


Abstract
The glucuronidation kinetics of the prototypic substrates 4-methylumbelliferone (4MU) and 1-naphthol (1NP) by human UDP-glucuronosyltransferases (UGT) 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15, and 2B17 were investigated. Where activity was demonstrated, inhibitory effects of diclofenac, probenecid, and the solvents acetone, acetonitrile, dimethyl sulfoxide, ethanol, and methanol were characterized. All isoforms except UGT1A4 glucuronidated 4MU, whereas all but UGT 1A4, 2B15, and 2B17 metabolized 1NP. However, kinetic models varied with substrate (for the same isoform) and from isoform to isoform (with the same substrate). Hyperbolic (Michaelis-Menten), substrate inhibition, and sigmoidal kinetics were variably observed for both 4MU and 1NP glucuronidation by the various UGTs. Km or S50 (sigmoidal kinetics) and Vmax values varied 525- (8–4204 μM) and 5535-fold, respectively, for 4MU glucuronidation, and 1360- (1.3–1768 μM) and 560-fold, respectively, for 1NP glucuronidation. The use of a two-site model proved useful for those reactions exhibiting non-Michaelis-Menten glucuronidation kinetics. The organic solvents generally had a relatively minor effect on UGT isoform activity. UGT 1A6, 2B15, and 2B17 were most susceptible to the presence of solvent, although solvent-selective inhibition was occasionally observed with other isoforms. Diclofenac and probenecid inhibited all isoforms, precluding the use of these compounds for the reaction phenotyping of xenobiotic glucuronidation pathways in human tissues. Diclofenac and probenecid Ki values, determined for selected isoforms, ranged from 11 to 60 μM and 96 to 2790 μM, respectively. Overall, the results emphasize the need for the careful design and interpretation of kinetic and inhibition studies with human UGTs.
PUBLISHER'S NOTE
The authors discovered that the recombinant UGT1A6 enzyme preparation used in this work also contained UGT1A9. Accordingly, experiments involving UGT1A6 were repeated with HEK293 cell lysate expressing only this enzyme. Data reported for all other isoforms are accurate. The corrected version of the paper, which is in departure from print, follows.
Glucuronidation is a synthetic reaction that involves covalent linkage (or “conjugation”) of a suitable functional group present on a substrate with glucuronic acid. This reaction, which requires UDP-glucuronic acid (UDPGA1) as cofactor, is catalyzed by the enzyme UDP-glucuronosyltransferase (UGT). Conjugation with glucuronic acid is responsible for the elimination of structurally diverse xenobiotics and endogenous compounds (Miners and Mackenzie, 1991; Radominska-Pandya et al., 1999; Tukey and Strassburg, 2000). In particular, glucuronidation is an essential clearance mechanism for drugs from all therapeutic classes. Glucuronidation additionally serves as an elimination pathway in humans for numerous dietary chemicals, environmental pollutants, and endogenous compounds (e.g., bilirubin, bile acids, and hydroxysteroids). Moreover, glucuronidation facilitates excretion of these compounds and the products of phase I metabolism in urine and bile as their hydrophilic conjugates, and generally results in detoxification, although a limited number of glucuronides possess biological activity (Ritter, 2000).
Consistent with its broad substrate profile, UGT exists as an enzyme superfamily. UGT genes have been classified into families and subfamilies based on the sequence identity of the encoded proteins (Mackenzie et al., 1997). Sixteen human UGT proteins have been identified to date: UGT 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2A1, 2B4, 2B7, 2B10, 2B11, 2B15, 2B17, and 2B28. Available evidence suggests that most isoforms exhibit distinct, but frequently overlapping substrate selectivities (Radominska-Pandya et al., 1999; Tukey and Strassburg, 2000). Furthermore, the individual isoforms differ in terms of regulation. Notably, whereas the majority of human UGTs are expressed in liver (and to a variable extent in other tissues), some isoforms are expressed only in extrahepatic tissues. The latter include UGT2A1 (predominantly the nasal epithelium), and UGT1A7, UGT1A8, and UGT1A10 (all gastrointestinal tract) (Tukey and Strassburg, 2000).
Given the importance of glucuronidation as a clearance and detoxification mechanism, there is growing interest in the identification of substrates that may be used to assess human UGT activity in vitro. 4MU and 1NP have been promoted in this regard since both form fluorescent glucuronides that are readily measurable (Bock et al., 1983; Miners et al., 1988). 4MU and 1NP are known to be metabolized by multiple human UGT isoforms, and hence these compounds represent convenient probes for comparing the glucuronidation activity and kinetics of tissues and recombinant enzymes. However, isoform selectivity has not been assessed in a systematic manner, and the kinetics of 4MU and 1NP glucuronidation by the individual human UGTs have not been characterized. Thus, optimal conditions for the measurement of UGT isoform activity using these substrates remain unknown.
There is increasing use of recombinant human UGTs to identify the isoform(s) responsible for the metabolism of new chemical entities and other compounds (i.e., “reaction phenotyping”) of pharmacological, toxicological, and physiological importance, as well as for determination of kinetic parameters. However, many UGT substrates are lipophilic and require organic solvents for effective solubilization in incubation mixtures. Organic solvents are known to variably affect cytochrome P450 (P450) isoform activity (Chauret et al., 1998; Hickman et al., 1998). Although it has been reported that a number of solvents do not significantly alter umbelliferone glucuronidation by cryopreserved human hepatocytes (Easterbrook et al., 2001), a selective effect on individual hepatic and extrahepatic human UGTs cannot be discounted.
In addition to the use of recombinant enzymes, the use of P450 isoform-selective inhibitors has proved invaluable for the reaction phenotyping of P450-catalyzed reactions in human tissues (Miners et al., 1994; Rodrigues 1999). Unlike P450, however, there are currently no UGT isoform-selective inhibitors available for this purpose (Miners et al., 2004). Diclofenac inhibits UGT1A9- and UGT2B7-catalyzed xenobiotic glucuronidation (Miners et al., 1997; Ammon et al., 2000; King et al., 2001), and probenecid has been implicated in the inhibition of numerous glucuronidated drugs in vivo (Miners and Mackenzie, 1991). Despite the known effects of diclofenac and probenecid on human UGT activities, studies that would permit the use of either of these compounds as selective inhibitors are lacking.
We report here a systematic investigation of the glucuronidation kinetics of 4MU and 1NP by the human xenobiotic-metabolizing isoforms UGT 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15, and 2B17. Where activity was demonstrated, inhibitory effects of diclofenac, probenecid, and the solvents acetone, acetonitrile, dimethyl sulfoxide (DMSO), ethanol, and methanol were characterized. Apart from characterizing the properties of human UGTs, which are generally poorly understood, the results provide important insights into the variable kinetic behavior of human UGTs. In particular, glucuronidation kinetic models were shown to vary between substrates for the same isoform and between isoforms with the same substrate.
Materials and Methods
Materials. 4MU, 4-methylumbelliferone-β-d-glucuronide (4MUG), 1NP, 1-naphthol-β-d-glucuronide (1NPG), UDP-glucuronic acid (UDPGA, trisodium salt), diclofenac (sodium salt), and probenecid were purchased from Sigma-Aldrich (St Louis, MO). Other reagents and organic solvents, including acetone, acetonitrile, DMSO, ethanol, and methanol, were of analytical reagent grade.
Methods.Expression of UGT proteins. Details of the UGT 1A3, 1A6, 1A8, 1A9, 1A10, and 2B7 cDNAs used here have been reported previously (Jin et al., 1993a; Mojarrabi et al., 1996; Miners et al., 1997; Mojarrabi and Mackenzie, 1997, 1998). cDNAs encoding UGT 1A1, 1A4, 1A7, 2B15, and 2B17 were polymerase chain reaction-amplified from Caco2 or HepG2 cells or from a human cDNA library, and the identity of the coding regions was confirmed by sequence analysis. The individual UGT cDNAs were stably expressed in a human embryonic kidney cell line (HEK293) as described previously (Sorich et al., 2002; Stone et al., 2003). Cells were separately transfected with the individual UGT cDNAs cloned into the pEF-IRES-puro6 expression vector (Hobbs et al., 1998). Following transfection, cells were incubated at 37°C in Dulbecco's modified Eagle's medium, which contained puromycin (1.5 mg/l), 10% fetal calf serum, and gentamicin (160 mg/l) in a humidified incubator with an atmosphere of 5% CO2. After growth to at least 80% confluency, cells were harvested and washed in phosphate-buffered saline. The cells were subsequently lysed by sonication using a Heat Systems-Ultrasonics sonicator set at a microtip limit of 4. Cells expressing UGT1A proteins were sonicated with four 2-s “bursts,” each separated by 3 min of cooling on ice. A similar method was applied to UGT2B subfamily enzymes, except that sonication was limited to 1-s bursts due to the greater instability of these proteins to sonication. Lysates were centrifuged at 12,000g for 5 min, and the supernatant fraction was separated and stored in phosphate buffer (0.1 M, pH 7.4) containing dithiothreitol (0.5 mM) at –80°C until use. The protein concentration of cell lysates was determined by the Lowry method (Lowry et al., 1951) using bovine serum albumin as the standard.
4MU and 1NP glucuronidation assays. 4MU and 1NP glucuronidation were measured using a previously published procedure (Miners et al., 1988). Briefly, incubations (total volume 0.6 ml) contained UDPGA (5 mM), MgCl2 (5 mM), cell lysate (protein concentrations given in Table 1), 4MU or 1NP, and phosphate buffer (0.1 M, pH 7.4). Substrate concentration ranges spanned the Km or S50 and hence varied for each isoform (Table 1). Incubations of UGT 1A1, 1A3, 1A8, 1A10, 2B7, and 2B15 with 1NP contained 0.5% DMSO, whereas incubations of UGT2B17 with both 4MU and 1NP contained acetonitrile (0.5%). After a 5-min preincubation at 37°C in a shaking water bath, reactions were initiated by the addition of substrate. Incubation times for each isoform activity are shown in Table 1. Rate of product formation was optimized for linearity with respect to protein concentration and incubation time, which varied for each UGT isoform (Table 1). Reactions were terminated by the addition of 0.6 M glycine-0.4 M trichloroacetic acid (0.14 ml) and cooling on ice. Following the addition of KH2PO4 (0.1 ml; 1 M, pH 7.4), mixtures were extracted with chloroform (7 ml) using a rotary mixer and then centrifuged at 1500g for 10 min. A 0.6-ml aliquot of the aqueous phase was separated for measurement of fluorescence (PerkinElmer model 3000 fluorescence spectrophotometer; PerkinElmer Life and Analytical Sciences, Boston, MA). Excitation/emission wavelengths for 4MUG and 1NPG fluorescence were 315/365 nm and 290/330 nm, respectively. Fluorescence readings were converted to 4MUG or 1NPG concentrations by comparison with 4MUG or 1NPG standard curves prepared over the concentration range 0.1 to 10 μM. The 4MUG and 1NPG standards were treated in the same manner as the incubation samples. Within-day overall 4MUG and 1NPG assay imprecision, determined by measuring product formation in five separate incubations using human liver microsomes as the enzyme source, was <4% for 4MU and 1NP concentrations in the range 20 to 2000 μM. All incubations were performed in duplicate; data points represent the mean (<10% variance) of the duplicate measurements.
Protein amount, incubation time, and substrate concentration ranges used for the measurement of 4MU and 1NP glucuronidation
Inhibition of 4MU glucuronidation by organic solvents. The potential inhibitory effects of acetone, acetonitrile, DMSO, ethanol, and methanol on the activity of each of the UGT isoforms studied here were investigated using 4MU as the substrate. Incubations were performed as described above with 4MU concentrations at the approximate apparent Km or S50 value (from fitting to the Michaelis-Menten, substrate inhibition, or Hill equation, respectively; see Table 2 and Fig. 3 legend) for the individual isoforms. Each organic solvent was added separately to incubations to give final concentrations of 0.5% and 1%, v/v. The effects of 2% and 4% methanol were additionally characterized, since this solvent is most widely used in incubations for measurement of xenobiotic glucuronidation activity. Data were compared with activities determined in the absence of solvent, and the inhibitory effects of organic solvents are reported as percentage of control activity for triplicate measurements.
Kinetic models and constants for 4MU glucuronidation by human UGT isoforms
Inhibition of 4MU glucuronidation by diclofenac and probenecid. Diclofenac and probenecid were screened as inhibitors of UGT isoform activities using 4MU as the substrate. Again, incubations were performed as described previously, with 4MU concentrations at the approximate apparent Km or S50 value for each isoform. Concentrations of diclofenac and probenecid used in screening experiments were 0, 10, 50, 100, and 500 μM and 0, 0.5, 1, 1.5, and 2 mM, respectively. Diclofenac (sodium salt) was dissolved in water, but the relatively high concentrations of probenecid necessary to characterize UGT isoform inhibition required addition of organic solvent; either 4% methanol (UGT 1A1, 1A7, 1A9, 1A10, and 2B17) or 1% DMSO (UGT 1A3, 1A6, 1A8, 2B7, and 2B15). Solvents were selected on the basis of effects on isoform activity (see Results). Control incubations contained the same concentration of solvent. Subsequent experiments that determined inhibitor constants for selected isoforms (viz. those exhibiting hyperbolic kinetics with 4MU as substrate) included four or five diclofenac/probenecid concentrations at each of three 4MU concentrations.
Immunoblotting. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% acrylamide gels) and transferred onto nitrocellulose (Bio-Rad, Hercules, CA). Blots were probed with a commercial UGT1A antibody (UGT1A isoforms) (BD Gentest, Woburn, MA) or a nonspecific UGT antibody raised against a purified mouse Ugt (Mackenzie et al., 1984) (UGT2B isoforms), followed by horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Southern Biotechnology Associates, Birmingham, AL). UGT protein was visualized by chemiluminescence (Roche Diagnostics, Mannheim, Germany), and intensities of UGT bands were measured with a Bio-Rad model GS-700 Imaging Densitometer.
Data Analysis. Kinetic constants for 4MU and 1NP glucuronidation by each isoform were obtained by fitting untransformed experimental data to the following kinetic models using GraFit (Erithacus Software, Horley, Surrey, UK): The Michaelis-Menten equation,  where v is the rate of reaction, Vmax is the maximum velocity, Km is the Michaelis constant (substrate concentration at 0.5 Vmax), and [S] is the substrate concentration.
where v is the rate of reaction, Vmax is the maximum velocity, Km is the Michaelis constant (substrate concentration at 0.5 Vmax), and [S] is the substrate concentration.
Substrate inhibition model (Houston and Kenworthy, 2000),  where Ksi is the constant describing the substrate inhibition interaction. The Hill equation, which describes sigmoidal kinetics (Cornish-Bowden, 1995),
where Ksi is the constant describing the substrate inhibition interaction. The Hill equation, which describes sigmoidal kinetics (Cornish-Bowden, 1995),  where S50 is the substrate concentration resulting in 50% of Vmax (analogous to Km in previous equations) and n is the Hill coefficient.
where S50 is the substrate concentration resulting in 50% of Vmax (analogous to Km in previous equations) and n is the Hill coefficient.
Autoactivation is characterized by the dependence of clearance on substrate concentration, and an alternative to intrinsic clearance (CLint), calculated as Vmax/Km, is required. For sigmoidal kinetic data, maximum clearance (CLmax) provides an estimate of the highest clearance attained, that is, when the enzyme is fully activated before saturation occurs (Houston and Kenworthy, 2000): 
Atypical kinetic data were additionally fitted to the two-site model described by Houston and Kenworthy (2000) and Galetin et al. (2002). The model is based on a steady-state rapid equilibrium approach and the assumption of no distinct difference between the binding sites,  where Ks is binding affinity and α and β are interaction factors that reflect changes in Ks and product formation (Kp), respectively. For positive homotropic cooperativity (sigmoidal kinetics), Vmax is equivalent to 2Kp[E]t, where [E]t is the total enzyme concentration, and hence β = 2 for this type of kinetics. Sequential binding of substrate molecules is incorporated into the model in the case of substrate inhibition, and the substrate inhibition site cannot be occupied until substrate is bound in the “catalytic” site (Vmax is equivalent to Kp[E]t). Interaction factor β (<1) defines the decrease in product formation from a two-substrate bound complex and is more important for the description of this type of atypical kinetic behavior.
where Ks is binding affinity and α and β are interaction factors that reflect changes in Ks and product formation (Kp), respectively. For positive homotropic cooperativity (sigmoidal kinetics), Vmax is equivalent to 2Kp[E]t, where [E]t is the total enzyme concentration, and hence β = 2 for this type of kinetics. Sequential binding of substrate molecules is incorporated into the model in the case of substrate inhibition, and the substrate inhibition site cannot be occupied until substrate is bound in the “catalytic” site (Vmax is equivalent to Kp[E]t). Interaction factor β (<1) defines the decrease in product formation from a two-substrate bound complex and is more important for the description of this type of atypical kinetic behavior.
Apparent Ki values for inhibition of 4MU glucuronidation by diclofenac and probenecid were determined by fitting experimental data to the expressions for competitive, noncompetitive, and mixed inhibition using Enzfitter (Biosoft, Cambridge, UK).
Goodness of fit to kinetic and inhibition models was assessed from the F statistic, r2 values, parameter standard error estimates, and 95% confidence intervals. Kinetic constants are reported as the value ± standard error of the parameter estimate. Kinetic curves are shown as Eadie-Hofstee plots from fitting to the Michaelis-Menten, substrate inhibition, and Hill equations.
Results
Expression of individual UGTs was confirmed by Western blot analysis using a commercial anti-UGT1A antibody (UGT1A proteins) and a nonspecific anti-UGT antibody raised against purified mouse Ugt (UGT2B proteins). Apart from UGT1A4, each of the isoforms studied here expressed as a protein of apparent molecular mass of approximately 52.5 kDa (Fig. 1). UGT1A4 migrated as a protein of marginally higher molecular mass (viz. 54 kDa), which has been noted previously (Bosma et al., 1994). Expression of UGT 1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, and 1A10 relative to UGT1A1 was 1:1.19:1.30:1.31:1.30:0.56:1.58:0.99, and expression of UGT 2B7, 2B15, and 2B17 (relative to UGT2B7) was 1:1.72:4.60.
With the exception of UGT1A4, all isoforms glucuronidated 4MU. [UGT1A4 activity was, however, confirmed with trifluoperazine as the substrate using a radiometric thin-layer chromatographic procedure (Jin et al., 1997) (data not shown).] The kinetic model of 4MU glucuronidation varied between isoforms (Table 2; Fig. 2). 4MU glucuronidation by UGT 1A1, 1A6, 1A7, and 1A10 followed Michaelis-Menten kinetics, with apparent Km values ranging from 14 to 113 μM (Table 2). In contrast, 4MU glucuronidation by UGT 1A3, 1A8, 1A9, 2B15, and 2B17 exhibited substrate inhibition (Table 2; Fig. 2). Substrate inhibition with UGT 1A9 and 2B15 was weak (Km 7- to 28-fold lower than Ksi), but Ksi and Km were similar for UGT1A8, and Ksi was lower than Km for UGT 1A3 and 2B17. UGT2B7-catalyzed 4MU glucuronidation followed sigmoidal kinetics (S50 = 335 μM), which manifests as a curvilinear Eadie-Hofstee plot (Fig. 2). 4MU glucuronidation kinetic data for UGT 1A3, 1A8, 1A9, 2B7, and 2B15, which exhibited substrate inhibition or sigmoidal kinetics, were additionally fitted to a two-site model with standard errors comparable with those for the substrate inhibition model or Hill equation (Table 2). Ks values generated using the two-site model were in the range 8.2 to 1914 μM. Although 4MU glucuronidation by UGT2B17 also exhibited substrate inhibition, limitations of assay sensitivity precluded generation of sufficient data at low substrate concentrations for reliable fitting to the two-site model. Vmax values for 4MU glucuronidation by the 10 human UGTs varied almost 5500-fold (Table 2). Since the relative recognition of each UGT protein by the antibodies used here is unknown, normalization of Vmax values for differences in expression was not undertaken. However, there was only 2.8-fold variability in the apparent expression of UGT1A isoforms. Apparent expression of UGT2B7 and UGT2B15 similarly differed less than 2-fold, but apparent expression of UGT2B17 was 4.6-fold higher than for UGT2B7.

Western blot analysis of protein expression levels in lysates of HEK293 cells stably expressing human UGT isoforms (UGT1A proteins, upper panel; UGT2B proteins, lower panel).
Each lane contained 20 μg of HEK293 lysate protein. Untransfected HEK293 cells served as the negative control.
UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, and 2B7 glucuronidated 1NP, whereas metabolism by UGT1A4, 2B15, and 2B17 could not be detected using the fluorescence assay employed in this work. 1NP glucuronidation by UGT 1A3 and 1A10 exhibited substrate inhibition and Michaelis-Menten kinetics, respectively (Fig. 2; Table 3). UGT 1A1-, 1A6-, 1A7-, 1A8-, 1A9-, and 2B7-catalyzed 1NP glucuronidation followed sigmoidal kinetics (Fig. 2; Table 3), with S50 values ranging from 1.3 μM (UGT1A9) to 345 μM (UGT1A1) and Hill coefficients in the range 1.3 (UGT1A1 and UGT1A6) to 2.3 (UGT1A7). As with 4MU, 1NP glucuronidation by those isoforms exhibiting atypical kinetics were also well described by fitting to the two-site model, with Ks values ranging from 6.7 to 1978 μM (Table 3). The range of Vmax values (560-fold; from empirical models) was lower than that observed with 4MU as the substrate.
Kinetic models and constants for 1NP glucuronidation by human UGT isoforms

Representative Eadie-Hofstee plots: 4MU glucuronidation by UGT1A6 (panel A), UGT1A8 (panel B), and UGT2B7 (panel C), and 1NP glucuronidation by UGT1A3 (panel D), UGT1A9 (panel E), and UGT1A10 (panel F).
Kinetic data were model-fitted as described under Materials and Methods (Data Analysis).
In vitro clearance for 4MU and 1NP glucuronidation was assessed as CLint (substrate inhibition and Michaelis-Menten kinetics) or CLmax (sigmoidal kinetics). 4MU clearance by UGT 1A6, 1A7, 1A9, and 1A10 was 2 to 3 orders of magnitude higher than for UGT 1A1, 1A3, and 1A8, and 3 to 5 orders of magnitude higher than for UGT 2B7, 2B15, and 2B17 (Table 4). 1NP clearance by UGT1A6 was more than 5-fold higher than for any other isoform (Table 4). 1NP clearances by UGT 1A7, 1A8, 1A9, and 1A10 were also substantially higher compared with clearance values for 1NP glucuronidation by UGT 1A1 and 1A3 and UGT2B7 (the only UGT2B subfamily isoform that metabolized this compound). As noted above, relative recognition of each UGT by the antibodies used here is unknown, and this precluded normalization of Vmax (and hence in vitro clearance) for protein expression. Even taking this limitation into account, the in vitro clearance data (Table 4) suggest that the UGT 1A6 to 1A10 cluster of enzymes preferentially metabolizes 4MU and 1NP.
In vitro clearance for 4MU and 1NP glucuronidation by human UGT isoforms
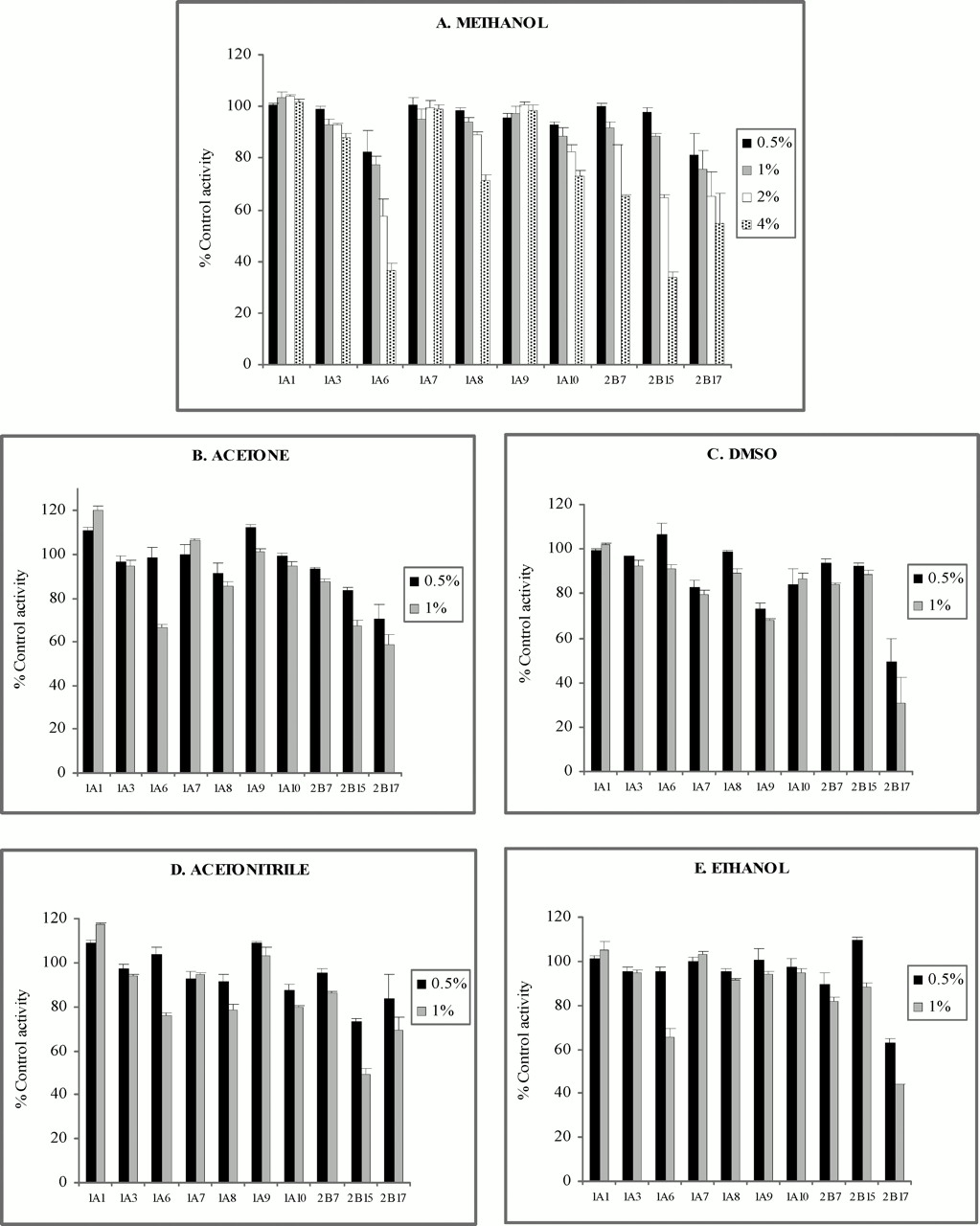
The effects of acetone, acetonitrile, DMSO, ethanol, and methanol on the activities of UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15, and 2B17 were assessed at the 4MU concentration corresponding to the approximate Km or S50. Results are shown in Fig. 3. With the exception of UGT 1A6, 2B15, and 2B17, solvent concentrations of 0.5 and 1% had relatively minor effects (≤20% change in control activity) on isoform activity, although some variability was observed. One percent acetonitrile reduced the activity of UGT1A8 by >20%, and DMSO (0.5 and 1%) reduced the activity of UGT 1A9 by >20%. Higher concentrations of methanol (viz. 2 and 4%, v/v) similarly had a relatively minor effect on the activity of most isoforms, although UGT 1A8, 1A10, and 2B7 activities were decreased by 24%, 23%, and 35%, respectively, in the presence of 4% methanol. Two percent DMSO, necessary for solubilization of 1NP in kinetic studies with UGT1A3, had a negligible influence on the activity of this isoform (data not shown). UGT 1A6, 2B15, and 2B17 were generally more sensitive to the effects of organic solvents (Fig. 3). Acetone, acetonitrile and ethanol (at a concentration of 1% v/v), and methanol (1, 2, and 4% v/v) decreased UGT1A6 activity by >20%, although DMSO (0.5 and 1% v/v) had a minor effect on UGT1A6 activity. Methanol, DMSO, and ethanol (0.5 and 1%) decreased UGT2B15 activity to the least extent, whereas methanol and acetonitrile (0.5 and 1%) had the least inhibitory effect on UGT2B17.
The effects of diclofenac (10–500 μM) and probenecid (0.5–2 mM) on the activities of UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15, and 2B17 were also determined at the 4MU concentration corresponding to the respective Km or S50 values of these isoforms. Both compounds variably inhibited the activity of all isoforms in a concentration-dependent manner (Fig. 4). Preincubation of diclofenac and probenecid with each isoform in the presence of UDPGA precluded irreversible inhibition. Inhibition by diclofenac ranged from 5 to 32% at 10 μM to 68 to 95% at 500 μM, whereas inhibition by probenecid ranged from <5 to 82% at 0.5 mM and 20 to 95% at 2 mM (Fig. 4). The kinetics of diclofenac and probenecid inhibition were investigated for those isoforms exhibiting Michaelis-Menten 4MU glucuronidation kinetics (viz. UGT 1A1, 1A6, 1A7, and 1A10) and for UGT1A9 (“weak” substrate inhibition). Apart from diclofenac and probenecid inhibition of UGT1A6-catalyzed 4MU glucuronidation, data were best fitted to a competitive inhibition model (Fig. 5; Table 5). Inhibition of UGT1A6 by diclofenac and probenecid were best fitted to mixed (competitive-noncompetitive) and noncompetitive inhibition models, respectively. Ki values for inhibition by diclofenac and probenecid ranged from 11 to 60 μM and 96 to 2790 μM, respectively.
Inhibitor constants for diclofenac and probenecid inhibition of 4MU glucuronidation by UGT isoforms a

Effects of organic solvents on 4MU glucuronidation by UGT isoforms.
Panels to the right show individual solvent concentrations. Concentrations of 4MU were: 100 μM, UGT1A1; 1000 μM, UGT1A3; 100 μM, UGT1A6; 15 μM, UGT1A7; 750 μM, UGT1A8; 10 μM, UGT1A9; 30 μM, UGT1A10; 350 μM, UGT2B7; 300 μM, UGT2B15; and 3400 μM, UGT2B17. Each bar represents the mean percentage activity relative to control from three or four replicates; error bars indicate standard deviations.

Inhibitory effects of diclofenac and probenecid on 4MU glucuronidation by human UGT isoforms.
Panels to the right show inhibitor concentrations. 4MU concentrations were as listed in Fig. 3. Each bar represents the mean percentage activity relative to control from duplicate measurements.
Discussion
Under the assay conditions employed in this study, UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, and 2B7 were shown to glucuronidate both 4MU and 1NP, whereas UGT 2B15 and 2B17 glucuronidated only 4MU. Other work in this laboratory has demonstrated that UGT2B4 also metabolizes both 4MU and 1NP (C. Rogers, P. I. Mackenzie, and J. O. Miners, unpublished data), although a more sensitive radiometric assay was necessary for the measurement of 4MUG and 1NPG formation by this enzyme. UGT1A4 lacked the capacity to glucuronidate 4MU and 1NP, consistent with a previous report of negligible activity (Green and Tephly, 1996). Of the remaining known human UGT proteins, UGT2B10 has previously been shown not to metabolize 4MU and 1NP (Jin et al., 1993b) and UGT2B11 is similarly devoid of xenobiotic glucuronidation activity (Beaulieu et al., 1998), whereas UGT2A1 (expressed only in nasal epithelium) and UGT2B28 were unavailable for study. Together with the clearance estimates (i.e., CLmax and CLint) reported here for 4MU and 1NP, it is apparent that UGT 1A6, 1A7, 1A8, 1A9, and 1A10 in general exhibit highest activity toward these phenolic substrates. Of the hepatically expressed isoforms, 1NP is a selective substrate for UGT1A6.
Using 4MU and 1NP as “probe” substrates, it was demonstrated that not only may glucuronidation kinetic models vary between substrates for the same isoform, but kinetic behavior varies between isoforms with the same substrate. Thus, 4MU glucuronidation by UGT 1A1 (Sorich et al., 2002), 1A6, 1A7, and 1A10 follows Michaelis-Menten (i.e., hyperbolic) kinetics; UGT 1A3, 1A8, 1A9, 2B4 (C. Rogers, P. I. Mackenzie, and J. O. Miners, unpublished data), 2B15, and 2B17 exhibit substrate-inhibition; and UGT2B7 follows sigmoidal kinetics. In contrast, 1NP glucuronidation exhibited sigmoidal kinetics with UGT 1A1 (Sorich et al., 2002), 1A6, 1A7, 1A8, 1A9, and 2B7. Hyperbolic kinetics and substrate inhibition were observed for 1NP glucuronidation by UGT1A10 and UGT1A3, respectively. The sigmoidal kinetics observed here for 1NP glucuronidation by UGT1A6 contrasts to the Michaelis-Menten kinetics (Km 72.5 μM) reported recently for this reaction by Soars et al. (2003). Different analytical methodologies and expression systems were used in the two studies, although this is unlikely to account for the wide variability. Numerous UGT1A6 sequences have been reported. The sequence of the UGT1A6 cDNA used here corresponds to GenBank accession number AY435141.
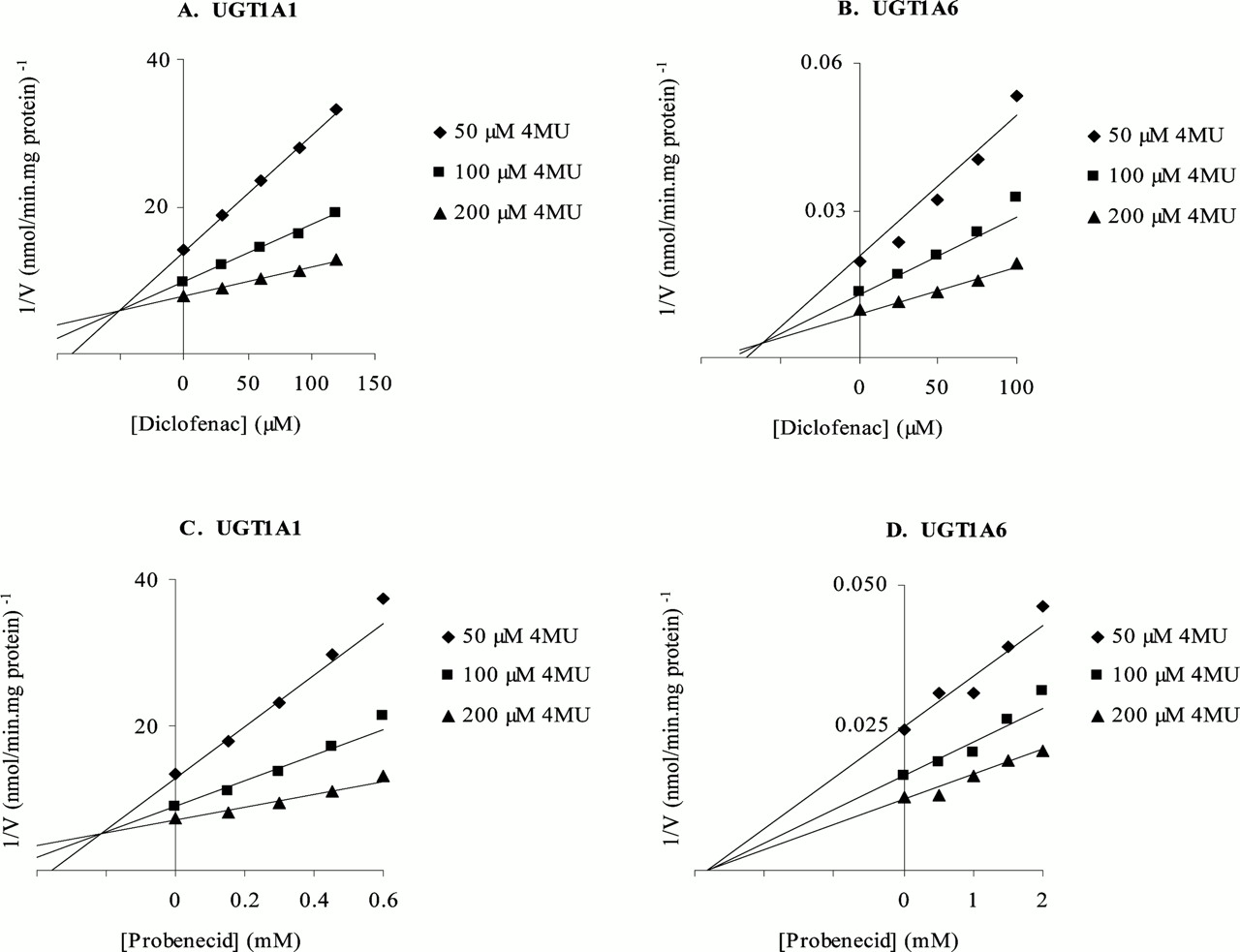
Representative Dixon plots for the inhibition of 4MU glucuronidation by diclofenac and probenecid.
Diclofenac inhibition of UGT1A1 (panel A) and UGT1A6 (panel B), and probenecid inhibition of UGT1A1 (panel C) and UGT1A6 (panel D). Kinetic data were model-fitted as described under Materials and Methods (Data Analysis).
A two-site kinetic model with an interaction between two identical binding sites provides an alternative approach to modeling data for those substrates and isoforms exhibiting atypical glucuronidation kinetics (i.e., substrate inhibition or sigmoidal kinetics). For 4MU glucuronidation by UGT2B7 and 1NP glucuronidation by UGT 1A1, 1A6, 1A7, 1A8, 1A9, and 2B7, an interaction between substrate molecules is associated with positive cooperativity and increased binding affinity of the second molecule (α <1 from fitting to two-site model), an observation that has been reported previously for numerous CYP3A4 substrates (Houston et al., 2003). In addition to the decrease in product formation associated with the substrate inhibition profiles obtained for 4MU and 1NP glucuronidation by UGT1A3 and 4MU glucuronidation by UGT1A8, fitting to the two-site model also indicates an increase in binding affinity (α <1). Recent work from this laboratory demonstrated that morphine 3- and 6-glucuronidation by UGT2B7 followed apparent biphasic kinetics that could be described by an interaction between two identical binding sites in a negative cooperative manner (Stone et al., 2003). In contrast, the UGT2B7-catalyzed glucuronidation of zidovudine exhibits Michaelis-Menten kinetics (Barbier et al., 2000; Boase and Miners, 2002; Court et al., 2003). Sigmoidal kinetics have also been described recently for numerous UGT1A1 substrates, including estradiol, buprenorphine, ethinylestradiol, and anthraflavic acid (Soars et al., 2003). Consistent with recent recommendations (Soars et al., 2003; Stone et al., 2003; Miners et al., 2004), it is apparent that multiple kinetic models are necessary for the kinetic characterization of glucuronidation reactions. Importantly, models may vary between substrates for the same isoform (e.g., differences in kinetic profiles for 4MU and 1NP glucuronidation by UGT1A8) and for the same reaction catalyzed by multiple isoforms (e.g., 4MU glucuronidation by UGT 1A6, 1A8, and 2B7; see Fig. 2). Although the Hill and Michaelis-Menten equations have been suggested for the modeling of glucuronidation kinetics (Soars et al., 2003), other models are clearly relevant. Indeed, the use of a multisite model (Houston and Kenworthy 2000; Galetin et al., 2002; Stone et al., 2003) may, in general, provide a more realistic approach for the analysis of nonhyperbolic glucuronidation kinetics. Where heterotropic activation occurs (Williams et al., 2002), the site for metabolism and activation by a modifier may be dissociated and involve different binding locations. Heterotropic effects require more elaborate modeling involving either the simultaneous binding of two different substrates to the active site or possibly a separate effector site (Houston et al., 2003). It should be noted that neither 4MU nor 1NP binds nonspecifically to human liver microsomes to a significant extent (J. O. Miners and J. A. McLure, unpublished data), precluding nonspecific microsomal binding as a cause of atypical kinetics. Moreover, kinetic models and Km or S50 values for 4MU and 1NP glucuronidation by UGT 1A1 and 1A9 were found not to vary when HEK293 cell lysates and microsomes were used as the enzyme source (data not shown), suggesting that other cellular constituents do not bind these substrates.
The mechanism responsible for the atypical glucuronidation kinetics observed here and reported in other recent publications (Soars et al., 2003; Stone et al., 2003) remains unknown. However, homotropic positive and negative cooperativity implicitly assumes the binding of more than one substrate molecule in the enzyme active site. This may occur in a single “active site” or, alternatively, UGTs may act as cooperative ligand-binding multimeric enzymes since there is increasing evidence indicating that at least some isoforms may form dimers (Meech and Mackenzie, 1997; Ikushiro et al., 1997; Ghosh et al., 2001; Kurkela et al., 2003). Irrespective of the mechanism, it is now clear that assumptions regarding the existence of one or more binding sites in a UGT active site cannot be inferred from kinetic studies with a limited number of substrates, as proposed recently for UGT2B7 (Rios and Tephly, 2002).
It has been reported previously that acetonitrile, DMSO, and methanol (0.1–2%, v/v) have at best a minor effect on umbelliferone glucuronidation by human hepatocytes (Easterbrook et al., 2001). Results presented here indicate that acetone, acetonitrile, DMSO, ethanol, and methanol (0.5 and 1%) generally caused ≤20% inhibition of UGT isoform activities, although a degree of selectivity was apparent. For example, >20% inhibition by acetone, acetonitrile, and DMSO was variably observed with UGT 1A6, 1A8, 1A9, 2B15, and 2B17, whereas ethanol selectively decreased UGT1A6 and UGT2B17 activity. Concentration-dependent inhibition of methanol (0.5–4%, v/v) was observed with UGT 1A6, 1A10, 2B7, 2B15, and 2B17, but the 4MU glucuronidation activities of other isoforms were relatively unaffected by this solvent. Overall, methanol and ethanol (0.5 and 1%) appear to affect UGT isoform activity to the least extent. These observations parallel the selective effects reported for organic solvents on P450 isoform activities (Chauret et al., 1998; Hickman et al., 1998) and indicate that solvent effects should be an important consideration for the design of in vitro kinetic and inhibition studies with recombinant human xenobiotic-metabolizing enzymes.
Previous reports from this laboratory and others indicate that diclofenac is a substrate of UGT 1A9 and 2B7 and may be used as an inhibitor of the glucuronidation of alternate substrates of these isoforms (Miners et al., 1997; Ammon et al., 2000; King et al., 2001). Studies performed here confirmed that diclofenac was an inhibitor of both isoforms but additionally demonstrated inhibition of UGT 1A1-, 1A3-, 1A6-, 1A7-, 1A8-, 1A10-, 2B15-, and 2B17-catalyzed 4MU glucuronidation. Apparent Ki values determined for UGT 1A1-, 1A6-, 1A7-, 1A9-, and 1A10 ranged from 11 to 60 μM. Diclofenac Km and Ki values reported previously for UGT2B7 were 13 μM and 9 μM, respectively (Miners et al., 1997; King et al., 2001). Probenecid is known to inhibit the metabolic clearance of numerous glucuronidated drugs in vivo (Miners and Mackenzie, 1991). Consistent with these known drug interactions, probenecid was demonstrated here to be a nonselective inhibitor of human UGTs. Kinetic studies with UGT 1A1, 1A6, 1A7, 1A9, and 1A10 were suggestive of competitive inhibition, with apparent Ki values in the range 96 to 2790 μM. It is noteworthy that the Ki values for most isoforms are considerably higher than the maximum unbound plasma concentrations (viz. 25–30 μM) reported for probenecid (Vree et al., 1993).
In conclusion, it has been demonstrated that 4MU and, to a lesser extent, 1NP represent convenient substrate probes for assessing the activity of most recombinant human UGTs. However, multiple models were necessary to describe the kinetics of 4MU and 1NP by UGT 1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, 2B15, and 2B17. Kinetic models varied with substrate (for the same isoform) and from isoform to isoform (with the same substrate). A two-site model proved useful for those reactions exhibiting atypical glucuronidation kinetics. Although organic solvents generally had a minor effect on UGT isoform activity, selective inhibition was occasionally observed, further emphasizing the need for the careful design and interpretation of kinetic studies with these enzymes. Finally, diclofenac and probenecid were shown to be nonselective UGT inhibitors, thereby precluding the use of these compounds for the reaction phenotyping of xenobiotic glucuronidation pathways in human tissues.
PUBLISHER'S NOTE
The authors discovered that the recombinant UGT1A6 enzyme preparation used in this work also contained UGT1A9. Accordingly, experiments involving UGT1A6 were repeated with HEK293 cell lysate expressing only this enzyme. Data reported for all other isoforms are accurate. The corrected version of the paper, which is in departure from print, follows.
Footnotes
-
↵1 Abbreviations used are: UDPGA, UDP-glucuronic acid; 4MU, 4-methylumbelliferone; 4MUG, 4-methylumbelliferone-β-d-glucuronide; 1NP, 1-naphthol; 1NPG, 1-naphthol-β-d-glucuronide; UGT, UDP-glucuronosyltransferase; P450, cytochrome P450; DMSO, dimethyl sulfoxide.
-
This work was supported by a grant from the National Health & Medical Research Council of Australia. V.U. is the recipient of a Flinders University International Postgraduate Scholarship.
- Received October 17, 2003.
- Accepted January 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}