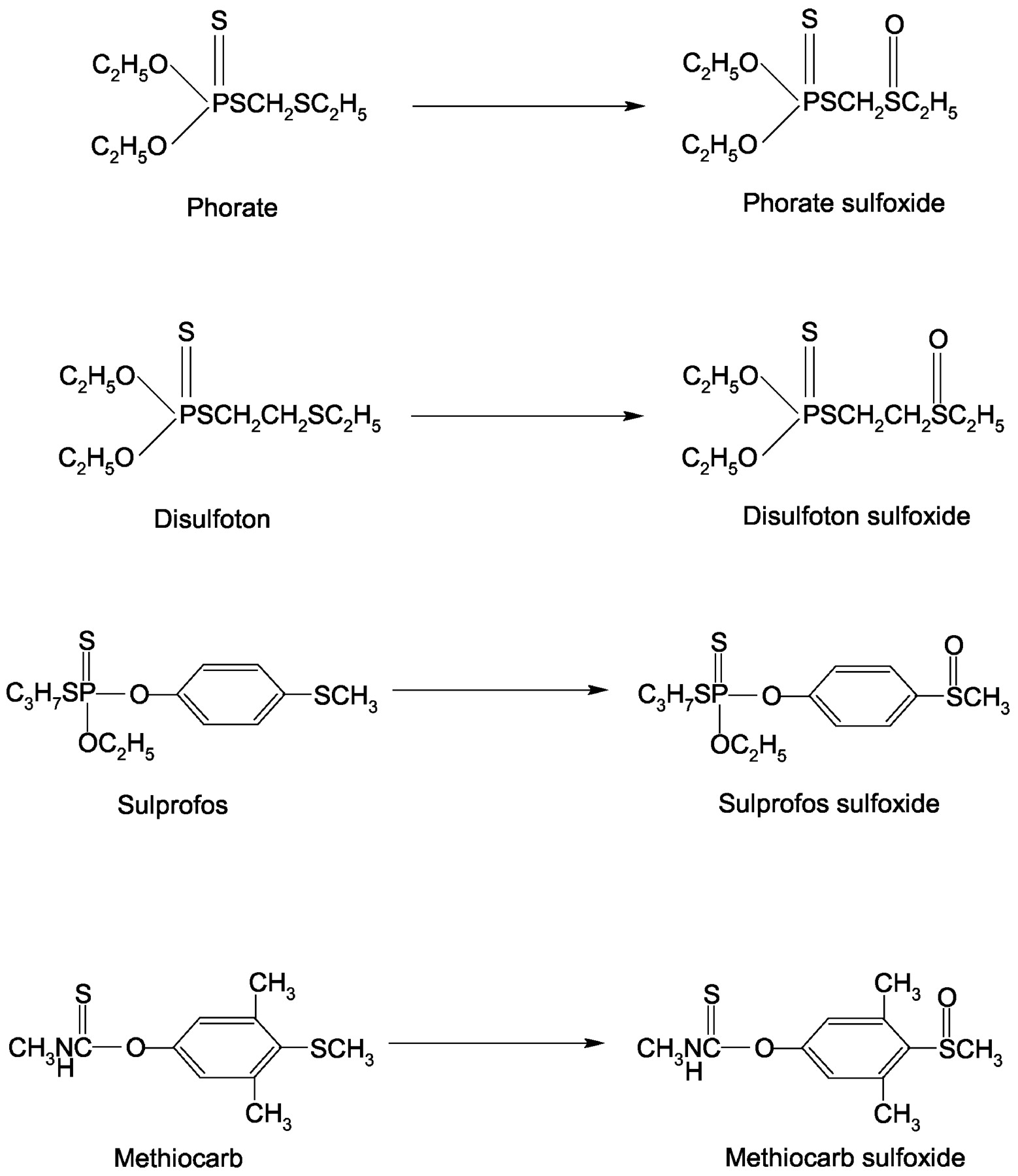
Thioether compounds investigated and their sulfoxidation products.
Kinetic parameters for the sulfoxidation of phorate, disulfoton, sulprofos, and methiocarb by human liver microsomes
Metabolism of thioether compounds to sulfoxides by HLMs were found to be predominantly P450-driven as compared with FMO when selective inhibitors for each enzyme system were utilized (Table 2). Incubation of HLMs with thioether compounds in the presence of a P450 inhibitor, 1-benzylimidizole (final concentration 1 mM), decreased sulfoxidation rates by 85 to 89%. In contrast to P450 inhibition by 1-benzylimidazole, heat treatments to inhibit the FMO component of metabolism in HLMs resulted in slight inhibition of the oxidation of these thioether compounds, ranging from 9 to 15% (Table 2).
Relative contributions of HLM P450s and FMOs toward sulfoxidation of phorate, disulfoton, sulprofos, and methiocarb Values are the mean ± S.E.M. (n = 3).
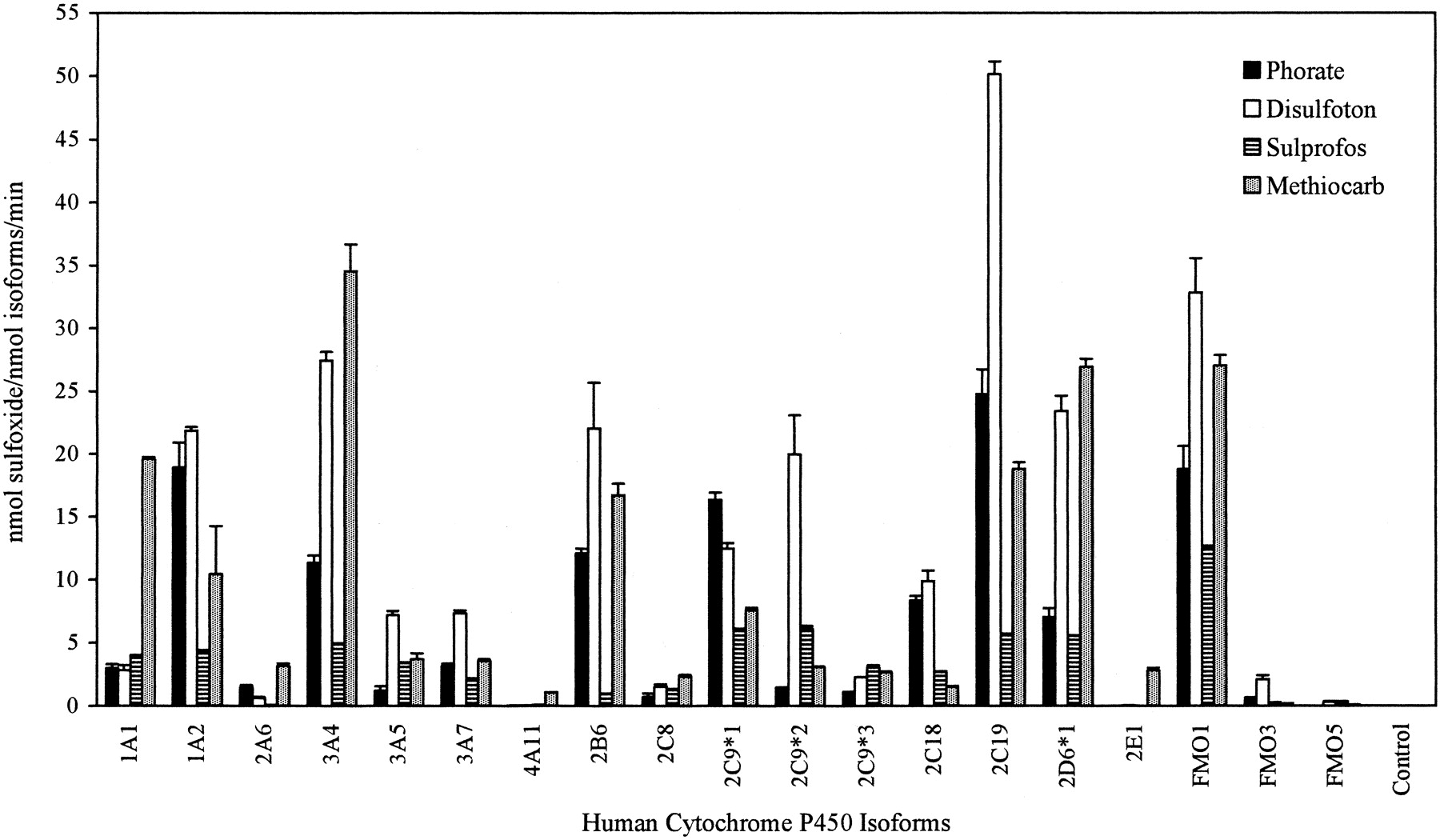
A screen of several P450 and FMO isoforms to investigate the sulfoxidation of thioether compounds was conducted (Fig. 2). Sulfoxidation of phorate to phorate sulfoxide was observed with all isoforms except CYP4A11, 2E1, and FMO5. Isoforms that showed high levels of phorate sulfoxide production were CYP1A2, 3A4, 2B6, 2C9*1, 2C18, 2C19, 2D6*1, and FMO1. As with phorate, all isoforms with the exception of 4A11 and 2E1 were able to oxidize disulfoton to disulfoton sulfoxide. Isoforms with elevated levels of disulfoton sulfoxide production included CYP1A2, 3A4, 2B6, 2C9*1, 2C9*2, 2C18, 2C19, 2D6*1, and FMO1. With sulprofos, all isoforms except 2E1 were able to oxidize sulprofos to sulprofos sulfoxide. Those with elevated levels of sulprofos sulfoxide formation included CYP1A1, 1A2, 3A4, 3A5, 2C9*1, 2C9*2, 2C9*3, 2C18, 2C19 2D6*1, and FMO1. With methiocarb, all isoforms with the exception of FMO5 were able to oxidize methiocarb to methiocarb sulfoxide. Isoforms that showed elevated levels of methiocarb sulfoxide production were CYP1A1, 1A2, 3A4, 2B6, 2C9*1, 2C19, 2D6*1, and FMO1.

Sulfoxidation of thioether compounds using human liver P450 and FMO isoforms.
Screening assays were run as recommended by the enzyme supplier, with a substrate concentration of 200 μM, an NADPH regenerating system, 30 min incubation, and P450 isoform final concentration of 50 pmol/ml or FMO isoform final concentration of 104-300 pmol/ml. Specific activities are expressed as nanomoles of product formed per nanomole of isoform per minute (n = 2).
Based on the preliminary screening assays described above (Fig. 1), detailed kinetic analyses were conducted for each thioether pesticide substrate with those P450 and FMO isoforms with appreciable metabolic capacity. Results of these analyses are shown for phorate, disulfoton, sulprofos, and methiocarb in Tables 3 to 6, respectively. Derivations of the Km and Vmax values for each substrate and isoform allowed calculations of individual intrinsic clearance rates and normalized rates of in vitro hepatic clearance as described by Rodrigues (1999a). Summation of the normalized rates then allowed for the determination of the %TNR for each isoform. These estimates of enzyme clearance take into account the relative differences in substrate affinity for the enzyme as well as the content and distribution of the isoforms involved in metabolism.
Sulfoxidation activity toward phorate with P450 and FMO isoforms Values are the mean ± S.E.M. (n = 3).
Sulfoxidation activity toward methiocarb with P450 and FMO isoforms Values are the mean ± S.E.M. (n = 3).
To investigate whether thioether compounds are metabolized at the same catalytic site as other CYP2C substrates, disulfoton was co- or preincubated with known CYP2C inhibitors (Fig. 3). Sulfaphenazole, a competitive inhibitor of CYP2C9, when coincubated with disulfoton and CYP2C9*1, inhibited disulfoton sulfoxidation with an IC50 value of 0.84 μM. Ticlopidine, a mechanism-based inhibitor of CYP2C19, when co- or preincubated with disulfoton and CYP2C19, inhibited disulfoton sulfoxidation with IC50 values of 43.5 μM and 4.30 μM, respectively.

Inhibition profiles of CYP2C9*1-dependent sulfoxidation of disulfoton by sulfaphenazole (A) and CYP2C19-dependent sulfoxidation of disulfoton by ticlodipine (B).
After 5 min of preincubation with HLMs, sulfoxidation of disulfoton was inhibited 29% by sulfaphenazole and 57% by ticlopidine. When both inhibitors were present, the combined inhibition was 71%. The results were not significantly different when the preincubation time was increased to 10 min.
Discussion
The mean metabolic intrinsic clearance rates obtained in human liver microsomes, as estimated by Vmax/Km, indicate that phorate and disulfoton are metabolized to phorate sulfoxide and disulfoton sulfoxide more efficiently than sulprofos and methiocarb (Table 1). Previous work in our laboratory determined that the relative contributions of each pathway responsible for phorate metabolism in mice could be established by selective inhibition of the alternate monooxygenase systems in microsomal preparations (Kinsler et al., 1988). Results of the current study have shown that in human liver microsomes, metabolism of thioether compounds to the corresponding sulfoxide is primarily P450-driven. Our experiments using a 1-min heat treatment at 50°C to inhibit the FMO contribution to metabolism decreased activity only slightly, to 85 to 90% of the control activity (Table 2), whereas the P450 inhibitor 1-benzylimidazole inhibited activity by 85 to 89%. Although heat treatments of this magnitude do not inhibit most P450 isoforms by more than 10%, a 55°C heat treatment has been shown to inhibit metabolic activity by the CYP2A6 and 2C9 isoforms by as much as 30% (Grothusen et al., 1996); thus, decreased contributions by these isoforms cannot be entirely ruled out. However, the fact that P450 inhibition by 1-benzylimidazole decreased sulfoxide formation by 85 to 89% and heat treatment by 9 to 15% is corroborative evidence that both P450s and FMOs have a role in this metabolic pathway. The results obtained with human liver microsomes are similar to previous results with mouse liver microsomes showing that phorate sulfoxide production due to P450 was 77% of the total, whereas that due to FMO was 23% (Kinsler et al., 1988). This is, however, different from inhibition results with rat liver microsomes in which the percentage of methiocarb sulfoxide production as compared with the control was approximately 50% each for P450 and FMO (Buronfosse et al., 1995). These metabolic differences are probably due to the fact that rats have higher expression of FMO1 than either mice or adult human liver (Cherrington et al., 1998).
Following the inhibition of the P450 or FMO monooxygenase systems, we were also interested in the relative contributions of individual P450 and FMO isoforms. Preliminary results using thin layer chromatographic methods had demonstrated that phorate was metabolized to phorate sulfoxide by the human P450 isoforms 1A2, 2E1, 3A4, 2C8, 2C9, 2C18, and 2C19 (Hodgson et al., 1998). These results suggested that members of the CYP2C subfamily were important in much of the phorate sulfoxide production in the liver, with isoforms 2C19 and 2C9 having the highest activity (although 2C18 was also high, this isoform is not expressed in liver to any significant extent). Our current results using three additional substrates and baculovirus-expressed Supersomes for all isoforms demonstrated that all of those tested, except CYP4A11 and 2E1, were capable of sulfoxidation of phorate and disulfoton. Likewise, for sulprofos, all isoforms tested, except 2E1, could facilitate this reaction and, of the FMOs tested, only FMO5 lacked the ability to metabolize methiocarb. Mean intrinsic clearance rates indicated that CYP2C9 and 2C19 were the predominant isoforms involved in sulfoxidation.
Previous studies of pesticide metabolism in rodents have demonstrated that phorate, disulfoton, sulprofos, and methiocarb were good substrates for FMO activity (Hajjar and Hodgson, 1982; Kinsler et al., 1988; Buronfosse et al., 1995). Our results confirm a role for FMO in these pesticide sulfoxidations in humans and further identify FMO1 as the primary FMO involved in the metabolism of thioether compounds. However, since FMO1 is poorly expressed in adult human liver (Phillips et al., 1995), the FMO contributions to metabolism of these pesticides in the liver are likely the result of FMO3, the predominant FMO isoform in adult human liver (Haining et al., 1997; Overby et al., 1997). Contributions of human FMO1 to the overall metabolism of these substrates will require further studies since FMO1 is well expressed in human kidney, has some expression in intestinal tissues (Yeung et al., 2000), and is the primary constitutive isoform found in human fetal livers (Dolphin et al., 1996).
Results of the isoform screening assays revealed that several P450 isoforms were capable of the sulfoxidation of these thioether substrates (Fig. 2). For each substrate examined, members of the CYP2C family, including CYP2C9, 2C18, and 2C19, had some of the highest affinities (lowest Km) of all the isoforms examined (Tables 3, 4, 5, 6). Other isoforms with high affinities for certain of these substrates included CYP1A1, CYP1A2, 2B6, and FMO1. Intrinsic clearance values among these isoforms tended to rank similarly to affinity determinations. However, the relative importance of individual isoforms to in vivo clearance is also dependent upon the relative abundance of each isoform. Thus, CYP2C18, which easily catalyzes phorate sulfoxidation, has essentially no role in phorate clearance in liver tissues because of its poor expression in liver samples (%TNR = 1.3). In contrast, CYP3A4 with one of the lowest intrinsic clearance rates, could contribute substantially to the metabolism of each substrate (TNR = 24-29%) because of its high relative abundance in liver tissue.
Sulfoxidation activity toward disulfoton with P450 and FMO isoforms Values are the mean ± S.E.M. (n = 3).
Sulfoxidation activity toward sulprofos with P450 and FMO isoforms Values are the mean ± S.E.M. (n = 3).
In these studies, the high %TNR observed for CYP2C9 clearly implicates an important role for this isoform in the metabolism of these thioether pesticides. It is interesting to note that the two polymorphic forms of CYP2C9 also retained the ability to metabolize sulprofos, although this was not true for the other substrates. Although isoforms such as CYP2C18 and 1A1 often had substrate affinities and clearances similar to 2C9, their absence in noninduced liver samples tends to negate their overall importance. Likewise, even though the intrinsic clearance rates for CYP2C19 were greater for each substrate than those observed for 1A2, the relative abundance of 1A2 relative to 2C19 always resulted in higher %TNR for 1A2.
The potential importance of the CYP2C subfamily in sulfoxidation reactions is further demonstrated by the results of inhibition studies carried out in HLMs using the specific 2C9 competitive inhibitor, sulfaphenazole, and the specific mechanism-based 2C19 inhibitor ticlopidine. These experiments, conducted at inhibitor concentrations shown to result in ca. 100% inhibition of disulfoton oxidation when preincubated with their respective isoforms, predict that ca. 30% of the overall HLM sulfoxidation of disulfoton is catalyzed by 2C9 and over 50% by 2C19. The inhibition of activity in HLMs by sulfaphenazole, the CYP2C9 inhibitor, reflects the percentage of total normalized rate for the contribution of CYP2C9 in sulfoxidation of disulfoton. In contrast, inhibition by ticlopidine may have overestimated the contribution of CYP2C19, although it is possible that in this particular sample, CYP2C19 may have greater than normal expression or that ticlopidine under the conditions utilized may have inhibited other contributing isoforms.
Inhibitors are important tools to study the topology of the active site of enzymes. Studies with sulfaphenazole, a well known competitive inhibitor of CYP2C9, and a high affinity substrate of CYP2C9 exhibited a Ki value of 0.3 μM and IC50 of 0.6 μM (Mancy et al., 1996; Ha-Duong et al., 2001a; Melet et al., 2003). The IC50 value (0.84 μM) we obtained for the inhibition of thioether sulfoxidation of disulfoton by sulfaphenazole indicates that these thioether substrates are competing for the same catalytic site as other high affinity CYP2C9 substrates. Similarly, it has been shown that ticlopidine, a selective mechanism-based inhibitor of CYP2C19, inhibits typical substrates of 2C19 with IC50 values of ca. 10 μM (Donahue et al., 1997; Ha-Duong et al., 2001a). The values we obtained by coincubation (IC50 43.5 μM) and preincubation (IC50 4.3 μM) with ticlopidine indicated that disulfoton sulfoxidation occurs at the same catalytic site as typical CYP2C19 substrates.
In the human liver, the CYP2C isoforms account for nearly 20% of the total P450 content (Shimada et al., 1994). Due to the involvement of the 2C isoforms in the oxidation of a relatively large number of pharmaceutical drug oxidations, there has been considerable interest in this P450 family. Much of this interest was initiated by the discovery of numerous polymorphisms and substrate recognition sites within these isoforms (Goth, 1992; Goldstein, 2001). A great deal of interest has also been focused upon molecular modeling techniques to derive a predictive model for substrates of the CYP 2C family, primarily CYP2C9 (Mancy et al., 1995, 1996; Poli-Scaife et al., 1997; Ha-Duong et al., 2001a; Melet et al., 2003). Even though there is currently no published crystal structure for a human membrane-bound P450, interest has recently been stimulated by the development of a crystal structure for the rabbit CYP2C5 (Cosme and Johnson, 2000; Williams et al., 2000).
It is generally accepted that most high affinity substrates or inhibitors of CYP2C9 possess at least one site for hydrogen bond formation between 5 and 10 Å from the site of metabolism, and many CYP2C9 substrates are weak acids containing an ionizable carboxylic acid moiety situated 7 to 11 Å from the known position metabolized specifically by this isoform (Lewis et al., 1998). Although some neutral substrates have been found that bind with high affinity to this enzyme, these compounds generally have aromatic rings associated with their structure (Miners and Birkett, 1998). Furthermore, Melet et al., (2003) reported that a phenylalanine 114 residue at the catalytic site of CYP2C9 plays an important role in recognition of aromatic substrates of 2C9 through π-stacking interactions. Additionally, although numerous compounds have been examined as potential substrates for CYP2C9 oxidation, the oxidation sites identified have been based on oxidation at carbon atoms. There has been no modeling work describing the S-oxidation of substrates by CYP2C9. Our results indicate that phorate, disulfoton, sulprofos, and methiocarb, which do not fit well with the predictive model substrate for 2C9, are also high affinity substrates for the CYP2Cs. These compounds are nonionizable and do not contain carboxylic acid moieties. Furthermore, although phorate and disulfoton do not contain an aromatic ring structure, they have the highest affinity and clearance values among these four substrates with either CYP2C9, 2C18, or 2C19. In addition, these substrates are relatively small molecules compared with the optimum substrate for oxidation at carbon. This leads us to believe that the substrate specificity may be broader than is currently believed and that a variety of related organophosphorus or carbamate compounds are also likely to be metabolized by members of the 2C family that do not necessarily fit well within the current predictive substrate modeling designs. Predictive substrate modeling work for the 2C family may need to be expanded to include a different substrate design for sulfoxidation reactions.
Footnotes
-
↵1 Abbreviations used are: P450, cytochrome P450; HPLC, high performance liquid chromatography; HLM, human liver microsomes; FMO flavin-containing monooxygenase.
-
This research was supported in part by National Institute for Occupational Safety and Health Grant OH07551-ECU and U.S. Army Agreement DAMD 17-00-2-0008.
-
Parts of these studies were presented at the 11th North American ISSX meeting in Orlando, Florida in October 2002, and at the 12th North American ISSX meeting in Providence, Rhode Island in October 2003. Some of the studies presented were included in the Ph.D. theses of E.D.K.
- Received September 2, 2003.
- Accepted December 8, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}