


Abstract
The leukotriene receptor antagonist montelukast was examined for its inhibition of the human drug-metabolizing enzyme cytochrome P4502C8 (CYP2C8). Montelukast was demonstrated to be a potent inhibitor of CYP2C8-catalyzed amodiaquine N-deethylase, rosiglitazone N-demethylase, and paclitaxel 6α-hydroxylase in human liver microsomes. Inhibition was also observed when the reaction was catalyzed by recombinant heterologously expressed CYP2C8. The mechanism of inhibition was competitive, with Ki values ranging from 0.0092 to 0.15 μM. Inhibition potency was highly dependent on the microsomal protein concentration. Increasing the microsomal protein concentration by 80-fold yielded a 100-fold decrease in inhibition potency. Preincubation of montelukast with human liver microsomes and NADPH did not alter the inhibition potency, suggesting that montelukast is not a mechanism-based inactivator. Montelukast was a selective inhibitor for human CYP2C8; inhibition of other human cytochrome P450 enzymes was substantially less. These in vitro data support the use of montelukast as a selective CYP2C8 inhibitor that could be used to determine the contribution of this enzyme to drug metabolism reactions. These data also raise the possibility that montelukast could have an effect on the metabolic clearance of drugs possessing CYP2C8-catalyzed metabolism as a major clearance pathway, thereby eliciting pharmacokinetic drug-drug interactions.
Cytochrome P450 (P450) enzymes represent a family of proteins that contribute predominantly in the metabolism of drugs and other xenobiotics. Alteration in the activity of these enzymes in vivo represents the major underlying mechanism behind pharmacokinetic drug-drug interactions (Clarke and Jones, 2002). In humans, some P450 enzymes have been designated as more predominant in the metabolism of drugs than others. These include CYP1A2, 2C9, 2C19, 2D6, and 3A. These five enzymes have been studied extensively with regard to their role in drug metabolic clearance as well as drug-drug interactions. However, recent investigations have uncovered roles for other cytochrome P450 enzymes, such as CYP2C8, in the metabolism of some drugs. Thus, CYP2C8 can also be the target of drug-drug interactions for those drugs in which it plays a predominant role in the clearance. Drugs for which CYP2C8 appears to play a major role in metabolic clearance include repaglinide, rosiglitazone, and cerivastatin. Drug interactions that arise by inhibition of CYP2C8 have been described recently, including interactions between cerivastatin and gemfibrozil (Backman et al., 2002), repaglinide, and gemfibrozil (Niemi et al., 2003b), and repaglinide and trimethoprim (Niemi et al., 2004).
In previous work, a vast number of drugs have been tested for their potential to inhibit CYP2C8 (Bun et al., 2003, Ong et al., 2000; Walsky et al., 2005). In a study conducted in our laboratories, we found that montelukast (Fig. 1), a leukotriene receptor antagonist used in the treatment of asthma, potently inhibited CYP2C8-catalyzed amodiaquine N-deethylase, with an IC50 value of 20 nM. Other agents used in the treatment of asthma, such as zafirlukast and salmeterol, were substantially less potent.

Structure of montelukast.
The studies described in this paper were undertaken with the following objectives: 1) to thoroughly characterize the inhibition of CYP2C8 by montelukast with respect to mechanism as well as testing versus more than one CYP2C8-catalyzed reaction; 2) to determine the selectivity of montelukast for CYP2C8 versus other human P450 enzymes; 3) to define the conditions under which montelukast could be used as a selective CYP2C8 inhibitor in P450 reaction phenotyping exercises; and 4) to use in vitro inhibition data in an attempt to predict the magnitude of in vivo drug interactions that could be caused by montelukast on CYP2C8-metabolized drugs. Previously, other investigators have reported that montelukast was only a weak inhibitor of human P450 enzymes (Chiba et al., 1997). (CYP2C8 was not examined in that investigation.) Through our investigation of the selectivity of montelukast for CYP2C8 versus other human P450 enzymes, we measured lower inhibition constants than those previously reported. Data are offered to suggest that montelukast is subject to nonspecific binding in liver microsomes, which can affect the measurement of inhibition constants.
Materials and Methods
Materials and Instrumentation. Substrates, metabolite standards, internal standards, inhibitors, and other materials were obtained from sources previously described (Walsky and Obach, 2004). Other reagents and solvents used were obtained from standard suppliers and were of reagent or HPLC grade. Human liver microsomes were prepared using standard procedures and represent a pool of samples from 54 individual donors. Human liver microsomes from individual donors were obtained from BD Gentest (Woburn, MA).
Analytical instrumentation included a Micromass Quattro Ultima tandem quadrupole mass spectrometer (Waters, Milford, MA) equipped with an electrospray ionization source. The HPLC consisted of Shimadzu 10ADvp HPLC pumps with a controller and a solvent degasser (Shimadzu, Columbia, MD), and a LEAP CTC PAL autosampler (LEAP Technologies Inc., Carrboro, NC).
Amodiaquine N-Deethylase Assay. Human liver microsomes (0.025 mg/ml) or rhCYP2C8 (1.7 pmol P450/ml) were incubated with amodiaquine (0.24–5.7 μM), NADPH (1.3 mM), and MgCl2 (3.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) in a total incubation volume of 0.2 ml. Incubations were commenced by the addition of NADPH and were carried out at 37°C for 10 min, followed by termination by addition of 0.02 ml of internal standard solution (0.75 μM [2H5]desethylamodiaquine in 92:5:3 H2O/CH3CN/HCOOH). The terminated incubation mixtures were filtered through a Millipore multiscreen mixed cellulose membrane (0.45-μm pore size) 96-well filtration plate (Millipore Corporation, Billerica, MA), and analyzed for N-desethylamodiaquine by HPLC-tandem mass spectrometry. In some experiments, the concentration of human liver microsomes was increased to up to 2 mg/ml (see below).
Samples were injected (10 μl) onto a YMC C4 column (3 × 50 mm, 5 μm; Waters) equilibrated in 2% organic mobile phase at a flow rate of 0.5 ml/min. The aqueous mobile phase comprised 5 mM ammonium formate containing 0.3% formic acid, and the organic mobile phase comprised 95:5 acetonitrile/methanol containing 0.3% formic acid. After injection, a linear gradient was applied to 8% organic at 0.5 min, then increased to 18% organic at 2.7 min. Detection was accomplished by selected reaction monitoring of the mass transitions m/z 328 → 283 for N-desethylamodiaquine and m/z 333 → 283 for internal standard. Instrument settings were as follows: capillary, 2.75 kV; cone, 40 V; source temperature, 110°C; desolvation temperature, 325°C; cone gas, 300 l/h; desolvation gas, 800 l/h; collision energy, 20 eV. Other potentials were adjusted to optimize the signal. Under these conditions, the retention times for analyte and internal standard were 1.9 min. Quantitation was done by extrapolation from a standard curve ranging from 0.01 to 1.5 μM with 1/x2 weighting.
Rosiglitazone N-Demethylase Assay. Human liver microsomes (0.05 mg/ml) or rhCYP2C8 (8.3 pmol P450/ml) were incubated with rosiglitazone (4.7–42 μM), NADPH (1.3 mM), and MgCl2 (3.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) in a total incubation volume of 0.2 ml. Incubations were commenced by the addition of NADPH, and were carried out at 37°C for 20 min or 10 min for human liver microsomes or rhCYP2C8, respectively, followed by termination by addition of 0.02 ml of internal standard solution (1 μM [2H4]desmethylrosiglitazone in 92:5:3 H2O/CH3CN/HCOOH). The terminated incubation mixtures were filtered through a Millipore multiscreen mixed cellulose membrane (0.45-μm pore size) and analyzed for N-desmethylrosiglitazone by HPLC-tandem mass spectrometry.
Samples were injected (10 μl) onto a Phenomenex LUNA 5-μm C8(2) column (3 × 30 mm; Phenomenex, Torrance, CA) equilibrated in 2% organic mobile phase at a flow rate of 0.5 ml/min. The aqueous mobile phase consisted of 5 mM ammonium formate containing 0.05% formic acid, and the organic mobile phase was composed of 95:5 acetonitrile/methanol containing 0.05% formic acid. After injection, a linear gradient was applied to 16% organic at 0.5 min, then increased to 30% organic at 2.6 min. Detection of N-desmethylrosiglitazone and internal standard was done by multiple reaction monitoring of m/z 344 → 121 and m/z 348 → 125, respectively. Instrument settings were as follows: capillary, 2.75 kV; cone, 60 V; source temperature, 110°C; desolvation temperature, 325°C; cone gas, 300 l/h; desolvation gas, 800 l/h; collision energy, 26 eV, with other potentials adjusted to optimize the signal. The retention time for analyte and internal standard was 2.0 min. The linear dynamic range of the assay was 0.001 to 1 μM using 1/x2 weighting.
Paclitaxel 6α-Hydroxylase Assay. Human liver microsomes (0.25 mg/ml) were incubated with paclitaxel (2.5–20 μM), NADPH (1 mM), in potassium phosphate buffer (100 mM, pH 7.4) in a total incubation volume of 0.2 ml. Incubations were commenced by the addition of NADPH and were carried out at 37°C for 30 min, followed by termination by addition of 0.6 ml of internal standard solution [1.3 μM 3-(4-(5-[1-(3-chloro-2,6-diflurobenzyloxyimino)-ethyl]-3,4-dihydroxytetrahydrofuran-2-yloxy)-3-hydroxyphenyl)-2-methyl-N-(4,6,7-trihydroxyhexahydrobenzo(1,3)dioxol-5-yl)-acrylamide] in methanol. Samples were vortexed on a Multi-tube Vortexer (Henry Troemner LLC, Thorofare, NJ) for 10 min, followed by centrifugation at 4000g for 20 min. The organic layer was removed by a Biomek FX robotic sample handler (Beckman-Coulter, Fullerton, CA) and evaporated under a gentle stream of nitrogen using a Turbo-Vap evaporator (Zymark, Hopkinton, MA) at 65°C. Samples were reconstituted in 50:50 CH3OH/H2O (150 μl).
Samples were injected (15 μl) onto a Synergi Polar-RP phenyl column (2 × 30 mm, 4 μm; Phenomenex) equilibrated in aqueous mobile phase at a flow rate of 0.8 ml/min. The aqueous mobile phase was composed of water containing 0.1% formic acid, and the organic mobile phase was composed of methanol containing 0.1% formic acid. After injection, a rapid linear gradient was applied from 0% to 95% organic in 1.5 min. Detection was accomplished by selected reaction monitoring of the mass transitions m/z 870 → 286 for 6α-hydroxytaxol and m/z 687 → 320 for the internal standard. Instrument settings were as follows: capillary, 3.50 kV; cone, 35 V; source temperature, 100°C; desolvation temperature, 350°C; cone gas, 100 l/h; desolvation gas, 800 l/h; and collision energies were 19 eV and 22 eV for 6α-hydroxytaxol and the internal standard, respectively. Other potentials were adjusted to optimize the signal. Under these conditions, the retention times for 6α-hydroxytaxol and internal standard were 1.5 and 1.4 min, respectively. Quantitation was done by extrapolation from a standard curve ranging from 0.0005 to 0.5 μM with 1/x2 weighting.
Time Dependent Inhibition Assay. All incubations were carried out at 37°C. Human liver microsomes were equilibrated for 5 min at 10-fold the final assay microsomal protein concentration with montelukast at 10-fold its IC25 concentration for each P450 enzyme evaluated, in potassium phosphate buffer (100 mM, pH 7.4) containing MgCl2 (3.3 mM) in an incubation volume of 0.18 ml. Preincubation was commenced by the addition of either 20 μl of NADPH (1.3 mM) or phosphate buffer (control) and continued for 30 min. Subsequently, 0.02 ml of the preincubation mixture was transferred to 0.18 ml of assay mixture (1:10 dilution), consisting of substrate (at Km), NADPH (1.3 mM), MgCl2 (3.3 mM) in potassium phosphate buffer (100 mM, pH 7.4) prewarmed to 37°C in a total incubation volume of 0.2 ml. Incubations were terminated at the appropriate time with acidified internal standard solution and analyzed as described previously (Walsky and Obach, 2004).
Data Analysis. Substrate saturation curves and inhibition data were analyzed using the Enzyme Kinetics module of Sigma Plot v8.0 (SPSS Inc., Chicago, IL). Best fit models were selected on the basis of the Akaike information criterion. Data for IC50 determinations were typically fit to the following equation: 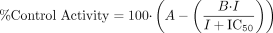 in which I is the inhibitor concentration, the IC50 represents the inflection point, and the value of 1–(A–B) is the maximum inhibition observed at an infinite inhibitor concentration. All data are reported as the mean ± standard error of n = 2 experiments.
in which I is the inhibitor concentration, the IC50 represents the inflection point, and the value of 1–(A–B) is the maximum inhibition observed at an infinite inhibitor concentration. All data are reported as the mean ± standard error of n = 2 experiments.
Results
Inhibition of CYP2C8 Activities by Montelukast. The inhibition of amodiaquine N-deethylase by montelukast was examined in pooled human liver microsomes, recombinant heterologously expressed human CYP2C8, and two liver microsomal samples from individual donors (possessing high and low CYP2C8 activities). Montelukast demonstrated competitive inhibition (Fig. 2) with Ki values ranging from 0.0092 to 0.038 μM (Table 1).

Dixon plots of the inhibition of CYP2C8 activities by montelukast. A, amodiaquine N-deethylase; B, rosiglitazone N-demethylase; C, paclitaxel 6α-hydroxylase.
Inhibition of CYP2C8 activities by montelukast
Montelukast also inhibited rosiglitazone N-demethylase. In human liver microsomes, inhibition of this activity was not complete, since rosiglitazone N-demethylase is also catalyzed by CYP2C9. Ki values were 0.019 and 0.11 μM in rhCYP2C8 and pooled human liver microsomes, respectively (Table 1). The higher-value Ki measured in human liver microsomes is likely due to the influence of the CYP2C9 contribution to rosiglitazone N-demethylase. Additionally, montelukast was tested as an inhibitor of paclitaxel 6α-hydroxylase, another CYP2C8-specific biotransformation pathway. The Ki for this activity was greater (0.15 μM) than the values observed for amodiaquine N-deethylase and rosiglitazone N-demethylase, likely due to the fact that the paclitaxel hydroxylase assay required a greater microsomal protein concentration (0.25 mg/ml), and montelukast is subject to nonspecific binding as microsomal concentrations are increased (see below).
Inhibition of Other Human Cytochrome P450 Enzymes by Montelukast. To determine the potential selectivity of montelukast for CYP2C8, the effects were measured on specific activities of other human P450 activities. In previously reported investigations, montelukast was shown to have little to no effect on other human P450 activities (Chiba et al., 1997). In the present investigation, montelukast was shown to be a more potent inhibitor of other human P450 enzymes than in the aforementioned previous report; however, the IC50 values for these other P450 activities were substantially greater than those measured for CYP2C8. A comparison of inhibitory potency of montelukast for CYP2C8 versus the other human P450 enzymes is shown in Table 2. Compared with CYP2C8, the next most potent interaction was with CYP2C9 (1.2 μM), which is 50-fold greater than the interaction with CYP2C8.
Inhibition of human cytochrome P450 activities in pooled human liver microsomes by montelukast
Relationship between Montelukast Preincubation and P450 Inhibition. Experiments were conducted in which montelukast was incubated with human liver microsomes in the presence and absence of NADPH, followed by addition of amodiaquine and measurement of the amodiaquine N-deethylase rate to determine whether montelukast requires activation (or possibly conversion to a metabolite that is more potent at CYP2C8 inhibition) to demonstrate its effect on CYP2C8. After preincubation of human liver microsomes with montelukast at concentration of 0.0066 μM and NADPH, no increase in inhibition was observed as compared with preincubation conducted in the absence of NADPH (79 and 71% of control activity with and without NADPH preincubation, respectively). This indicates that montelukast is not a time-dependent inhibitor or mechanism-based inactivator of CYP2C8. Similar preincubation experiments were conducted with the other human P450 enzymes, and no evidence of time-dependent inhibition was observed with these enzymes either (data not shown).
Relationship between Microsomal Protein Concentration and Inhibitory Potency of Montelukast. One of the potential reasons underlying the observation of differences in inhibition constants for the same enzyme and inhibitor is the use of different microsomal protein concentrations in enzymatic incubations. This phenomenon is mostly associated with lipophilic inhibitors, which can be reversibly bound in the phospholipid portion of liver microsomes; the binding effectively reduces the intended inhibitor concentration available to bind to the enzyme (Margolis and Obach, 2003). Since montelukast is a lipophilic drug and could potentially bind to liver microsomes, a test of the relationship between inhibitory potency and liver microsomal protein concentration in the incubation was conducted. This is of particular importance since, in the earlier work in which montelukast was not shown to be an inhibitor of P450 enzymes, the microsomal protein concentration was 1.0 mg/ml (Chiba et al., 1997), whereas in the work reported in the present paper, the microsomal protein concentration was 0.01 to 0.2 mg/ml. Plots of percentage of CYP2C8 control activity versus montelukast concentration were constructed using inhibition data from incubations using five different microsomal protein concentrations (Fig. 3). The microsomal protein concentrations selected for study range from 0.025 to 2.0 mg/ml. The IC50 value increased with increasing microsomal protein concentration and ranged over 100-fold, from 0.020 to 2.0 μM (Table 3). Determination of the fraction unbound of montelukast to microsomes (fu,mic) and, hence, free Ki, under these incubation conditions was attempted using equilibrium dialysis as previously described (Margolis and Obach, 2003); however, technical limitations did not allow for an accurate assessment. Recovery of montelukast from the dialysis apparatus was too low to permit reliable measurement, and solubility limitations at neutral pH did not allow for the use of high montelukast concentrations in an attempt to saturate binding to the apparatus to increase recovery. A similar relationship was observed between microsomal concentration and the inhibitory potency of montelukast for CYP2C9-catalyzed diclofenac 4′-hydroxylation, albeit the IC50 values were substantially greater than the corresponding values for CYP2C8 (Fig. 3).
Inhibition of amodiaquine N-deethylase activity in human liver microsomes by montelukast at different microsomal protein concentrations [0.025 (▴), 0.10 (▪), 0.30 (•), 1.0 (▾), and 2.0 (♦) mg/ml). A, percentage of control activity versus montelukast concentration; B, relationship between IC50 and microsomal protein concentration for CYP2C8 (▴) and CYP2C9 (•).
Inhibition of human liver microsomal amodiaquine N-deethylase activity by montelukast at varying microsomal protein concentrations
Comparison of the Selectivity of Montelukast and Quercetin for Inhibition of CYP2C8. The flavonoid quercetin has been used as a CYP2C8 inhibitor in P450 reaction phenotyping studies (Rahman et al., 1994; Komatsu et al., 2000; Marill et al., 2002; Projean et al., 2003). However, there have also been some reports describing the ability of quercetin to inhibit human P450 enzymes besides CYP2C8 (Obach, 2000; Zou et al., 2002). The inhibition of a panel of P450-specific activities by montelukast at 0.2 μM and quercetin at 30 μM, which are the concentrations of these two inhibitors needed to obtain approximately 90% inhibition of CYP2C8, was examined in pooled human liver microsomes (Fig. 4). Montelukast at 0.2 μM inhibited none of the other human P450-specific activities above 10%, except for CYP2C8. However quercetin inhibited most of the other activities to at least some extent (∼40%), with some activities (CYP1A2, CYP3A) inhibited potently. For example, CYP3A-mediated felodipine dehydrogenase was very profoundly inhibited by 30 μM quercetin, to an extent greater than that observed for CYP2C8 (98% inhibition). These results suggest that montelukast would be a tool superior to quercetin for use in a panel of human P450-specific inhibitors in reaction phenotyping studies.

Inhibitory effects of montelukast and quercetin at their CYP2C8 IC90 concentrations on P450 activities in pooled human liver microsomes. CYP3A activity was evaluated using three substrates: felodipine (F), midazolam (M), and testosterone (T).
Discussion
The data presented in this report support the claim that montelukast is a potent and selective inhibitor of human CYP2C8. These findings have ramifications in two areas. First, it appears that montelukast could be useful as a tool in in vitro drug metabolism experiments aimed toward determining whether CYP2C8 is involved in the metabolism of new drugs. Second, the high inhibitory potency of montelukast for CYP2C8 suggests a potential, in the clinic, for this drug to cause drug interactions with those agents known to be metabolized by CYP2C8 as an important clearance pathway.
Montelukast is a potent inhibitor of human CYP2C8 as demonstrated in both human liver microsomes (pooled and individual) and recombinant expressed enzyme. It was also demonstrated using three different CYP2C8-catalyzed reactions: amodiaquine N-deethylase, rosiglitazone N-demethylase, and paclitaxel 6α-hydroxylase. The mechanism of inhibition was competitive in all cases, and the inhibitory potency ranged from 0.0092 to 0.15 μM. (This range is hypothesized to be due to the different microsomal concentrations used in the assays.) Montelukast possesses a carboxylic acid substituent and, compared with most other drugs, has a large molecular weight (mol. wt. = 586). As such, it is consistent with the substrate binding pocket described from recent X-ray crystallographic studies of CYP2C8 (Schoch et al., 2004). Determination of the structural entities of montelukast that are responsible for the high potency for CYP2C8, and structure-activity relationships, will be the focus of further research.
The data presented support the claim that montelukast could serve as a CYP2C8-selective chemical inhibitor in P450 reaction phenotyping experiments. The inhibition of other human drug-metabolizing P450 enzymes was significantly less potent, such that concentrations of montelukast could be used to inhibit CYP2C8 by 90% (Table 3) without causing significant inhibition of other human P450 enzymes in human liver microsomes. The use of isoform-selective P450 chemical inhibitors represents a powerful technique by which the contributions of individual P450 enzymes to the metabolic clearance of drugs can be assessed. However, to date, a high quality selective chemical inhibitor of CYP2C8 was lacking. Others have used quercetin as a CYP2C8-selective inhibitor (Rahman et al., 1994; Komatsu et al., 2000; Marill et al., 2002; Projean et al., 2003); however, quercetin can also inhibit other human P450 enzymes, making its application suboptimal (Fig. 4). It should be noted that in the use of montelukast as a P450 reaction phenotyping tool, careful attention must be paid to the microsomal protein concentration used to study the reaction. The potency of montelukast for CYP2C8 decreases with increasing microsomal protein concentration. We hypothesize that this is due to nonspecific microsomal binding, as has been demonstrated for other lipophilic drugs (Obach, 1999; Austin et al., 2002; Margolis and Obach, 2003); however, technical limitations prohibited the measurement of microsomal binding for montelukast. Such nonspecific microsomal binding would decrease the potency of montelukast for all microsomal enzymes (as was shown for CYP2C9, the next most potently inhibited enzyme; Fig. 3), so that the selectivity would remain intact, irrespective of the microsomal concentrations used in these experiments. We have listed concentrations of montelukast that would yield 90% inhibition of CYP2C8 at various microsomal protein concentrations for other investigators to use as a guide when using this compound in P450 reaction phenotyping studies (Table 3).
In a previous report, the inhibition of human P450 enzymes by montelukast (but not including CYP2C8) was described (Chiba et al., 1997). These investigators described the inhibition by montelukast as weak, with IC50 values for CYP1A2, 2A6, 2C19, and 2D6 above 500 μM. This is in marked contrast to the data in Table 2, in which IC50 values of 1.2 to 180 μM were measured for the other human P450 enzymes (besides CYP2C8). Based on our findings of the dependence of montelukast's inhibitory potency on microsomal protein concentration, it is likely that the previous investigation overestimated IC50 values due to nonspecific binding, since the microsomal protein concentration used in the earlier investigation was 1.0 mg/ml (Chiba et al., 1997), whereas the protein concentrations used in the present investigation were lower.
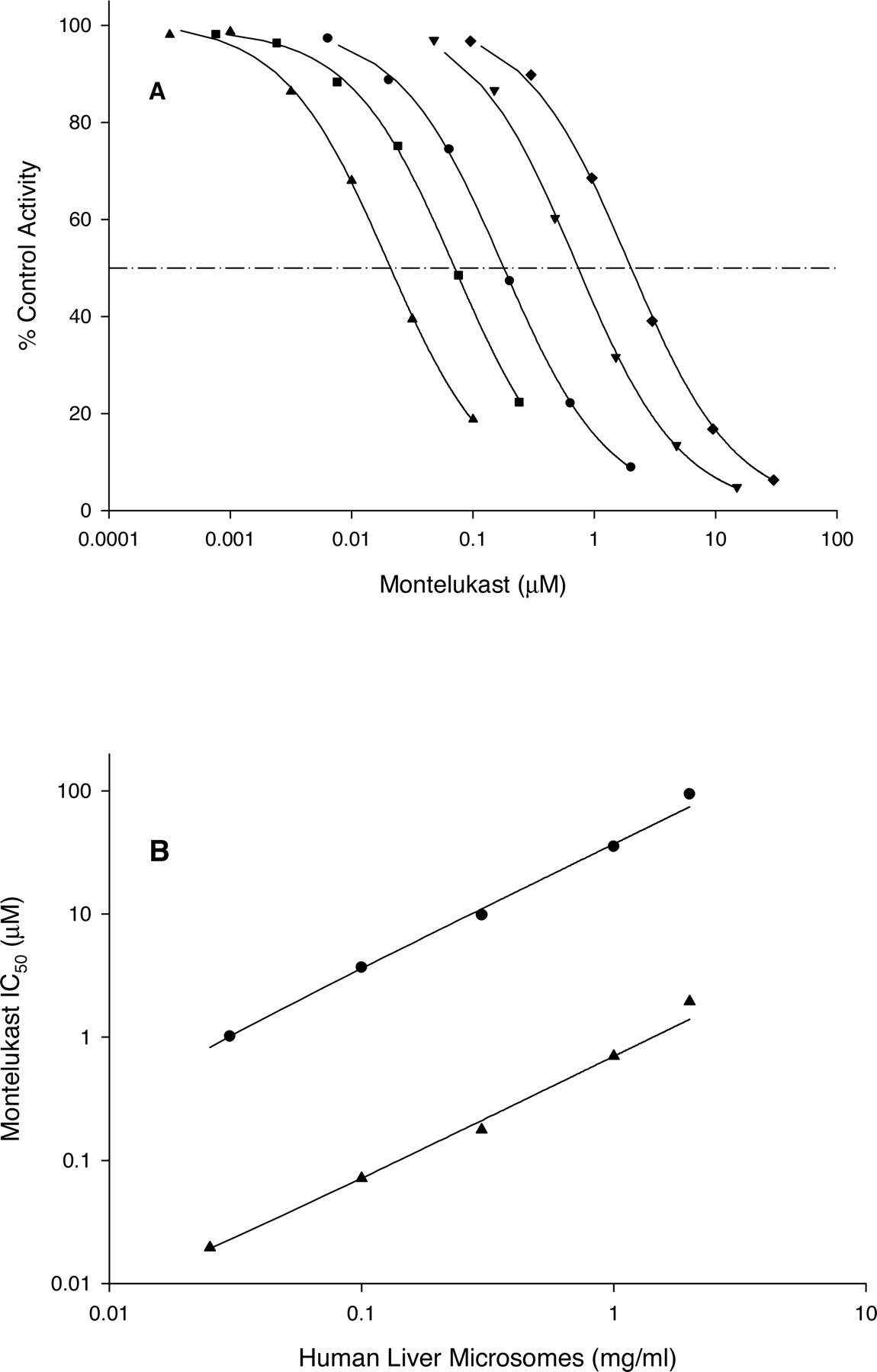
Since the potency of montelukast as an inhibitor of CYP2C8 is high (0.0092–0.15 μM), the question arises whether it could cause inhibition of this enzyme in vivo and, hence, pharmacokinetic drug interactions with those agents primarily cleared via CYP2C8-mediated metabolism. Unlike many of the other major drug-metabolizing human P450 enzymes, such as CYP3A4 or CYP2D6, CYP2C8 has not received much attention as an enzyme involved in drug clearance or the target of drug-drug interactions. However, some studies have recently been reported. Gemfibrozil has been shown to cause marked increases in the exposure to repaglinide (Niemi et al., 2003b), cerivastatin (Backman et al., 2002), and rosiglitazone (Niemi et al., 2003a) via inhibition of CYP2C8 (either by the parent drug gemfibrozil or its glucuronide metabolite, or both; Shitara et al., 2004). Attempts to predict the magnitude of drug interactions from in vitro inhibition data have met with mixed success (Davit et al., 1999; Yao and Levy, 2002; Ito et al., 2004). The greatest barrier appears to be making a reliable estimation of the relevant in vivo concentration of inhibitor to use ([I]in vivo) in the expression: 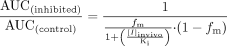 in which fm is the fraction of the affected drug that ordinarily goes through the inhibited enzyme. The most appropriate inhibitor concentration to use in such estimates could be systemic concentrations, estimations of portal vein concentrations incident to the liver during absorption (Kanamitsu et al., 2000), estimations of hepatic concentrations (Von Moltke et al., 1998), and either total or unbound concentration. Utilizing these in vitro inhibition data in predicting drug-drug interactions that could be caused by montelukast on a CYP2C8-cleared compound yields the predictions listed in Table 4. Estimates of the magnitude of drug-drug interactions for a CYP2C8-cleared drug range from 2 to 119, largely due to whether the unbound or total montelukast concentrations are the ones most relevant for enzyme inhibition in vivo. The uncertainty inherent in predicting the magnitude of in vivo drug interactions from in vitro inhibitory potency data, and the numerous variables, known and unknown, possessing their own variability, that factor into such a prediction, require that in vitro data be confirmed or refuted with an actual drug interaction study in humans. Nevertheless, based on these in vitro data, the potential effect of montelukast on the pharmacokinetics of a CYP2C8-cleared drug such as repaglinide warrants investigation.
in which fm is the fraction of the affected drug that ordinarily goes through the inhibited enzyme. The most appropriate inhibitor concentration to use in such estimates could be systemic concentrations, estimations of portal vein concentrations incident to the liver during absorption (Kanamitsu et al., 2000), estimations of hepatic concentrations (Von Moltke et al., 1998), and either total or unbound concentration. Utilizing these in vitro inhibition data in predicting drug-drug interactions that could be caused by montelukast on a CYP2C8-cleared compound yields the predictions listed in Table 4. Estimates of the magnitude of drug-drug interactions for a CYP2C8-cleared drug range from 2 to 119, largely due to whether the unbound or total montelukast concentrations are the ones most relevant for enzyme inhibition in vivo. The uncertainty inherent in predicting the magnitude of in vivo drug interactions from in vitro inhibitory potency data, and the numerous variables, known and unknown, possessing their own variability, that factor into such a prediction, require that in vitro data be confirmed or refuted with an actual drug interaction study in humans. Nevertheless, based on these in vitro data, the potential effect of montelukast on the pharmacokinetics of a CYP2C8-cleared drug such as repaglinide warrants investigation.
Estimation of possible drug interactions that could be caused by montelukast on CYP2C8-cleared drugs
Values used for montelukast include: D = 10 mg; Fa = 0.62; ka = 0.008/min; fu = 0.01; Cmax = 603 ng/ml = 1.0 μM (Zhao et al., 1997); Ki = 0.0092 μM. The fraction of the affected drug metabolized by CYP2C8 is assumed to be unity in the expression. The estimate for Fa = 0.62 was made from the observation that clearance after intravenous administration was reported at 0.7 ml/min/kg (Cheng et al., 1996); hence, hepatic extraction is less than 5%, and oral bioavailability is 62%. The estimate for ka was made from the expression Tmax = (ln(ka/kel))/(ka–kel) in which kel was calculated from the reported t1/2 and Tmax values of 4.7 h and 3.5 h, respectively (Cheng et al., 1996).
In conclusion, these data have shown that montelukast is a potent and selective reversible competitive inhibitor of CYP2C8. Inhibition was demonstrated with three different CYP2C8 marker activities and in different in vitro systems (i.e., pooled and individual human liver microsomes and recombinant CYP2C8). The data suggest that montelukast could be useful as a selective CYP2C8 inhibitor in P450 reaction phenotyping studies, and montelukast appears to be vastly superior to quercetin, another CYP2C8 inhibitor, as a tool for this objective. Finally, the high potency of montelukast for CYP2C8 inhibition warrants its investigation as an agent that could cause pharmacokinetic drug-drug interactions in humans.
Acknowledgments
We acknowledge Dr. Caroline Lee (Pfizer, La Jolla, CA) for supporting the testing of montelukast as an inhibitor of paclitaxel 6α-hydroxylase.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002766.
-
ABBREVIATIONS: P450, cytochrome P450; HPLC, high-performance liquid chromatography; rh, recombinant human.
- Received October 26, 2004.
- Accepted December 16, 2004.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}