


Abstract
The function of hepatic transporters is to move organic substances across sinusoidal and canalicular membranes. During extrahepatic cholestasis, transporters involved in the movement of substances from blood to bile, such as sodium/taurocholate-cotransporting polypeptide (Ntcp) and multidrug resistance protein 2 (Mrp2), are down-regulated, whereas others that transport chemicals from liver to blood, such as Mrp3, are up-regulated. Unlike extrahepatic cholestasis, where transporter expression responds to the stress of accumulating bile constituents, lipopolysaccharide (LPS)-induced intrahepatic cholestasis may be directly caused by alterations in transporter expression. The aim of this study was to quantitatively determine the effect of LPS on transporter expression and study the mechanism(s) by which LPS alters mRNA levels of major hepatic transporters in Sprague-Dawley rats. Hepatic mRNA levels of Mrp2, Mrp6, multiple drug resistance protein 1a (Mdr1a), organic anion-transporting polypeptide 1 (Oatp1), Oatp2, Oatp4, Ntcp, bile salt export pump, organic cation transporter 1 (Oct1), and organic anion transporter 3 (Oat3) were dramatically decreased, beginning approximately 6 h after LPS administration, whereas Mrp5 and Oat2 levels were unchanged. In contrast, LPS increased mRNA levels of Mrp1, Mrp3, and Mdr1b concurrently with the down-regulated transporters. Pretreatment with dexamethasone, which decreases the release of cytokines, reversed the reduction of Mdr1a, Oatp1, Oatp2, Oct1, and Ntcp mRNA following LPS administration. Furthermore, dexamethasone pretreatment also prevented the LPS-mediated increase in Mrp1, Mrp3, and Mdr1b, whereas pretreatment with aminoguanidine or gadolinium chloride, an inhibitor of inducible nitric oxide synthetase and a Kupffer cell toxicant, respectively, had no effect on the LPS-induced changes. The concurrent repression and induction of various transporters, as well as dexamethasone abatement of both LPS-mediated repression and induction, indicates that these responses may be mediated through similar pathways.
The liver plays an important role in the enterohepatic circulation of bile acids, as well as the detoxication of endogenous and exogenous compounds through the biotransformation and biliary excretion of these compounds. Recent cDNA cloning and immunohistochemical localization of several transporters for both the sinusoidal uptake and canalicular excretion of endogenous and exogenous compounds has provided insight into hepatic transport. Organic anions are efficiently taken up into hepatocytes by secondary active transporters, accounting for the first-pass effect (Jansen, 2000). Canalicular transport is typically concentrative, as is the case with bile acids (1,000-fold gradient), and is therefore an active process. Several transporters are constitutively expressed in liver, including uptake transporters such as organic anion-transporting polypeptides 1, 2, and 4 (Oatp1, 2, and 4; Li et al., 2002), the bile salt transporter sodium/taurocholate-cotransporting polypeptide (Ntcp; Ananthanarayanan et al., 1994), organic cation transporter 1 (Oct1; Slitt et al., 2002), and organic anion transporters 2 and 3 (Oat2 and 3; Buist et al., 2002), as well as the excretion transporters, such as multidrug resistance proteins (Mrps; Cherrington et al., 2002), multiple drug resistance proteins (Mdrs; Brady et al., 2002), and the bile salt export pump (Bsep; Weymann et al., 1997) (Table 1). The varied affinities of these transporters for different compounds in portal blood provide both specific and redundant means for extracting bile acids and other compounds from the blood and excreting them from the hepatocyte (Kullak-Ublick et al., 2000).
Nomenclature for hepatic xenobiotic transporters
During extrahepatic cholestasis, such as obstructive cholestasis or bile duct ligation, bile flow is greatly diminished and bile constituents such as bile acids begin to accumulate in the liver. The buildup of biliary constituents in hepatocytes causes an increase in oxidative stress and inflammation, and subsequently leads to hepatotoxicity (Lopez et al., 2000). These complex and overlapping events initiate several secondary responses, including the down-regulation of various transporters, thus making mechanistic details regarding the regulation of transporters during extrahepatic cholestasis difficult to determine.
LPS administration is a particularly useful model of cholestasis because the mechanism of cholestasis appears to be the direct alteration of transporter expression that leads to the reduced Na+-dependent bile flow, rather than a secondary event following the accumulation of biliary constituents (Lee and Boyer, 2000). During LPS-induced cholestasis, Gram-negative bacterial cell wall components trigger acute inflammation. It is thought that LPS-induced intrahepatic cholestasis is caused by down-regulation of the sinusoidal uptake and canalicular excretion transporters. Therefore, LPS provides a useful model to determine mechanistic events leading to transporter regulation.
When stimulated by LPS, Kupffer cells produce inflammatory cytokines such as tumor necrosis factor α (TNF-α). These cytokines stimulate multiple signal transduction pathways that lead to activation of factors such as nuclear factor of the κ-enhancer in B cells and inducible nitric oxide synthetase. Hepatocytes respond to these factors by altering gene expression, predominantly at the level of transcription (Moshage, 1997). Three lines of evidence suggest that LPS-mediated regulation of hepatic transporters is mediated by cytokines. First, reduced bile flow and bile acid secretion by LPS can be prevented by administration of TNF-α antibodies (Whiting et al., 1995). Second, down-regulation of Ntcp and Mrp2 can also be produced by treatment with recombinant TNF-α and interleukin 6 (Green et al., 1996; Kim et al., 2000). Third, pretreatment with dexamethasone (Dex), which blocks the release of cytokines, partially blocks the decrease of Mrp2 by LPS (Roelofsen et al., 1995; Kubitz et al., 1999).
The effect of LPS administration on the regulation of mRNA expression of several hepatic transporters is not known. In addition, a quantitative and comparative study of the effects of LPS on the regulation of all major hepatic transporters has not been done. Therefore, the aim of this study was to quantitatively determine the effect of LPS on the mRNA levels of hepatic transporters over time, and whether these LPS-mediated changes could be blocked by inhibitors of specific mediators of LPS signaling in Sprague-Dawley rats. In the present study, three chemical pretreatments that block some aspect of the LPS signaling cascade were used to determine the important events in transporter regulation during intrahepatic cholestasis. First, Dex is a highly effective anti-inflammatory and immunosuppressive chemical that blocks the release of cytokines (Schimmer and Parker, 1996). Second, aminoguanidine (Agn) is a potent inhibitor of inducible nitric oxide synthetase, reducing the levels of nitric oxide by 50% following LPS administration (Picard et al., 2001). Third, gadolinium chloride(III) hexahydrate (GdCl) is a toxicant known to destroy Kupffer cells, which are the resident macrophages in liver, and thus limits cytokine release in liver (Hardonk et al., 1992).
Materials and Methods
Chemicals. Agn was purchased from Cayman Chemical (Ann Arbor, MI). Lipopolysaccharide (LPS; from Escherichia coli serotype 0111:B4), GdCl, Dex, and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Animals. Male Sprague-Dawley rats (200–250 g; Sasco, Omaha, NE) were acclimated to the housing facility (2–3 rats/cage, 50% relative humidity, 12-h light/dark cycle) for 1 week and fed Teklad 8604 rodent chow (Harlan, Indianapolis, IN). LPS (4 mg/kg) was administered to male Sprague-Dawley rats (200–250 g) in a volume of 5 ml/kg i.p., and livers were excised at 0, 1.5, 3, 6, 9, 12, and 16 h thereafter. In a separate experiment, pretreatments with Dex (10 mg/kg i.p. in corn oil, 1 h), Agn (100 mg/kg i.p. in saline, 1 h), or GdCl (10 mg/kg i.v. in saline to restrained animals, 24 h) were administered in a volume of 3 ml/kg before treatment with LPS (4 mg/kg) or saline. These treatments have previously been shown to block the release of cytokines, inhibit inducible nitric oxide synthetase, and destroy all large Kupffer cells, respectively (Hardonk et al., 1992; Schimmer and Parker, 1996; Picard et al., 2001). Livers were excised 16 h after LPS administration and stored at -80°C. Total RNA was isolated using RNAzol B reagent (Tel-Test Inc., Friendswood, TX) per the manufacturer's protocol. Total RNA concentrations were determined by UV spectrophotometry and integrity was examined by ethidium bromide staining after agarose gel electrophoresis.
Branched DNA Assay. Specific transporter probe sets were designed as previously described: Mrp1, 2, and 3 (Cherrington et al., 2002), Mrp5; Mrp6, Ntcp, and Bsep (Leazer and Klaassen, 2003); Mdr1a and Mdr1b (Brady et al., 2002); Oatp1, 2, and 4 (Li et al., 2002); Oct1 (Slitt et al., 2002); and Oat2 and 3 (Buist et al., 2002). Oligonucleotide probes were diluted in lysis buffer supplied in the QuantiGene HV Signal Amplification Kit (Bayer Corp.-Diagnostics Div., Tarrytown, NY). All reagents for analysis (i.e., lysis buffer, capture hybridization buffer, amplifier/label probe buffer, and substrate solution) were supplied by the manufacturer. Total RNA (1 μg/μl; 10 μl) was added to each well of a 96-well plate containing capture hybridization buffer and 50 μl of each diluted probe set. Total RNA was allowed to hybridize to each probe set overnight at 53°C. Subsequent hybridization steps were carried out per the manufacturer's protocol, and luminescence was measured with a Quantiplex 320 bDNA luminometer interfaced with Quantiplex Data Management Software Version 5.02 for analysis of luminescence from 96-well plates.
Statistics. Data are expressed as mean ± standard error. For multiple comparisons, analysis of variance was performed followed by Duncan's multiple range test. The level of significance was set at p ≤ 0.05.
Results
Time Course of LPS on Transporter Regulation. The expression levels of several hepatic transporters were determined in male Sprague-Dawley rats following LPS administration using the branched DNA signal amplification assay. The mRNA expression of each of the Oatp family members studied was significantly reduced in the liver by LPS administration (Fig. 1). Oatp1 was significantly reduced at 9, 12, and 16 h after LPS administration by 41, 56, and 73%, respectively. Oatp2 expression was also decreased beginning at 3 h by 60%, and 48, 69, 85, and 73% at 6, 9, 12, and 16 h after LPS administration, respectively. Oatp4 mRNA levels were also dramatically reduced at 6, 9, 12, and 16 h after LPS administration by 41, 60, 76, and 69%, respectively.

Hepatic Oatp mRNA expression following LPS administration. Total hepatic RNA from male Sprague-Dawley rats treated with LPS (4 mg/kg) for the time indicated was analyzed by the bDNA assay. Data are expressed as relative light units (RLU) ± standard error of the mean. Asterisk indicates significance from time 0 (p ≤ 0.05).
Of the various families of hepatic transporters studied, the organic cation and anion transporters exhibited the least dramatic alterations in mRNA levels by LPS administration (Fig. 2). Oct1 was significantly reduced at 9, 12, and 16 h after LPS administration by 34, 37, and 33%, respectively. Oat2 mRNA levels were not significantly altered at any time, but Oat3 was significantly reduced at 3, 9, 12, and 16 h (47, 60, 64, and 47%, respectively), but not 6 h after LPS treatment.

Hepatic Oct and Oat mRNA expression following LPS administration. Total hepatic RNA from male Sprague-Dawley rats treated with LPS (4 mg/kg) for the time indicated was analyzed by the bDNA assay. Data are expressed as relative light units (RLU) ± standard error of the mean. Asterisk indicates significance from time 0 (p ≤ 0.05).
Ntcp and Bsep, the transporters predominantly responsible for the uptake and excretion of bile acids, were both decreased at the mRNA level following LPS administration (Fig. 3). Ntcp was decreased significantly at 9 h (64% of control), and fell to 24 and 6% at 12 and 16 h after LPS administration, respectively. After an initial increase of 24% at 1.5 h, Bsep was decreased significantly from control values at 6, 9, 12, and 16 h after LPS administration, having decreased 21, 42, 64, and 57%, respectively.

Hepatic bile acid transporter mRNA expression for hepatic uptake (Ntcp) and biliary excretion (Bsep) following LPS administration. Total hepatic RNA from male Sprague-Dawley rats treated with LPS (4 mg/kg) for the time indicated was analyzed by the bDNA assay. Data are expressed as relative light units (RLU) ± standard error of the mean. Asterisk indicates significance from time 0 (p ≤ 0.05).
The mRNA levels of the multidrug resistance genes encoding P-glycoproteins were differentially regulated by LPS (Fig. 4). Mdr1a was significantly reduced by 67, 62, 69, and 67% at 6, 9, 12, and 16 h after LPS administration, respectively. Mdr1b was the transporter most robustly increased, reaching 1500% of normal levels by 16 h. Mdr1b expression appeared to be increased at 3 h but was not statistically significant until 9, 12, and 16 h after LPS administration, when it was increased to 1100, 1000, and 1500% of control values, respectively.
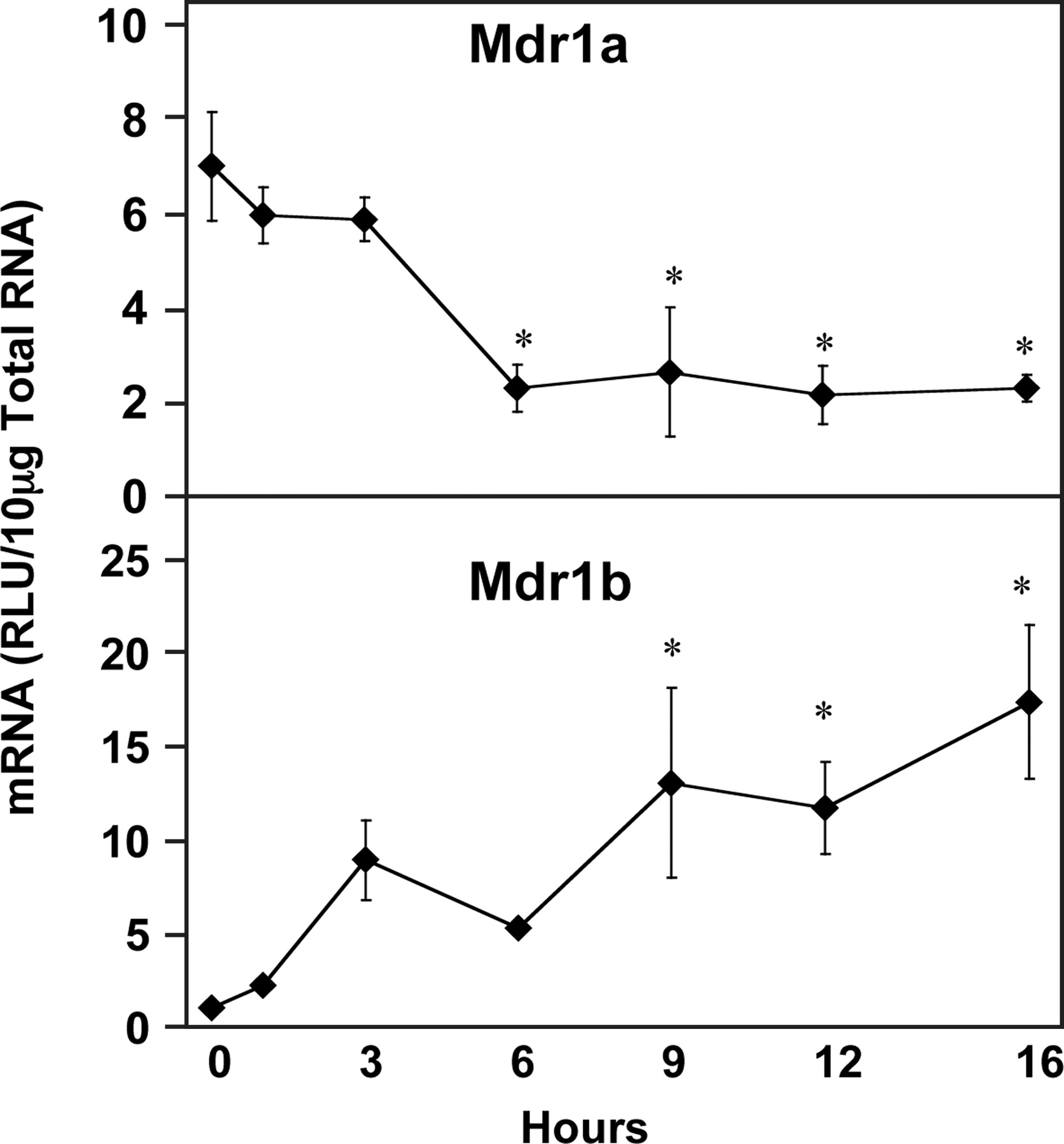
Hepatic multiple drug resistance gene expression (Mdr) following LPS administration. Total hepatic RNA from male Sprague-Dawley rats treated with LPS (4 mg/kg) for the time indicated was analyzed by the bDNA assay. Data are expressed as relative light units (RLU) ± standard error of the mean. Asterisk indicates significance from time 0 (p ≤ 0.05).
Among transcripts for the multidrug resistance protein family, expression of some individual transporters was increased, whereas others were decreased by LPS (Fig. 5). Mrp1 mRNA levels were significantly induced at 9, 12, and 16 h after LPS administration by 99, 120, and 115%, respectively. In contrast, Mrp2 expression was dramatically decreased by 68, 81, 94, and 93% at 6, 9, 12, and 16 h after LPS administration, respectively. Mrp3 expression began to increase at 9 h and was increased significantly at 16 h (124%) after LPS administration. Mrp5 was not changed by LPS administration, whereas Mrp6 was decreased (44 and 41%) at 9 and 12 h, respectively. Interestingly, Mrp6 mRNA levels returned to control levels by 16 h after LPS administration.
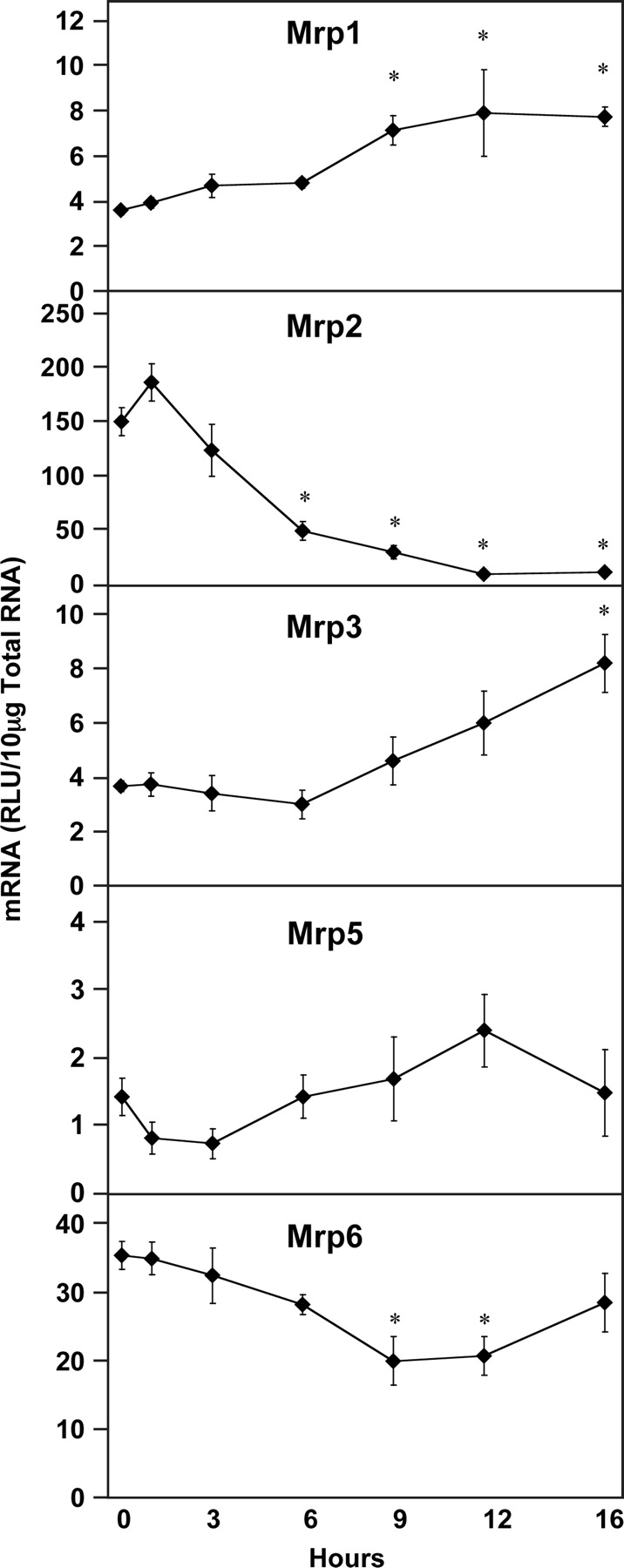
Hepatic multidrug resistance protein (Mrp) mRNA expression following LPS administration. Total hepatic RNA from male Sprague-Dawley rats treated with LPS (4 mg/kg) for the time indicated was analyzed by the bDNA assay. Data are expressed as relative light units (RLU) ± standard error of the mean. Asterisk indicates significance from time 0 (p ≤ 0.05).
Effects of Chemical Pretreatment on LPS-Mediated Changes in Transporter Expression. The effect of LPS administration on transporter mRNA levels was measured in response to three chemical pretreatments: the immunosuppressant Dex, the inducible nitric oxide synthetase inhibitor Agn, and the Kupffer cell toxicant GdCl. The expression levels of all transporters examined were not altered by any of these chemical pretreatments alone (data not shown). The reduction in the mRNA levels of two Oatp family members by LPS was prevented by Dex (Fig. 6). Dex pretreatment blocked the reduction of Oatp1 mRNA levels, whereas Agn or GdCl did not block the reduction of hepatic Oatp1 expression by LPS. Similarly, the LPS-induced decrease in Oatp2 expression was blocked by Dex pretreatment but was unaffected by Agn and GdCl pretreatment. None of the pretreatments significantly blocked the decreased expression of Oatp4 following LPS administration.

Pretreatment effects on hepatic Oatp mRNA expression. Male Sprague-Dawley rats were treated with LPS or saline for 16 h and hepatic total RNA was analyzed by the bDNA assay. Additionally, dexamethasone (10 mg/kg), Agn (100 mg/kg), or GdCl (10 mg/kg) was administered prior to LPS or saline. Data are expressed as relative light units (RLU) ± standard error of the mean. *, significance from control, †, significance from LPS treatment alone.
Dex pretreatment did not similarly block the LPS-mediated effects on the expression of hepatic Octs and Oats following LPS administration. None of the pretreatments significantly altered the reduction of Oct1 mRNA levels by LPS (Fig. 7). Oat2 expression was not significantly altered by LPS administration. However, Dex pretreatment resulted in a higher expression of Oat2 than in rats treated with LPS alone. The reduction in Oat3 expression by LPS was not affected by the Dex pretreatment (64% reduction). However, the Agn and GdCl pretreatments produced Oat3 expression levels that were not significantly higher than that of LPS treatment alone, nor significantly lower than that of vehicle treatment (19 and 33% decrease, respectively).
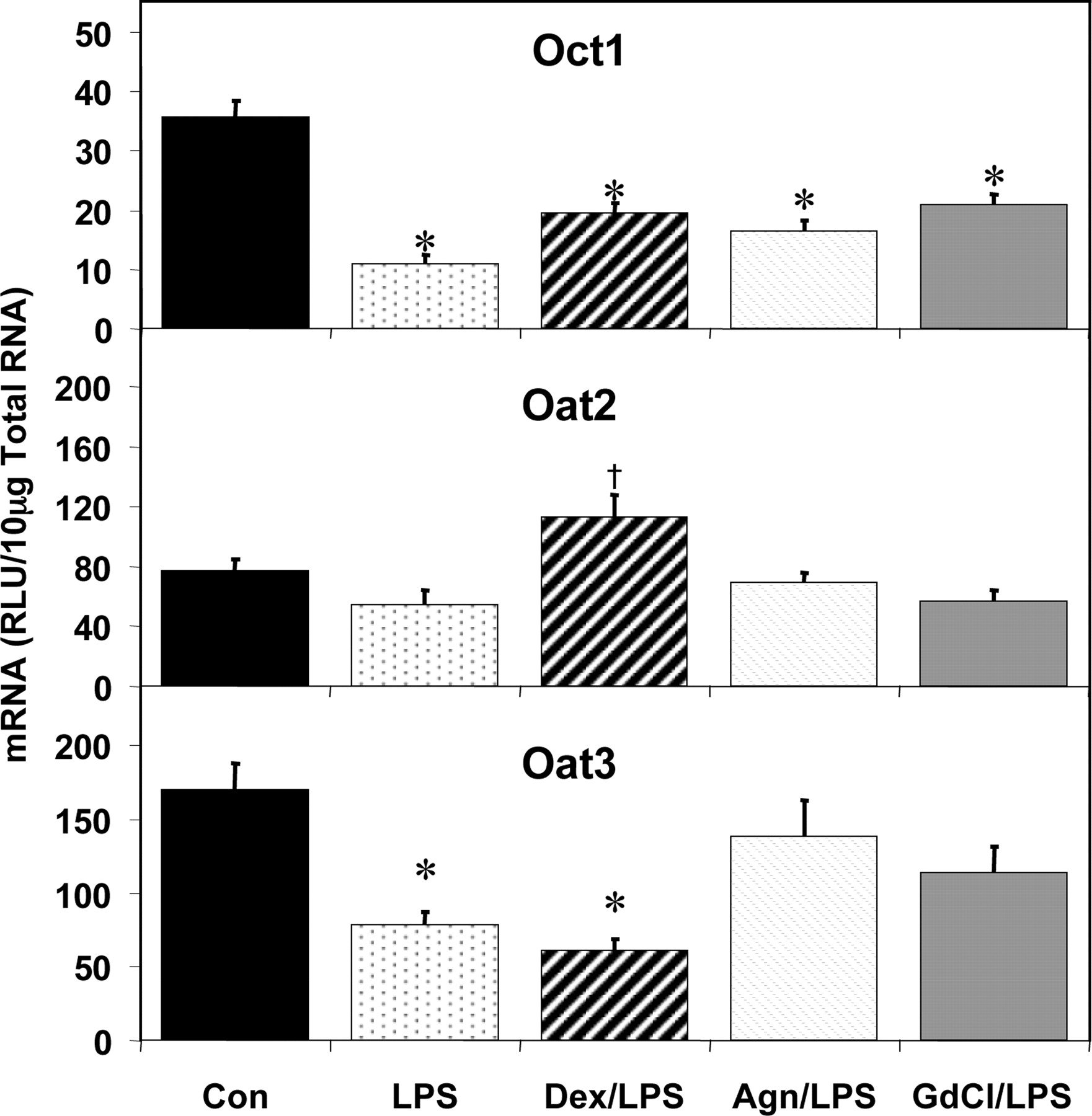
Pretreatment effects on hepatic Oct and Oat mRNA expression. Male Sprague-Dawley rats were treated with LPS or saline for 16 h and hepatic total RNA was analyzed by the bDNA assay. Additionally, dexamethasone (10 mg/kg), Agn (100 mg/kg), or GdCl (10 mg/kg) was administered prior to LPS or saline. Data are expressed as relative light units (RLU) ± standard error of the mean. *, significance from control; †, significance from LPS treatment alone.
The decrease in expression of bile acid transporters following LPS administration was also blunted by Dex (Fig. 8). Ntcp, which was dramatically down-regulated by LPS (95%), was only decreased by 52% following Dex pretreatment. In contrast, Agn or GdCl pretreatment did not prevent the LPS-mediated decrease in Ntcp levels (91 and 95% decrease, respectively). The decrease in expression of Bsep was similarly blunted by Dex (only 21% reduction) but not by Agn or GdCl pretreatments (64 and 48% decrease, respectively).

Pretreatment effects on hepatic bile acid transporter for uptake (Ntcp) and biliary excretion (Bsep) mRNA expression. Male Sprague-Dawley rats were treated with LPS or saline for 16 h and hepatic total RNA was analyzed by the bDNA assay. Additionally, Dex (10 mg/kg), Agn (100 mg/kg), or GdCl (10 mg/kg) was administered prior to LPS or saline. Data are expressed as relative light units (RLU) ± standard error of the mean. *, significance from control; †, significance from LPS treatment alone.
Dex also had a profound influence on the LPS-induced effects on the expression of the Mdr genes (Fig. 9). The reduction of Mdr1a expression by LPS was completely blocked by Dex but was not affected by Agn or GdCl pretreatment. The robust induction of Mdr1b by LPS was decreased by Dex treatment (from a 1000 to a 250% increase). However, Agn and GdCl did not block the increase of Mdr1b by LPS, remaining 800 and 1300% higher than in control rats.

Pretreatment effects on hepatic multiple drug resistance (Mdr) gene expression. Male Sprague-Dawley rats were treated with LPS or saline for 16 h and hepatic total RNA was analyzed by the bDNA assay. Additionally, Dex (10 mg/kg), Agn (100 mg/kg), or GdCl (10 mg/kg) was administered prior to LPS or saline. Data are expressed as relative light units (RLU) ± standard error of the mean. *, significance from control; †, significance from LPS treatment alone.
Dex pretreatment had the most profound effects of blocking LPS-mediated transporter regulation among the Mrp family of transporters (Fig. 10). The increase in Mrp1 mRNA levels by LPS administration was completely blocked by Dex but not by Agn and GdCl. Although the marked decrease of Mrp2 mRNA by LPS was blunted by Dex from a 95 to a 72% reduction, none of the pretreatments significantly blocked the LPS-mediated decrease in Mrp2. The increase in Mrp3 levels by LPS was completely blocked by Dex, whereas Agn or GdCl had no effect on LPS-mediated induction of Mrp3. LPS did not significantly alter either Mrp5 or Mrp6 at the 16-h time point, although GdCl treatment along with LPS did increase Mrp5 180% over LPS alone.
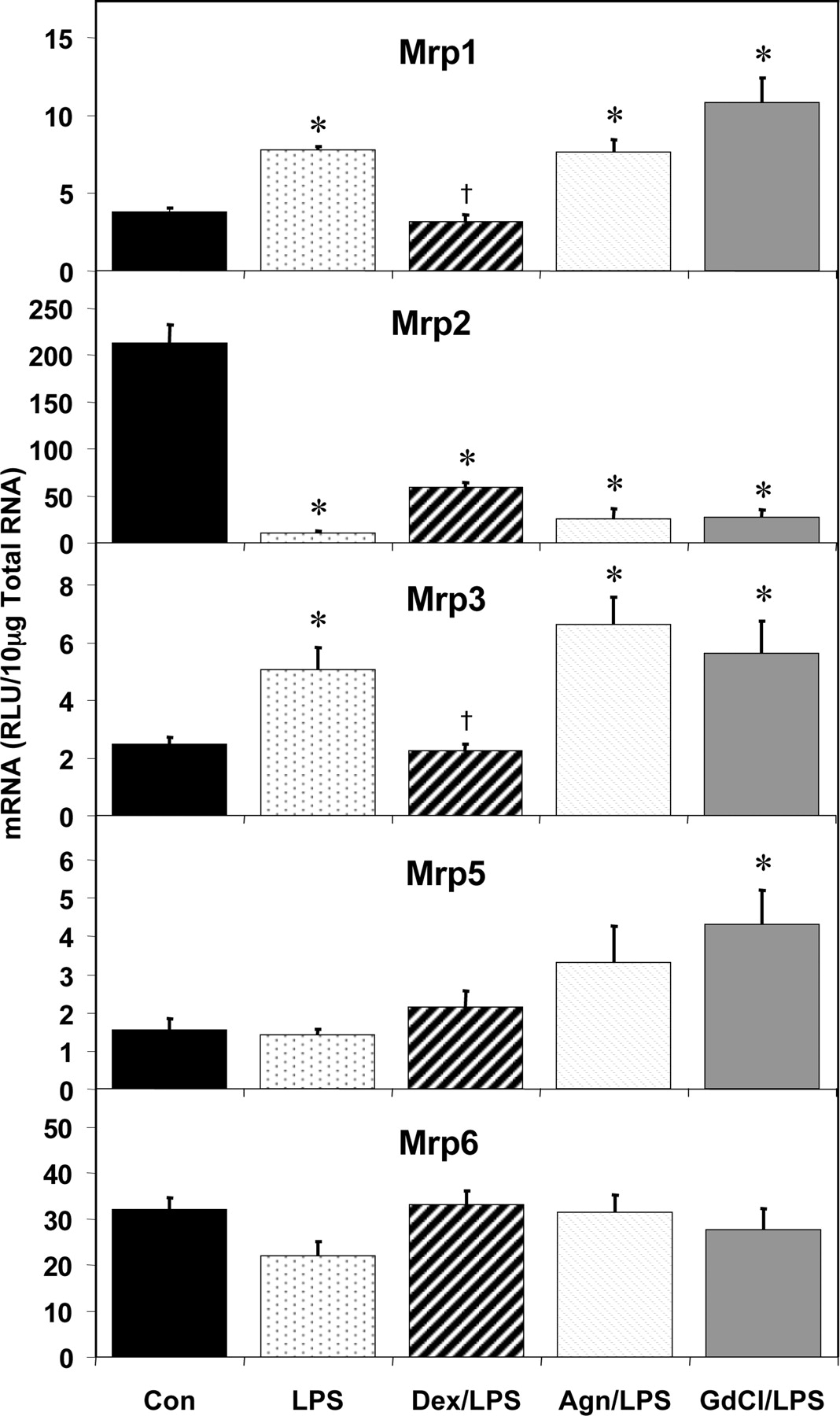
Pretreatment effects on hepatic multidrug resistance protein (Mrp) mRNA expression. Male Sprague-Dawley rats were treated with LPS or saline for 16 h and hepatic total RNA was analyzed by the bDNA assay. Additionally, Dex (10 mg/kg), Agn (100 mg/kg), or GdCl (10 mg/kg) was administered prior to LPS or saline. Data are expressed as relative light units (RLU) ± standard error of the mean. *, significance from control; †, significance from LPS treatment alone.
Discussion
In the present study, the effect of LPS administration on mRNA levels of 15 different hepatic xenobiotic transporters is detailed over time in the liver of male Sprague-Dawley rats. The overall results show that LPS decreases many of the transporters involved in the transport of chemicals from blood to hepatocyte, and from the hepatocyte into bile. Specifically, the hepatocyte uptake transporters Ntcp; Oatp1, 2, and 4; Oct1 and Oat3; and the biliary efflux transporters Mrp2, Mdr1a, and Bsep are all decreased following LPS administration, whereas sinusoidal efflux transporters (hepatocyte to blood) including Mrp1 and Mrp3 are increased.
The down-regulation of several transporters, including sinusoidal uptake transporters, may be one compensatory mechanism to protect the liver against damage during sepsis. The present data confirm previously published reports that LPS produces dramatic decreases in the mRNA levels of Ntcp and Mrp2 beginning at 6 and 9 h, respectively (Trauner et al., 1997; Kubitz et al., 1999; Zollner et al., 2002). Additionally, the present study demonstrates the time course of the Oatp1, 2, and 4, as well as Bsep, response to LPS administration. Although LPS has been reported to down-regulate Bsep, Oatp1, and Oatp2 in rats at single time points, with similar findings for Mdr1a in mice and Oatp4 in cecum-punctured rats (Vos et al., 1998; Kakyo et al., 1999; Lund et al., 1999; Hartmann et al., 2001; Hartmann et al., 2002; Tygstrup et al., 2002), this is the first study to quantify and compare the effects of LPS on the regulation of each of the major hepatic xenobiotic transporters over time. Furthermore, the observations that Oct1 and Oat3 are down-regulated are novel findings.
The hepatic transporters that are increased by LPS administration serve to export organic anions back into the blood from the hepatocyte, with the exception of Mdr1b. This is the first report of the up-regulation of Mrp3 in response to LPS administration in rat, whereas Mrp1 and Mdr1b have previously been shown to be upregulated by LPS only at a single time point (Vos et al., 1998). This alternate vectoral excretion from hepatocyte to blood may serve as a second compensatory mechanism to reduce hepatic exposure to bile acids as well as other toxic biliary constituents during sepsis. Clinically, jaundice is common among septic patients; however, the circulating bilirubin in these patients is predominantly present in the relatively nontoxic conjugated forms (Pirovino et al., 1989). To appear in the circulating blood, conjugated bilirubin requires an active transport process across the sinusoidal membrane out of the hepatocyte. Given the affinity of Mrp1 and Mrp3 for conjugated substrates (Hirohashi et al., 1999), it is likely that the coordinated down-regulation of Mrp2 and up-regulation of Mrp1 and Mrp3 is responsible for the elevated plasma levels of conjugated bilirubin.
The majority of transporters that are altered (either suppressed or induced) by LPS begin to show significant changes in expression concurrently between 6 and 9 h after administration. This would indicate that the up-regulation of sinusoidal efflux transporters, such as Mrp1 and Mrp3, is not dependent upon prior down-regulation of canalicular efflux transporters and the subsequent accumulation of their substrates. Rather, the time courses for both the induction and suppression of transporter mRNA levels are similar, which indicates that regulation of both hepatic uptake and export systems may use a common signaling pathway.
Bolder et al. (2002) suggest that LPS-induced down-regulation of transporter expression is not directly altered by the cholestatic accumulation of biliary constituents but, rather, is mediated by direct transcriptional signaling. They demonstrated that stress, 2 h before LPS administration, maintained sodium-dependent and -independent transport of cholyltaurine, as well as Ntcp and Bsep protein levels. However, even without transport deficiency, mRNA levels for both Ntcp and Bsep were still down-regulated, indicating that LPS mediates a direct transcriptional regulation of transporters. Additionally, the accumulation of bile acids does not appear to be responsible for the down-regulation of hepatic transporters, because bile acid administration does not replicate the transcriptional down-regulation of mouse anion transporters following LPS administration (Hartmann et al., 2002).
A role for inflammatory cytokines in the transcriptional regulation of hepatic transporters has been suggested (Geier et al., 2003). Within 1 to 4 h after LPS administration, bile acid-dependent and -independent bile flow is reduced (Utili et al., 1977; Whiting et al., 1995), as is the transport of bromosulphalein (Utili et al., 1977), taurocholate (Whiting et al., 1995), and dinitrophenylglutathione (Roelofsen et al., 1995). At this early time point, there is dramatic efflux of glutathione into bile and decreased intracellular content of glutathione. Cytokines, and particularly TNF-α, appear to mediate this short-term response because reductions in bile flow, Na-dependent taurocholate transport, and Mrp2 expression at these time points are protected by pretreatment with TNF-α antibodies (Whiting et al., 1995) and elicited by treatment with TNF-α (Whiting et al., 1995; Nakamura et al., 1999). However, TNF-α and other acute phase response cytokines may only mediate the less specific acute effects, whereas other factors, which are downstream of the acute response, may cause the later effects of LPS, including the suppression and induction of hepatic transporters. For example, in the current study, transporter mRNA levels reach maximal suppression or induction between 12 and 16 h after LPS administration, at which time plasma cytokine levels and hepatic glutathione storage have returned to normal, suggesting the involvement of other factors (Chen et al., 1994; Roelofsen et al., 1995).
The LPS-induced impaired excretion of numerous substrates, as well as the down-regulation of Ntcp and Mrp2, have been attributed to TNF-α acute phase cytokine formation (Green et al., 1996; Nakamura et al., 1999). Dex, which is a potent immunosuppressant (Waage, 1987), blocks the LPS-induced production of TNF-α and interleukin 1 (Staruch and Wood, 1985). Dex has previously been shown to block LPS-induced inhibition of 2,4-dinitrophenyl-S-glutathione transport in rats by approximately 50% (Roelofsen et al., 1995). Dex pretreatment has also been shown to completely abolish the LPS-induced inhibition of bromosulphalein excretion and to maintain canalicular localization of Mrp2 following LPS administration (Kubitz et al., 1999). In that study, LPS was shown to redistribute Mrp2 protein away from the canaliculi into intracellular vesicles within the first 3 h after administration, putatively accounting for the early alterations in excretion of Mrp2 substrates. As noted previously, Mrp2 mRNA levels are not reduced until later time points, suggesting separate mechanisms for the transcriptional regulation of hepatic transporters and the early transport deficiencies. Additionally, Dex pretreatment completely maintained Mrp2 localization to the canaliculus but had only minimal effects of the Mrp2 mRNA levels (Kubitz et al., 1999).
The present study shows that Dex protects against the LPS-induced changes in expression of many, but not all, hepatic transporters. Dex pretreatment significantly blocked the effects of LPS on several transporters that were down-regulated, maintaining expression in most instances near control values. These transporters include Mdr1a, Oatp1, Oatp2, Ntcp, and Bsep. Dex pretreatment also blocked the increase of the three transporters that were up-regulated by LPS, namely Mrp1, Mrp3, and Mdr1b. Conversely, Dex did not block the LPS-induced decrease in Mrp2. Similarly, Oatp4 was also dramatically decreased by LPS and only slightly protected by Dex pretreatment. Other transporters that are down-regulated by LPS but not protected by Dex include Oct1 and Oat3.
The Dex abatement of both LPS-mediated suppression and induction of transporters indicates that these responses may be mediated through similar pathways. Underscoring the complex nature of hepatic transporter regulation is the observation that Dex pretreatment can prevent the LPS-mediated effects on some, but not all transporters.
In contrast to Dex, Agn and GdCl pretreatments had little effect on the regulation of hepatic transporters by LPS. Because nitric oxide levels are still increased by LPS administration following Agn pretreatment, although only to 50% that of LPS treatment alone, nitric oxide signaling cannot be definitively ruled out as the mechanism of altered regulation (Picard et al., 2001). However, the absence of any effect in these data indicates that LPS-induced regulation of hepatic transporters is most likely not mediated through inducible nitric oxide synthetase or by factors solely produced by Kupffer cells. In a previous report, Kupffer cell-conditioned media elicited a 30% reduction of Mrp2 expression in cultured hepatocytes, suggesting that inflammatory modulators produced by the resident macrophages of the liver can trigger the down-regulation of transporters in hepatocytes (Nakamura et al., 1999). However, because GdCl destroys all Kupffer cells (Hardonk et al., 1992), these data indicate that other factors, not Kupffer cells alone, mediate the effects of LPS on the regulation of hepatic transporters. This possibility is supported by an earlier study showing that both the activation of Kupffer cells, as well as some undetermined circulating plasma factors in animals where Kupffer cells had been destroyed, were capable of causing the occurrence and development of LPS-induced liver injury (Fujita et al., 1995).
In conclusion, the current data provide the first complete study to quantify and comparatively examine the effects of LPS on the regulation of the major rat hepatic xenobiotic transporters over time. Alterations in the mRNA levels of these transporters may represent two separate means of hepatoprotection during endotoxemia. First, down-regulation of most of the sinusoidal uptake transporters may reduce the accumulation of organic anions in the liver; and second, the up-regulation of key sinusoidal efflux transporters may serve to export organic anions from the liver and thereby reduce exposure of the liver to these toxic compounds. The observation that both the up- and down-regulation of these transporters occur simultaneously indicates that the mechanism(s) of regulation for these compensatory processes may be related. Additionally, Dex pretreatment diminished the effects of LPS administration on the mRNA levels of many export pumps that are increased, as well as decreased, by LPS administration. The concurrent repression and induction of transporter expression by LPS administration, as well as Dex abatement of both LPS-mediated suppression and induction of transporters, indicates that these responses may be mediated through similar pathways.
Footnotes
-
This work was supported by National Institutes of Health Grants ES-09716 and ES-03192.
-
ABBREVIATIONS: Oatp, organic anion-transporting polypeptide; Ntcp, sodium/taurocholate-cotransporting polypeptide; Oct, organic cation transporter; Oat, organic anion transporter; Mrp, multidrug resistance protein; Mdr, multiple drug resistance protein; Bsep, bile salt export pump; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor α; Dex, dexamethasone; GdCl, gadolinium chloride(III) hexahydrate; Agn, aminoguanidine; bDNA, branched DNA.
-
↵1 Present address: Department of Pharmacology and Toxicology, University of Arizona, Tucson, AZ 85721.
- Received December 10, 2003.
- Accepted April 15, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}