


Abstract
Multiple drug resistance (mdr) genes encode P-glycoprotein, which is responsible for resistance to some cancer chemotherapeutic drugs and efflux of xenobiotics of cells. Thus, mdr can protect organs from xenobiotics. In rats, there are two mdr1 genes capable of xenobiotic transport, mdr1a and mdr1b. The purpose of this study was to determine the tissue distribution of rat mdr1a and mdr1b mRNA and whether microsomal enzyme inducers that increase phase I and II drug-metabolizing enzymes coordinately regulate mdr1a and/or mdr1b. The mRNA levels of mdr1a and mdr1b were determined using branched-DNA signal amplification technology. The highest level of expression of mdr1a mRNA was observed in the gastrointestinal tract, with levels increasing, respectively, from duodenum, jejunum, and ileum to large intestine. Expression levels of mdr1a mRNA in the cerebral cortex, cerebellum, kidney, lung, and liver were less than one-tenth of that in the ileum. The tissue distribution of mdr1b mRNA was similar to mdr1a with highest expression in the gastrointestinal tract but only about 3-fold higher than in most other tissues. The induction of mdr1a and mdr1b mRNA transcripts in liver, kidney, and ileum by treatment of rats with 18 chemicals representing aryl hydrocarbon receptor ligands, constitutive androstane receptor ligands, pregnane X receptor ligands, peroxisome proliferator-activated receptor ligands, electrophile-response-element activators, and CYP4502E1 inducers was assessed. Hepatic, renal, and intestinal expression of mdr1a and mdr1b mRNA were not significantly altered by treatment of rats with any of these classes of ligands. In conclusion, the primary expression of rat mdr1 genes is in the gastrointestinal tract where they are thought to function to decrease the absorption of some xenobiotics. Rat mdr1 gene expression is not readily increased by microsomal enzyme inducers in rats through coordinate mechanisms with phase I and II drug-metabolizing enzymes.
The xenobiotic transporters enable ionic and water-soluble xenobiotics to enter cells, as well as aid the excretion of these xenobiotics and/or their phase I (oxidative and reductive) and/or phase II (conjugative) metabolites of cells. Therefore, the presence or absence of these transporters is important in determining the concentration a xenobiotic will attain in a tissue. Furthermore, knowledge of when and where transporters are expressed may aid the prediction of xenobiotic toxicities.
The P-glycoproteins (P-gp2) are encoded by a family of related genes, the multiple drug resistance (mdr) genes. Group I mdr genes encode P-gps that have been shown to mediate drug transport and clinical tumor drug resistance. When transfected into drug-sensitive cells, group I gene products confer the mdr phenotype (Ueda et al., 1987). Human and monkey genomes contain a single group I gene (denoted MDR1), the protein of which is capable of drug transport. Rodents contain two group I genes (denoted mdr1a and 1b), the proteins of which are capable of drug transport. Rodent P-gps are associated with chemotherapeutic drug resistance and intestinal excretion. Additionally, they are present in the blood-brain barrier (Fromm, 2000a). Thus, the function of P-gp may be to protect cells from naturally occurring toxins. The group II gene product, mdr2, containing extensive sequence identity to group I, is associated with phospholipid transport and excretion of lipid into bile rather than with drug resistance (Smit et al., 1993). The fourth member of the P-gp family, sister of P-glycoprotein, or bile salt excretory protein, is associated with hepatic bile salt excretion and is implicated in the disease progressive familial intrahepatic cholestasis 2 (Gerloff et al., 1998; Strautnieks et al., 1998).
Early studies of gene expression and induction of human and mouse mdr genes were performed using long cDNA probes which cross-hybridized with other mdr genes than those of interest (Fojo et al., 1987; Croop et al., 1989). However, more recent studies of rodent mdr gene expression have been performed which used short cDNA probes more specific for their respective mRNAs because they do not cross-hybridize with characterized gene family members. These studies have been restricted to subjective descriptions of expression levels (reviewed inSantoni-Rugiu and Silverman, 1997). To understand the importance of xenobiotic transporters in pharmacology and toxicology requires more detailed information regarding the tissue distribution of individual mdr genes and the mechanisms controlling mdr gene regulation and induction.
In addition to defining the sites of mdr gene expression, this is the first study to assess the expression of rodent mdr genes in distinct sections of the intestine, an important site for absorption of xenobiotics and nutrients. Furthermore, the differential expression of mdr genes in various tissues will be described in a quantitative manner. Mechanisms involved in mdr gene regulation are described in whole animal studies. The effect of multiple chemicals belonging to six classes of microsomal enzyme inducers will be assessed. The mechanistic classes and chemicals are as follows: 1) the aryl hydrocarbon receptor (AhR) including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), indole-3-carbinol (I3C), β-naphthaflavone (BNF), and polychlorinated biphenyl (PCB) 126; 2) the constitutive androstane receptor (CAR) including phenobarbital (PB), PCB 99, and diallyl sulfide (DAS); 3) the pregnane X receptor (PXR) such as pregnenolone-16α-carbonitrile (PCN), spironolactone (Spir), and dexamethasone (Dex); 4) the peroxisome proliferator-activated receptor (PPAR) including clofibrate (Clof), diethylhexylphthalate (DEHP), and perfluorodecanoic acid (PFDA); 5) the electrophile response element (EpRE) such as ethoxyquin (EQ) and oltipraz (OPZ); and 6) the cytochrome P4502E1 inducers such as isoniazid (INH), acetylsalicylic acid (ASA), and streptozotocin (STZ). The purpose of this study was to determine the tissue distribution of rat mdr1a and mdr1b mRNAs and whether microsomal enzyme inducers that increase phase I and II drug-metabolizing enzymes coordinately regulate mdr1a and/or mdr1b genes.
Materials and Methods
Animals.
Male Sprague-Dawley rats (200–250g; Charles River Laboratories inc., Wilmington, MA) were acclimated to the housing facility (2–3 rats/cage, 50% relative humidity, 12-h light/dark cycle) for 1 week and fed Teklad 8604 rodent chow (Harlan Labs, Madison, WI). Rats were euthanized in a CO2 atmosphere. Tissues were snap-frozen in liquid nitrogen (intestinal epithelia was obtained by scraping prior to freezing) and stored at −80°C.
RNA Isolation.
Total RNA was isolated using RNAzol B reagent (Tel-Test Inc., Friendswood, TX) per the manufacturer's protocol. Briefly, 0.2 g of tissue was added to 2.0 ml of RNAzol B, placed in sterile polypropylene vials, and subjected to homogenization (30 s) with a Polytron (Brinkman Instruments Inc., Westbury, NY). To each homogenate, 0.2 ml chloroform was added, and the vials were vigorously shaken for 45 s followed by incubation at 4°C for 7 to 8 min. The vials were then subjected to centrifugation at 10,000g for 15 min. The aqueous (upper) phase containing the RNA was removed, and total RNA was precipitated for 30 min at −20°C in 3 to 4 ml isopropanol. After precipitation, the vials were centrifuged at 12,000g for 15 min. The supernatant was removed and each pellet washed with 3.0 ml of 75% ethanol and centrifuged again at 7,500g for 10 min. After centrifugation, the supernatant was discarded and the residual ethanol evaporated. Each pellet was redissolved in 0.2 ml of 0.1% SDS in 10 mM Tris (pH 7.5). RNA concentrations were assessed by ultraviolet absorbance at 260 nm. The integrity of the RNA samples (intact 18S and 28S bands) was assessed by agarose gel electrophoresis and visualization by ethidium bromide staining.
Chemicals.
TCDD was a gift from Dr. Karl Rozman (University of Kansas Medical Center, Kansas City, KS). Oltipraz was a gift of Dr. Ronald Lubet (National Cancer Institute, Bethesda, MD). 2,2′,4,4′,5-Pentachlorobiphenyl (PCB 99) and 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) were purchased from AccuStandard (New Haven, CT). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Microsomal Enzyme Inducers.
Each treatment group consisted of five male Sprague-Dawley rats (200–250g). Treatments were administered as follows: TCDD (3.9 μg/kg, i.p. in corn oil, 1 day), I3C (56 mg/kg, p.o. in corn oil), BNF (100 mg/kg, i.p. in corn oil), PCB 126 (40 μg/kg, i.p. in corn oil, 7 days), PB (80 mg/kg, i.p. in saline), PCB 99 (16 mg/kg, i.p. in corn oil, 7 days), DAS (500 mg/kg, i.p. in corn oil), PCN (50 mg/kg, i.p. in corn oil), Spir (75 mg/kg, i.p. in corn oil), Dex (40 mg/kg, i.p. in corn oil), Clof (200 mg/kg, i.p. in saline), DEHP (1200 mg/kg, p.o. in corn oil), PFDA (40 mg/kg, i.p. in corn oil, 1 day), EQ (150 mg/kg, p.o. in corn oil), OPZ (150 mg/kg, p.o. in corn oil), INH (200 mg/kg, i.p. in saline), ASA (500 mg/kg, p.o. in corn oil), STZ (100 mg/kg, i.p. in 100 mM sodium citrate, 1 day), corn oil (i.p.), corn oil (p.o.), and saline (i.p.). All animals were treated for 4 days unless otherwise noted and injections were in a volume of 5 ml/kg. Chemical induction was validated by measuring the induction of cytochrome P450s (Cherrington et al., 2002).
Development of Specific Oligonucleotide Probe Sets for bDNA Analysis.
The mdr gene sequences of interest were accessed from GenBank. These target sequences were analyzed by ProbeDesigner software version 1.0 (Bayer Corp., Diagnostics Div., Tarrytown, NY). Multiple and specific probes were developed to each mdr mRNA transcript (Table1). Individual oligonucleotide probes serve either as capture probes, which attach the specific transcript to the plate, as label probes, which attach the signal amplifier to the specific transcript, or as blocker probes, which bind to the specific transcript to reduce background. Oligonucleotide probes complimentary to specific regions of the mdr transcripts were specific to a single mRNA transcript (i.e., mdr1a or mdr1b). All oligonucleotide probes were designed with a Tm of approximately 63°C. This feature enables hybridization conditions to be held constant (i.e., 53°C) during each hybridization step and for each oligonucleotide probe set. Every probe developed in ProbeDesigner was submitted to the National Center for Biotechnological Information for nucleotide comparison by the basic logarithmic alignment search tool (BLASTn), to ensure minimal cross-reactivity with other known rat sequences and expressed sequence tags. Oligonucleotides with a high degree of similarity (≥80%) to other rat gene transcripts were eliminated from the design.
Oligonucleotide probes generated for analysis of mdr expression by bDNA signal amplification
Branched DNA Assay.
Specific mdr oligonucleotide probe sets (capture, label, and blocker probes), were combined and diluted to 50, 100, and 200 fmol/μl, respectively in the lysis buffer supplied in the Quantigene bDNA signal amplification kit (Bayer Corp., Diagnostics Div.) with modifications according to Hartley and Klaassen (2000). All reagents for analysis (i.e., lysis buffer, capture hybridization buffer, amplifier/label probe buffer, wash A and wash D, and substrate solution) were supplied in the Quantigene bDNA signal amplification kit. Total RNA (1 μg/μl; 10 μl) was added to each well of a 96-well plate containing capture hybridization buffer and 100 μl of each diluted probe set. Total RNA was allowed to hybridize to each probe set containing all probes for a given transcript (blocker probes, capture probes, and label probes) overnight at 53°C in a Quantiplex bDNA heater. Subsequently, the plate was removed from the heater, cooled to room temperature, and rinsed with wash A. Samples were hybridized with a solution containing the bDNA amplifier molecules (50 μl/well) diluted in amplifier/label probe buffer and incubated for 30 min at 53°C. The plate was again cooled to room temperature. The amplifier solution was aspirated and wells were washed with wash A (3×). Label probe, diluted in amplifier/label (same as above) probe buffer, was added to each well (50 μl/well), and hybridized to the bDNA-RNA complex for 15 min at 53°C. The plate was cooled to room temperature, and each well was rinsed with wash A (2×) followed by wash D (3×). Alkaline phosphatase-mediated luminescence was triggered by the addition of a dioxetane substrate solution (50 μl/well). The enzymatic reaction was allowed to proceed for 30 min at 37°C and luminescence measured with the Quantiplex 320 bDNA Luminometer (Bayer Corp., Diagnostics Div.) interfaced with Quantiplex data management software version 5.02 (Bayer Corp., Diagnostics Div.) for analysis of luminescence from 96-well plates (Hartley and Klaassen, 2000).
Statistics.
Data from the inducer studies were analyzed using ANOVA followed by a Duncan's multiple range post hoc test. Asterisks (★) represent a statistical difference (p ≤ 0.05) from controls.
Results
To determine the tissues where mdr1a and mdr1b mRNA transcripts are expressed, total RNA from five male rats was individually isolated from liver, kidney, lung, stomach, duodenum, jejunum, ileum, large intestine, cerebellum, and cerebral cortex and analyzed by the Quantigene signal amplification assay. To ensure the major tissues of expression were reported, single determinations were performed using pooled RNA from five male or female rats isolated from the above tissues as well as the following: heart, blood vessel, spleen, thymus, muscle, skin, adrenal, lymph node, thyroid, eye, pituitary, thalamus, brain stem, caudate, frontal cortex, hippocampus, olfactory bulb, spinal cord, urinary bladder, testes, ventral prostate, dorsal prostate, ovary, and uterus (data not shown). The tissues assayed were selected based on the known expression pattern and sites of transport activity. To determine whether gender differences occur in mdr1 mRNA expression, individual RNA samples from five male or female rats were analyzed from the 10 major tissues listed above. No major differences were observed between male and female expression of mdr1a and mdr1b mRNAs (data not shown).
The tissue distribution of rat mdr1a mRNA is shown in Fig.1. The highest level of expression of rat mdr1a mRNA was observed in the gastrointestinal tract, with levels increasing, respectively, from stomach (1%), duodenum (20%), jejunum (36%), to ileum (100%), and large intestine (73%). Expression levels of mdr1a mRNA in the cerebral cortex (6%), cerebellum (4%), kidney (3%), lung (2%), and liver (1%) were all less than one-tenth of that in the ileum.
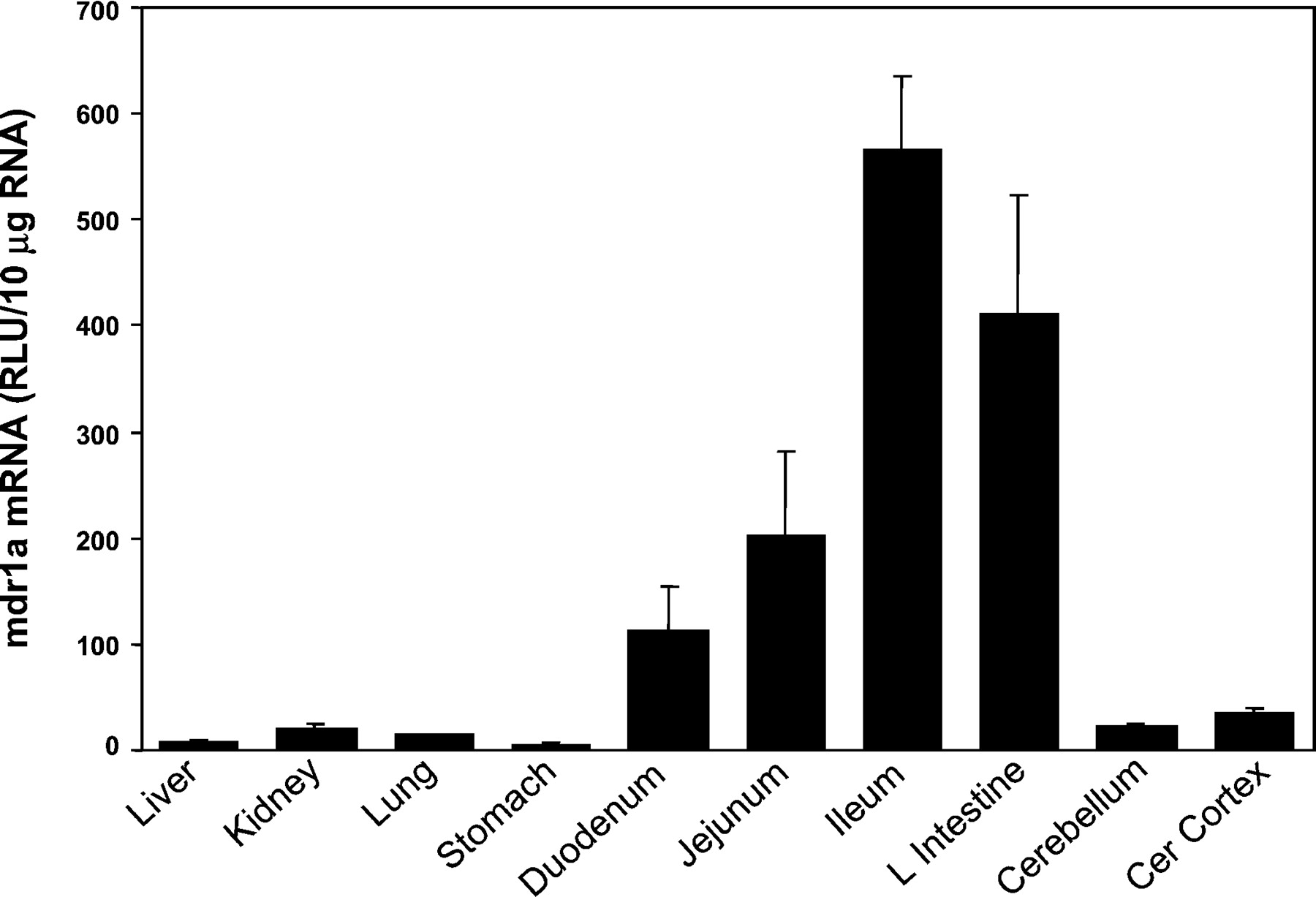
The tissue distribution of rat mdr1a mRNA.
Ten micrograms of total RNA from various tissues of male Sprague-Dawley rats were analyzed with the mdr1a-specific probe set described underMaterials and Methods. Results are the means ± S.E.M. of determinations from five rats per group.
The tissue distribution of mdr1b mRNA is shown in Fig.2. The tissue distribution of mdr1b mRNA was similar to mdr1a with highest expression in the gastrointestinal tract but only about 3-fold higher than in most other tissues. In general, the highest level of expression of rat mdr1b mRNA was observed in the gastrointestinal tract, with levels increasing, respectively, from stomach (11%), duodenum (25%), jejunum (39%), to ileum (100%) and large intestine (80%). Moderate expression levels of mdr1b mRNA were seen in the lung (47%) and kidney (21%). Lower levels of mdr1b mRNA were seen in the cerebellum (15%), cerebral cortex (12%), and liver (7%).

The tissue distribution of rat mdr1b mRNA.
Ten micrograms of total RNA from various tissues of male Sprague-Dawley rats were analyzed with the mdr1b-specific probe set described underMaterials and Methods. Results are the means ± S.E.M. of determinations from five rats per group.
Chemicals and drugs that increase gene expression for phase I and II drug-metabolizing enzymes may coordinately regulate mdr1a and/or mdr1b genes. Chemicals that act through the same mechanism should have a similar effect on mdr gene regulation. Therefore, the effect of multiple chemicals belonging to six classes of microsomal enzyme inducers was assessed. These include chemicals that act through 1) the AhR including TCDD, I3C, BNF, and PCB 126; 2) the CAR including PB, PCB 99, and DAS; 3) the PXR such as PCN, Spir, and Dex; 4) the PPAR including Clof, DEHP, and PFDA; 5) the EpRE such as EQ and OPZ; and 6) the CYP4502E1 inducers such as INH, ASA, and STZ. The predicted chemical activation of these transcriptional mechanisms was confirmed by measuring the induced gene expression of an appropriate cytochrome P450 (Cherrington et al., 2002). CYP1A1 induction by AhR ligands ranged from 7- to 693-fold. CYP2B1/2 induction by CAR activators ranged between 54- and 96-fold. PXR ligands induced CYP3A1/23 from 14- to 33-fold, and PPAR ligands induced CYP4A2/3 between 4- and 9-fold. EpRE activators induced quinone reductase mRNA expression by 3- to 5-fold, whereas ARE activators increased CYP2E1 mRNA expression levels 2-fold (Cherrington et al., 2002). When one group of inducers has a common effect, it suggests a mechanism by which transporter expression is modulated.
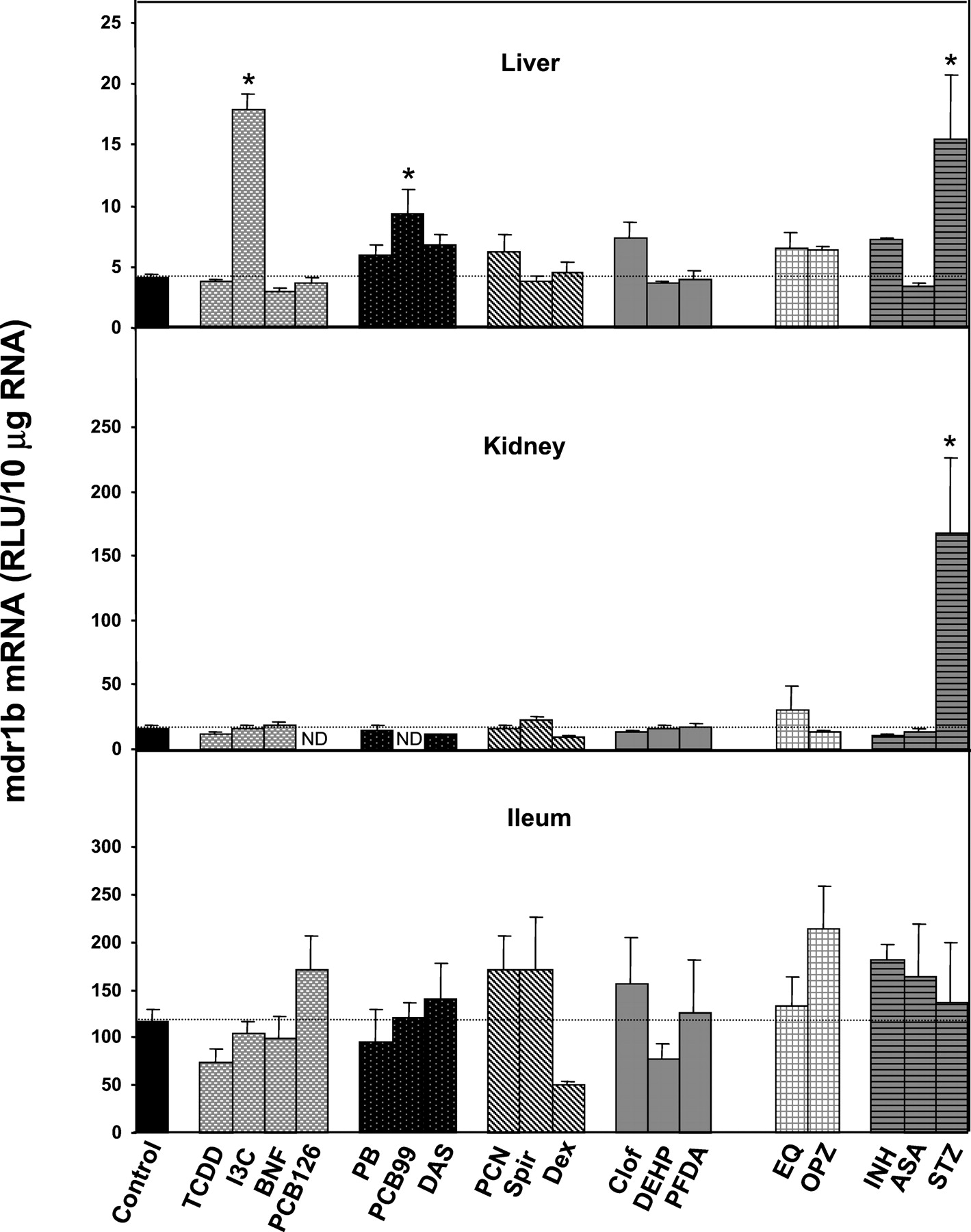
The induction of rat mdr1a mRNA (Fig. 3) was analyzed in liver (top panel), kidney (middle panel), and ileum (bottom panel). None of the six groups of microsomal enzyme inducers known to activate specific ligand-activated nuclear receptors elicited a group-specific induction of mdr1a mRNA by all chemicals within a group nor was there consistent induction in more than one tissue by any of the chemicals as shown in Fig. 3. Hepatic, renal, and intestinal expression of mdr1a mRNA was not consistently altered by treatment of rats with any of these classes of ligands. Treatment with the CAR ligand DAS and the EpRE ligand OPZ induced mdr1a in liver, and the CYP4502E1 inducer INH induced mdr1a in kidney. However, none of the treatments induced mdr1a mRNA in the ileum.

The induction of mdr1a mRNA in liver (a), kidney (b), and ileum (c), by treatment of rats with 18 chemicals representing AhR ligands, CAR ligands, PXR ligands, PPAR ligands, EpRE activators, and CYP4502E1 inducers was assessed.
Ten micrograms of total RNA from various tissues of male Sprague-Dawley rats were analyzed with the mdr1a-specific probe set described underMaterials and Methods. Results are the means ± S.E.M. of determinations from five rats per group. Statistical differences between groups were compared by a two-way ANOVA followed by a Duncan's Multiple Range posthoc test. Asterisks (★) represent a statistical difference (p ≤ 0.05) between treatment groups.
The induction of rat mdr1b mRNA (Fig. 4) was analyzed in liver (top panel), kidney (middle panel), and ileum (bottom panel). None of the six classes of ligands, AhR ligands, CAR ligands, PXR ligands, PPAR ligands, EpRE activators, and CYP4502E1 inducers showed consistent induction of mdr1b in the three tissues analyzed nor did all chemicals within any one of the six groups cause a group-specific induction of mdr1b, as shown in Fig. 4. Hepatic, renal, and intestinal expression of mdr1b mRNA was not significantly altered by treatment of rats with any of these classes of ligands. Although no group effects were observed, treatment with the AhR ligand I3C and CAR ligand PCB 99 increased mdr1b mRNA level in the liver, and the CYP4502E1 inducer STZ induced mdr1b in both liver and kidney. However, none of the treatments induced mdr1b mRNA in the ileum.

The induction of mdr1b mRNA in liver (a), kidney (b), and ileum (c), by treatment of rats with 18 chemicals representing AhR ligands, CAR ligands, PXR ligands, PPAR ligands, EpRE activators, and CYP4502E1 inducers was assessed.
Ten micrograms of total RNA from various tissues of male and female Sprague-Dawley rats were analyzed with the mdr1b-specific probe set described under Materials and Methods. Results are the means ± S.E.M. of determinations from five rats per group. Statistical differences between groups were compared by a two-sided ANOVA followed by a Duncan's Multiple Range post hoc test. Asterisks (★) represent a statistical difference (p ≤ 0.05) between treatment groups.
Discussion
The bDNA signal amplification system has many advantages over other contemporary methods of gene expression analysis. A critical component of the bDNA signal amplification system is that multiple, highly specific oligonucleotides are generated to a target region of a particular mRNA transcript. This novel approach is advantageous as it uses the specificity inherent in short oligonucleotide probes combined with the sensitivity of longer oligonucleotides or cDNA probes. Because this methodology requires the use of multiple oligonucleotides at one time for each sample, and it relies on complete hybridization between the probe and the target (which is subjected to stringent washing following hybridization), nonspecific hybridization is decreased. Additionally, the differential expression of mdr genes is described in a semiquantitative manner. The bDNA signal amplification system requires less initial setup and validation of probes and gives consistent reproducibility due to its linear amplification of signal as compared with template amplification of quantitative polymerase chain reaction.
Many of the gene expression studies on mdr genes were previously performed with cDNA probes, which cross-hybridized to different mdr gene family members. Thus, Northern-blot analyses with longer probes should be interpreted with caution. In contrast, shorter oligonucleotide probes are more specific for their cognate mRNA and exhibit less nonspecific binding. However, these oligonucleotide probes have lower specific activity, which makes them less sensitive for use on low abundance mRNAs. In rats, mdr1a mRNA has previously been detected at a high level in small intestine; moderate levels in liver, kidney, lung, and brain; and low levels in spleen and heart. Skeletal muscle was also analyzed, but no mdr1a mRNA was detected. Tissues that were not previously analyzed include adrenal, stomach, various sections of the small intestine, large intestine, and colon (reviewed inSantoni-Rugiu and Silverman, 1997). Potential problems with cross-hybridization of probes are minimized with bDNA analysis through the use of multiple short oligonucleotide probes. Short oligonucleotide probes that are specific for their respective mRNAs have been used in the present study to show that in rats the mdr1a mRNA is present at the highest level of expression in the gastrointestinal tract, with levels increasing, respectively, proximally to distally [i.e., stomach, duodenum, jejunum, to ileum and large intestine (Fig. 2)]. Expression levels of mdr1a mRNA in cerebral cortex, cerebellum, kidney, lung and liver were all less than one-tenth of that in the ileum. The present study has assessed the expression of rodent mdr genes in various sections of the intestine, which were scraped to enrich for epithelial cells. These results show that the mdr1a mRNA is expressed at a high level at the distal end of the intestine.
Rat mdr1b mRNA has previously been detected, using a 313 base pairs cDNA probe, at a high level in lung; moderate level in liver; and at low levels in small intestine, kidney, and spleen. Several tissues (adrenal gland, brain, heart, and skeletal muscle) were also previously analyzed by the RNase protection assay and were reported to lack mdr1b mRNA. Tissues that were not analyzed include stomach, large intestine, and colon (Brown et al., 1993). The tissue distribution of mdr1b mRNA reported here showed highest expression in the distal gastrointestinal tract and moderate expression in the lung, kidney, stomach, brain, and liver (Fig. 2). Brown et al. (1993) found mdr1b mRNA at high levels in the lung and low levels in the intestine. The present study detected mdr1b mRNA at higher levels in the ileum and large intestine than the lung but lower levels in duodenum and jejunum than the lung. It is not apparent which section of the intestine Brown et al. (1993) used or if they used only epithelial cells as were used in the present study. Our method of tissue collection of the intestine led to the isolation and analysis of mRNA from only epithelial cells of the intestine. Expression of mdr genes may have appeared higher in other tissues if only capillary endothelium were analyzed (e.g., in the brain).
The physiological importance of P-gp in the intestine, where it is detected in high abundance, has been supported by the use of knockout animals. Although P-gp has a function in many tissues, its function in decreasing the absorption across the gastrointestinal tract has received the most attention. P-gp functions as an absorption barrier by transporting drugs from intestinal cells into the lumen (Terao et al., 1996). The hypersusceptibility to some xenobiotics in mdr1a knockout mice supports a normal physiological role of P-gp to protect vital cells from toxic challenges (Leveille-Webster and Arias, 1995). Furthermore, mice in which both mdr1a and mdr1b genes have been disrupted show reduced intestinal and hepatobiliary clearance of cationic amphiphilic drugs (Sparreboom et al., 1997; Smit et al., 1999). After oral administration of HIV-1 protease inhibitors, plasma concentrations were 2- to 5-fold higher in P-gp knockout mice in comparison with control animals (Kim et al., 1998, Choo et al., 2000; Fromm, 2000a). Bioavailability of the chemotherapeutic agent paclitaxel is increased in P-gp knockout mice in comparison with control animals (Sparreboom et al., 1997). These knockout mice have slower elimination, and higher tissue concentrations, of vinblastine and ivermectin than do wild-type mice (Schinkel et al., 1995). Finally, these P-gp null mice have elevated tissue levels of the cardiac glycoside digoxin (Kawahara et al., 1999), cyclosporin A (Kwei et al., 1999), dexamethasone (Meijer et al., 1998), rifampicin (Schuetz et al., 1996a), and morphine (Zong and Pollack, 2000) than do control mice treated by oral administration. These examples, in conjunction with intestinal mRNA levels presented here, demonstrate that intestinal P-gp limits drug absorption and contributes to drug elimination.
Studies on the induction of rat mdr1a and mdr1b genes have been hampered by the lack of probe specificity. Moreover, there have been conflicting results from studies performed in different labs using different techniques. Chemical treatment of rats in vivo with the AhR ligand TCDD showed that the mRNA level of mdr genes in liver increases; however, these early studies used long cDNA probes that did not distinguish mdr1a from mdr1b (Burt and Thorgeirsson, 1988). Subsequent studies with more specific probes verified the induction of mdr1b mRNA and showed that it involved a different receptor than the AhR (Thorgeirsson et al., 1991). However, other reports did not show rat mdr1a and mdr1b mRNA induction in vivo upon TCDD treatment when analyzed with short specific probes (Gant et al., 1991; Teeter et al., 1991). In primary human hepatocytes, Schuetz et al. (1995) have shown that TCDD treatment will both induce and suppress human MDR1 mRNA. There is significant individual variability in this response, which has been concluded to act through a receptor other than the AhR (Schuetz et al., 1995). Results presented here show induction of neither mdr1a or mdr1b mRNAs with TCDD treatment (Figs. 3 and 4). Chemical treatment of rats in vivo with the AhR ligands TCDD, I3C, BNF, and PCB 126 showed no consistent induction of mdr1a or mdr1b mRNAs in the three tissues analyzed: liver, kidney, and ileum (Figs. 3 and 4). However, I3C treatment increased mdr1b mRNA level in the liver but not in the other tissues. Therefore, it can be concluded that mdr1a and mdr1b in rats are not typically induced through the AhR.
The CAR ligand PB has been reported to stimulate the human MDR1 gene in the human colon adenocarcinoma cell line LS180 (Schuetz et al., 1996b). Yet in human primary hepatocytes, PB treatment had no effect on MDR1 (Runge et al., 2000). Results presented here also show no significant changes in rat mdr1a or mdr1b mRNA levels upon in vivo treatment with PB. None of the CAR ligands (PB, PCB 99, and DAS) showed consistent induction of rat mdr1a or mdr1b mRNAs in the three tissues analyzed, although DAS treatment increased mdr1a mRNA in liver and PCB 99 slightly induced mdr1b mRNA in liver (Figs. 3 and 4).
Chemicals that act through the PXR include PCN and Dex in rodents as well as rifampin and, to a lesser extent, Dex in humans. In the male rat liver, Salphati and Benet (1998) observed that PCN and Dex treatments resulted in no effect on mdr1a or mdr1b mRNAs. In the female rat liver, they reported no effect on mdr1a but could not detect mdr1b (Salphati and Benet, 1998). When rat primary hepatocytes are placed in culture, the level of mdr1b mRNA increases with time in culture. However, Dex treatment abolishes this effect (Fardel et al., 1993;Schuetz et al., 1995). In rat, mouse, and human hepatoma cell lines as well as human liver, Dex increased mdr1 mRNA (Zhao et al., 1993;Schuetz et al., 1995; Seree et al., 1998). In nonhepatoma cell lines NIH3T3 and HeLa cells, Dex did not induce mdr genes (Zhao et al., 1993). In the mouse, in vivo Dex treatment showed various effects in different tissues. In liver, heart, brain, and colon, there was an increase in mdr1b mRNA, whereas in the adrenal gland, lung, and kidney, mdr1b mRNA decreased upon Dex treatment (Seree et al., 1998). Chemical treatment of rats in vivo with the PXR ligands PCN, Spir, and Dex in the present studies showed no consistent induction of rat mdr1a or mdr1b mRNAs in the three tissues analyzed, namely liver, kidney, and ileum (Figs. 3 and 4).
The antibiotic rifampin is a PXR ligand in humans but not rodents. Rifampin treatment of humans in vivo increased intestinal P-gp content 3-fold with increased clearance of digoxin (Greiner et al., 1999). Rifampin in vitro up-regulated P-gp in human colon carcinoma cells (Schuetz et al., 1996b). There are two recent descriptions of the mechanism involved in rifampin induction of the human MDR1 gene. The human colon carcinoma cell lines LS174T and LS180 have been used as intestinal models to study induction because in these cells the endogenous MDR1 gene is highly inducible by rifampin (Geick et al., 2001; Synold et al., 2001). MDR1 gene regulation has been shown to be mediated by a DR4 motif in the upstream enhancer of the human MDR1 gene at about −8 kilobase, to which PXR binds (Geick et al., 2001).
However, in primary human hepatocytes from two donors, Runge et al. (2000) did not see an increase in MDR1 protein with rifampin treatment. Whereas the report of Synold et al. (2001) indicates that rifampicin causes a moderate induction of hepatic MDR1 gene expression in primary human hepatocytes. These discrepancies can in part be explained by the observed interindividual variation in the content of P-gp between human livers as shown by Schuetz et al. (1995), which contributes to individual variability in rifampicin disposition (Schuetz et al., 1996a). Therefore, induction of MDR1 by PXR ligands in humans and human cell lines appear in most but not all studies, whereas PXR ligands consistently do not induce mdr in rats in vivo. Thus, there appear to be species differences in the ability of PXR ligands to induce P-gp in humans and rats. Rifampin treatment induced another member of the ATP-binding cassette transporter family, MRP2 at both the mRNA and protein levels in intact human intestine (Fromm et al., 2000b). This result was extended to primary cultures of human hepatocytes, in which PXR ligands, rifampicin and HIV protease inhibitors, caused a significant increase in MRP2 mRNA level (Dussault et al., 2001).
There are no previously published studies on the effects of PPAR ligands, EpRE ligands, nor CYP4502E1 inducers on the induction of rat mdr1a or mdr1b mRNAs. None of the PPAR ligands used in the present study (Clof, DEHP, and PFDA) induced rat mdr1a nor mdr1b mRNAs in the three tissues analyzed. EpRE ligands (EQ and OPZ) showed no consistent induction of rat mdr1a or mdr1b mRNAs in the three tissues analyzed. None of the CYP4502E1 inducers (INH, ASA, and STZ) caused consistent induction of rat mdr1a and mdr1b mRNAs in the three tissues analyzed (Figs. 3 and 4).
The primary expression of rat mdr1 genes is in the gastrointestinal tract where they are thought to function to decrease the absorption and enhance the excretion of some xenobiotics. Rodent mdr1a and mdr1b mRNAs are most abundant in the large intestine. In contrast to phase I and II drug-metabolizing genes, mdr1 gene expression is not readily increased by microsomal enzyme inducers. Previous studies showing induction by one chemical of a class of inducers are not conclusive and do not define the mechanism involved. To define the mechanism(s) of mdr gene regulation requires the demonstration of similar in vivo effects for multiple chemical treatments, which act through a common mechanism. Although human P-gp appears to be in part regulated by the PXR, further mechanistic studies of the regulation of P-gp expression are needed to increase our understanding of pharmacological interactions between various drugs and chemicals.
Footnotes
-
↵1 Current address: Department of Drug Metabolism, Merck Research Laboratories, Mail Stop RY80D-100, Rahway, NJ 07065.
-
Supported by National Institutes of Health Grants ES-09716, ES-03192, ES-05883011, training Grant ES-07079, and Lied Foundation Grant 559086.
- Abbreviations used are::
- P-gp
- P-glycoproteins
- mdr
- multiple drug resistance
- AhR
- aryl hydrocarbon receptor
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- I3C
- indole-3-carbinol
- BNF
- β-naphthaflavone
- PCB
- polychlorinated biphenyl
- CAR
- constitutive androstane receptor
- PB
- phenobarbital
- DAS
- diallyl sulfide
- PXR
- pregnane X receptor
- PCN
- pregnenolone-16α-carbonitrile
- Spir
- spironolactone
- Dex
- dexamethasone
- PPAR
- peroxisome proliferator-activated receptor
- Clof
- clofibrate
- DEHP
- diethylhexylphthalate
- PFDA
- perfluorodecanoic acid
- EpRE
- electrophile response element
- EQ
- ethoxyquin
- OPZ
- oltipraz
- INH
- isoniazid
- ASA
- acetylsalicylic acid
- STZ
- streptozotocin
- ANOVA
- analysis of variance
- Received January 2, 2002.
- Accepted April 11, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}