


Abstract
Caco-2 cell lysate, and intestinal and liver microsomes derived from female humans and rats were used to compare and contrast the metabolism and disposition of raloxifene. In Caco-2 cell lysate, raloxifene 6-β-glucuronide (M1) was the main metabolite, although raloxifene 4′-β-glucuronide (M2) was formed in comparable abundance (58% versus 42%). In rat liver and intestinal microsomes, M1 represented about 76 to 86% of glucuronidated metabolites. In contrast, raloxifene 4′-β-glucuronide (M2) was the predominant metabolite in expressed UGT1A10 (96%) and human intestinal (92%) microsomes. Intrinsic clearance for M2 (CLint, M2) in human intestinal microsomes was 33- to 72-fold higher than in rat microsomes, whereas intrinsic clearance for M1 (CLint, M1) was 3- to 4-fold lower. Taken together, total intrinsic clearance (CLint, M1 + CLint, M2) in human intestinal microsomes was 3- to 6-fold higher than that in rat intestinal microsomes, but was similar in liver microsomes. In addition, intrinsic clearance in small intestinal microsomes was 2- to ∼5-fold higher than that in hepatic microsomes, regardless of species. To account for the difference in species- and disposition model-dependent intestinal metabolism, we probed the presence of various UGT1A isoforms in Caco-2 cells using real-time reverse transcriptase-polymerase chain reaction and, as expected, detected no UGT1A10. In conclusion, the lack of UGT1A10 may explain why Caco-2 cell and rat intestinal microsomes metabolized raloxifene differently from human intestinal microsomes. The presence of human intestinal UGT1A10 and the higher overall intrinsic clearance value in the human intestine as the result of UGT1A10 expression could explain why raloxifene has much lower bioavailability in humans (2%) than in rats (39%).
Raloxifene, a member of the selective estrogen receptor modulators, is a drug for the treatment and prevention of osteoporosis in postmenopausal women (Heringa, 2003). It is also undergoing clinical trials for breast cancer prevention. Sixty-two to 70% of postmenopausal women are potential candidates for selective estrogen receptor modulators, and the percentage of actual users is expected to rise inasmuch as a hormone replacement therapy trial recently yielded disappointing negative results (Rossouw et al., 2002; Lacey et al., 2002).
The bioavailability of raloxifene was reported to be very low (2%) in humans, even though it has oral efficacy in osteoporosis. This may represent a significant challenge to its possible future use as a chemopreventive agent. It is generally believed that absorption is not the main reason for poor bioavailability since approximately 60% of the dose was absorbed after oral administration (Eli Lilly 1998 Raloxifene package insert; Hochner-Celnikier, 1999; Snyder et al., 2000). However, we found recently that raloxifene was effluxed by various transporters such as multidrug resistance-related protein and organic anion transporter, which could contribute to its poor bioavailability (Jeong et al., 2004). More importantly, we also found that raloxifene is extensively conjugated (about 90% at concentrations less than 10 μM) during transport across the Caco-2 cell monolayer (Jeong et al., 2004), consistent with previous observations that raloxifene undergoes extensive phase II metabolism in the gut (Kemp et al., 2002). However, the main metabolites in Caco-2 cells were sulfates, whereas the primary metabolite in humans is raloxifene 4′-β-glucuronide or M2. In humans, plasma concentration of 4′-β-glucuronide is 8-fold higher than that of 6-β-glucuronide or M1 (Jones, 1997).
In contrast to its poor bioavailability in humans, raloxifene has fairly good bioavailability in rats (39%) (Lindstrom et al., 1984). This is somewhat unexpected since the major metabolic pathway in rats is also glucuronidation. Upon further examination, the major metabolite in rats was found to be M1 (Lindstrom et al., 1984), similar to what we found for glucuronidation of raloxifene in Caco-2 cells (Jeong et al., 2004). Because many of the pharmacodynamic studies of raloxifene are conducted in rats (Merchenthaler et al., 1998; Kubatka et al., 2001; Cao et al., 2002; Ozgonul et al., 2003; Zheng et al., 2004), it would be important to resolve why there were these important species differences in raloxifene disposition. Therefore, the main purpose of the present study is to determine how species and disposition model choice affect the intestinal and hepatic disposition of raloxifene. Our results indicate for the first time that lack of UGT1A10 expression is the main reason why 4′-β-glucuronide is not the main metabolite in rats and Caco-2 cells, and may explain the species-dependent disposition of raloxifene.
As a part of characterization work on raloxifene metabolism, we also measured the drug interaction potentials between raloxifene and flavonoids. Flavonoids, especially isoflavones, are taken as dietary supplements by women who experience symptoms associated with menopause, which can be worsened by raloxifene administration. Therefore, a combination of flavonoids and raloxifene is expected to be taken by postmenopausal women (Cotter and Cashman, 2003; Goodman-Gruen and Kritz-Silverstein, 2003; Crisafulli et al., 2004a,b; Kreijkamp-Kaspers et al., 2004). A concern arising from this possible combination is that metabolism of raloxifene is similar to that of flavonoids. Both are extensively metabolized in gut via phase II conjugations (Crespy et al., 1999; Walle et al., 1999; Andlauer et al., 2000; Liu and Hu, 2002; Chen et al., 2003; Jia et al., 2004). Therefore, we determined the potential for interactions between selected flavonoids (e.g., genistein and apigenin) and raloxifene using the microsomes. Our results indicated that there is a moderate chance of beneficial interactions in the gut wall but not in liver, which could lead to a higher raloxifene bioavailability in vivo.
Materials and Methods
Materials. Raloxifene was extracted from Evista tablets (Eli Lilly & Co., Indianapolis, IN) using 100% ethanol, and concentration was then verified by using authentic raloxifene hydrochloride purchased from the National Cancer Institute Chemical Standard Repository managed by Midwest Research Institute (Kansas City, MO). Uridine diphosphoglucuronic acid, alamethicin, disaccharic-1,4-lactone monohydrate, magnesium chloride, Tris, and Hanks' balanced salt solution were products of Sigma-Aldrich (St. Louis, MO). Female human liver microsomes and human UGT1A10 Supersomes were purchased from BD Gentest (Woburn, MA), and female human jejunal and ileal microsomes were obtained from Tissue Transformation Technologies (Edison, NJ). Cloned Caco-2 cells (TC7) were a kind gift from Dr. Moniqué Rousset (Institute National de la Santé et de la Recherche zU178, Villejuit, France). All other materials were analytical grade or better and used as received.
Cell Culture. Cell culture conditions for growing Caco-2 cells have been described previously (Liu and Hu, 2002; Hu et al., 1994a,b; Chen et al., 2003; Jeong et al., 2004). The seeding density was 100,000 cells/cm2 (4.2 cm2 per monolayer), and Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum was used as the growth medium. Quality control criteria were the same as described previously (Hu et al., 1994a,b). Cell monolayers from 19 to 22 days past seeding were used for the experiments.
Preparation of Caco-2 Cell Lysate. After six mature (19–22 days post-seeding) Caco-2 cell monolayers were washed twice with 3 ml of 37°C Hanks' balanced salt solution (pH 7.4), they were cut out together with the porous polycarbonate membranes, immersed in 6 ml of 50 mM potassium phosphate buffer (pH 7.4), and sonicated in an ice bath (4°C) for 30 min as described previously (Jeong et al., 2004). Afterward, the cell lysate was centrifuged at 1000 rpm for 5 min to remove the polycarbonate membrane. The protein concentrations of the cell lysate were then determined using a commercial protein assay kit (Bio-Rad, Hercules, CA).
Real-Time Quantitative RT-PCR Analysis of UGT1As. Standard reagents and methods were used to perform quantification of UGT1A mRNA levels using reverse-transcriptase and SYBR Green dye. The following is a brief description of the procedures.
RNA Isolation. Small amounts of total RNA were extracted from Caco-2 cells using a commercial RNA isolation kit (Ambion, Austin, TX), whereas large amounts of total RNA from 5 × 108 cells were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). RNA concentration and purity were determined by OD260 and OD280 readings, and RNA integrity was monitored by agarose gel electrophoresis. Prior to RT-PCR and real-time PCR, contaminating DNA in RNA samples and excess DNase were removed using DNA-free DNase Treatment and Removal Reagents (Ambion). To monitor for possible DNA contamination, PCR amplification of β-actin cDNA was performed using a primer pair (5′-ggcggcaccaccatgtaccct-3′ and 5′-cgatccacacggagtacttgc-3′) that spans intron 5 of that gene. Genomic DNA template leads to a 312-bp product and the cDNA template generates a 202-bp product. In the absence of genomic DNA in the RNA samples, the 202-bp PCR product is only expected in the RT-PCR sample. Extracted RNA samples free from DNA were stored at -80°C until real-time PCR analysis.
Reverse Transcription. Synthesis of first-strand cDNA was carried out using SuperScript III RNase H- Reverse Transcriptase (Invitrogen).
Oligonucleotide Primers for Real-Time RT-PCR. Primers for the amplification of the human UGT RNA transcripts as well as for β-actin sequences were generated using standard molecular biology software (Table 1). The UGT primers are specific either for regions in each of the unique exon 1 sequences or for sequences that are common to several (e.g., UGT1A7–UGT1A10) or all of the UGT1A RNAs (Table 1).
PCR primers for house-keeping gene and human UGT1A family Some primers were designed based on the work of Tukey and his coworkers (e.g., Strassburg et al, 1999).
Real-Time PCR. Real-time quantitative PCR was performed using the GeneAmp 5700 Sequence Detection System (Applied Biosystems, Foster City, CA) and carried out using SYBR Green as the quantification tool according to the manufacturer's instruction. The final concentration of each forward or reverse oligonucleotide primer in the PCR mixture was 200 nM. PCR assays for constructing standard curves were performed in triplicate, whereas the remaining PCR assays were performed in duplicate, both in three separate replicate runs. The concentrations of the primers and templates were optimized, and the final PCR conditions were as follows: 95°C for 10 min, followed by 40 cycles consisting of 95°C for 15 s, and 60°C for 1 min. Data acquisition and analysis were handled by the built-in software (Applied Biosystems).
Data Analysis in RT-PCR and Real-Time PCR Methods.Semiquantitative RT-PCR for UGT Detection. To detect the different isoforms of UGT, PCR was conducted using UGT-specific sense and antisense primers (β-actin primers were added as an internal reference). PCRs for experiments with cultured cells were performed with 0.5 U of High Fidelity TaqDNA polymerase (Invitrogen) on 4 to 10 μl of cDNA under the following conditions: 94°C for 5 min, followed by 30 cycles consisting of a 30-s denaturing step at 94°C, a 45-s annealing step at 58°C, and a 45-s elongation step at 72°C in a thermal cycler (MJ Research, Watertown, MA). The products were separated in a 1.5% agarose gel that contained ethidium bromide. The bands were visualized under UV light and photographed with a computer-assisted camera. The specificity of all primer pairs was confirmed through sequencing or restriction analysis of the PCR products.
Real-Time RT-PCR. Briefly, a threshold was assigned to the log phase of product accumulation. The point at which the threshold crosses the amplification curve is defined as a cycle threshold value, termed CT. With increasing target quantity in the PCRs, the CT value decreases linearly, and thus, CT values can be used as relatively quantitative measurements of the input target amount. For the present determination, the threshold value was set at 0.4 (for UGT in Caco-2 cells), which was significantly above the background noise, and the numbers of cycles required to reach this CT value were determined.
Animals. Female Sprague-Dawley rats (70∼110 days old, body weight 240∼260 g; Simonsen Laboratories, Gilroy, CA) were fed with Teklad F6 rodent diet (Harlan Teklad, Madison, WI). The rats were fasted overnight before the day of the experiment.
Preparation of Rat Intestinal and Liver Microsomes. The protocols for preparing rat intestinal and liver microsomes were similar to those described previously (Chen et al., 2003).
UGT Metabolism Studies. Glucuronidation of raloxifene by Caco-2 cell lysate as well as intestinal and liver microsomes was measured using procedures described previously (Chen et al., 2003; Jeong et al., 2004). The microsome fraction or Caco-2 cell lysate (final concentration ∼0.05 mg protein/ml in reaction mixture) was mixed with magnesium chloride (0.88 mM), saccharolactone (4.4 mM) and alamethicin (0.022 mg/ml). Raloxifene (final concentration 0∼34.7 μM) or raloxifene (4.34 μM) plus apigenin or genistein (final concentration 0∼100 μM) in 50 mM potassium phosphate buffer (pH 7.4) was then added. Uridine diphosphoglucuronic acid (3.5 mM) was added last to the reaction mixture (total volume 200 μl) to initiate reaction. The mixture was incubated in a 37°C shaking (200 rpm) water bath for 30∼60 min for intestinal and liver microsomes and 4 h for Caco-2 cell lysate. The final organic solvent concentration was 1% (1% ethanol for raloxifene or 0.95% ethanol plus 0.01% dimethyl sulfoxide for raloxifene with flavonoids). The reaction was stopped by the addition of a 40-μl solution of 6% glacial acetic acid in acetonitrile containing 100 μM testosterone as the internal standard.
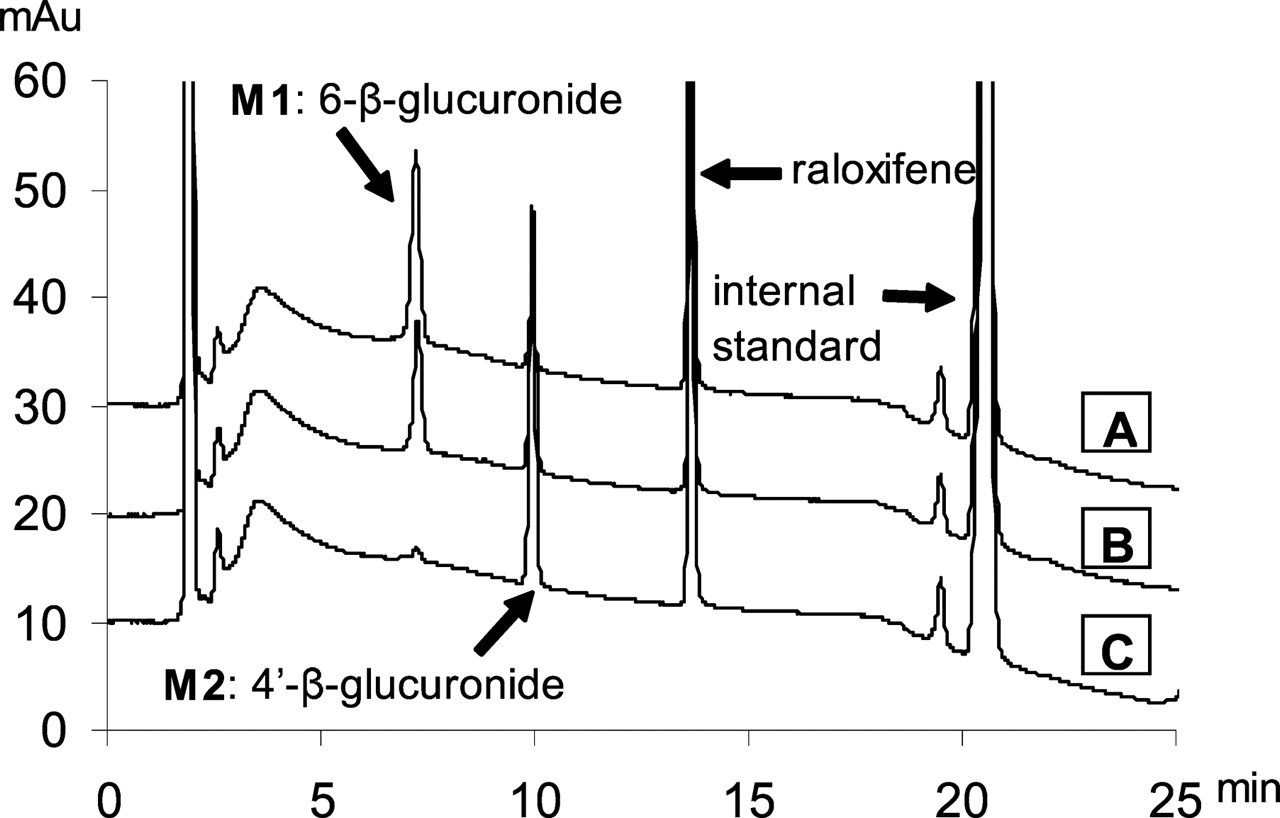
HPLC Analysis of Raloxifene and Its Conjugates. The HPLC conditions were the same as those described before (Jeong et al., 2004). The retention times for raloxifene, 6-β-glucuronide, 4′-β-glucuronide, and internal standard (testosterone) were 13.6, 7.2, 9.8, and 21.4 min, respectively (Fig. 1).
Data Analysis in Microsome Model. Metabolic rates (V) were plotted against raloxifene concentration (C) in the final reaction mixture. Apparent Km, Vmax, and intrinsic clearance (CLint) values were obtained via nonlinear regression analysis of the Michaelis-Menten equation (or eq. 1) and eq. 2. 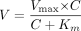
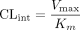

Representative HPLC profiles of raloxifene and its metabolites. A, B, C show the peaks of major metabolites formed by female rat and human liver microsomes and UGT1A10, respectively. Two metabolites, M1 and M2, were identified as 6-β-glucuronide and 4′-β-glucuronide, since UGT1A10 was reported to produce only 4′-β-glucuronide (Kemp et al., 2002). The retention times for raloxifene, 6-β-glucuronide, 4′-β-glucuronide, and internal standard (testosterone) are 13.6, 7.2, 9.8, and 21.4 min, respectively.
For rat intestinal microsomes, which showed substrate inhibition on Eadie-Hofstee plots, the kinetic parameters were calculated according to eq. 3, using the ADAPT II program (D'Argenio and Schumitzky, 1997). 
In eq. 3, Ksi is the constant describing the substrate inhibition (Houston and Kenworthy, 2000), whereas other terms are the same as described previously. Equation 3 was used by Houston and Kenworthy (2000) to describe substrate inhibition in a two-binding site model.
For the inhibition study with flavonoids, percentage of control of the metabolic rates (V) was plotted against flavonoid concentration in the final reaction mixture. Apparent IC50 values, or the inhibitor concentration at which the metabolic rate was decreased 50% when compared with the control, were calculated by fitting the data to a classical IC50 equation using nonlinear regression (a SigmaPlot function).
Statistical Analysis. Student's t test (Microsoft Excel) was used to analyze the data. The prior level of significance was set at 5%, or p < 0.05.
Results
Metabolism of Raloxifene in Caco-2 Cell Lysate. Caco-2 cell lysates were incubated with raloxifene (4.34 μM) for 4 h in glucuronidation reaction, and the results indicated that there were two main glucuronide peaks (M1 and M2), as previously reported (Jeong et al., 2004). Sulfates were not observed using Caco-2 cell lysate since coenzymes for sulfotransferases were not added to the incubation mixture. Between the two glucuronides [6-β-glucuronide and 4′-β-glucuronide (Jones, 1997)], M2 was identified as 4′-β-glucuronide, because M2 was the main metabolite formed by UGT1A10 (Fig. 1 and later). In comparing the rates of metabolism, we found that raloxifene 6-β-glucuronide (M1) was formed at a rate of 0.626 ± 0.075 pmol/min/mg protein, 40% faster than the formation rate of raloxifene 4′-β-glucuronide (M2). The actual percentages of metabolites found at the end of the 4-h experiment were 58% (M1) versus 42% (M2).
Relative Expression of UGT Isoforms in the Caco-2 Cells. To explain the difference seen in Caco-2 cell lysate and what is expected based on reported rapid metabolism in human intestinal microsomes (Kemp et al., 2002), we determined the relative expression levels of UGT isoforms in Caco-2 cell lysate using a real-time RT-PCR method. The results indicated that UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, and UGT1A8, and UGT1A9 isoforms were expressed in Caco-2 cells, whereas UGT1A7 and UGT1A10 were not (Fig. 2). Among the UGT1A isoform mRNAs expressed by Caco-2 cells, UGT1A6 was the most abundant, followed by UGT1A3, UGT1A1, UGT1A5, UGT1A4, and UGT1A9. Based on CT values, the relative expression levels of these isoforms are UGT1A6 (26): UGT1A3 (1.5):UGT1A1 (1.0):UGT1A5 (0.72):UGT1A4 (0.54): UGT1A9 (0.24):UGT1A8 (0.01).
Metabolism of Raloxifene in Human Microsomes. Raloxifene glucuronidation was determined using pooled female human intestinal (jejunal and ileal) and liver microsomes along with expressed human UGT1A10 microsomes to further define the role of UGT1A10 in organ-specific metabolism of raloxifene.

RT-PCR analysis of UGT isoform-specific RNAs in the Caco-2 cells. The bands (from left to right) correspond to house-keeping gene (β-actin) (lane 1), UGT1A (lane 2), UGT1A1 (lane 3), UGT1A3 (lane 4), UGT1A4 (lane 5), UGT1A5 (lane 6), UGT1A6 (lane 7), UGT1A7 (lane 8), UGT1A8 (lane 9), UGT1A9 (lane 10), UGT1A10 (lane 11), UGT1A3-5 (lane 12), UGT1A7-10 (lane 13), positive control (lane 14), and DNA ladder (lane 15).
Human jejunal and ileal microsomes were found to mainly metabolize raloxifene to M2, different from raloxifene glucuronidation in Caco-2 cell lysate. M2 is also the main metabolite in expressed human UGT1A10 (96%) (Fig. 3). Previously, Kemp et al. (2002) showed that UGT1A10 displayed complete selectivity for M2 formation. In contrast, substantial amounts of M1 were found in liver microsomes (41% M1 versus 59% M2; Fig. 3).
In intestinal microsomes, M2 formation displayed significantly larger Vmax value (9.1∼11-fold) and higher intrinsic clearance (14∼16-fold) values than did M1 formation (Table 2; Fig. 4). In liver microsomes, M2 formation was associated with faster intrinsic clearance (CLint, M2, 2.5-fold) but smaller Vmax values (34% less) when compared with M1 formation (Table 2; Fig. 4).
Metabolic parameters of raloxifene glucuronidation by liver and intestinal microsomes in human and rat Raloxifene was added to the microsomes with cofactors as described under Materials and Methods. Apparent Km, Vmax, and CLint values were obtained by nonlinear regression of the data points in Fig. 4 and Fig. 6.
When comparing M1 formation by microsomes prepared from different organs/tissues, we found that liver microsomes displayed the highest apparent Km and Vmax values. As consequence, it has the largest apparent CLint, M1 (1.2∼1.7-fold) values (Table 2; Fig. 4A). For M2 (the main metabolite) formation, intestinal microsome displayed apparent Km values similar to those of liver, but had significantly higher Vmax values (5.0∼5.2-fold) and apparent CLint, M2 (3.9∼4.9-fold) values when compared with liver (Table 2; Fig. 4B).
Metabolism of Raloxifene in Rat Microsomes. Rates of glucuronidation were also determined using female rat intestinal microsomes prepared from four different regions (duodenum, jejunum, ileum, and colon) and liver microsomes to determine the possible regional difference in the metabolism of raloxifene and to contrast the results with those obtained from humans.
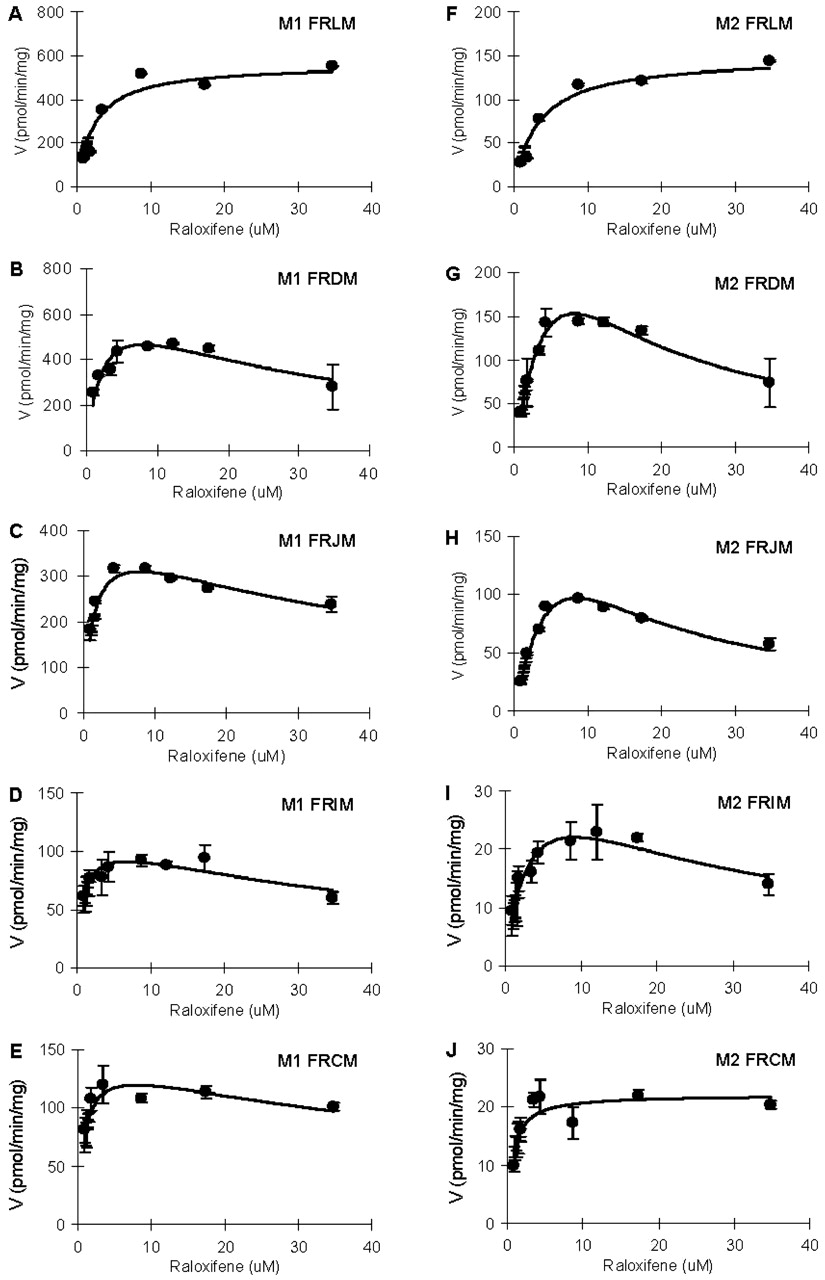
In rat microsomes, the main metabolite was M1 (82% in liver; 76∼86% in intestine) (Fig. 2). The apparent Km values for M1 formation were lower than those for M2 formation in intestinal microsomes other than colonic microsomes, and the difference ranged from approximately 4- to 10-fold. Km values in colonic microsomes were similar. In all intestinal microsomes, the apparent Vmax values for M1 formation were higher (14% to ∼700%) than those for M2 formation. In liver microsomes, M1 formation displayed apparent Km values that were 44% lower and apparent Vmax values that were 3.8-fold higher than those for M2 formation (Table 2).

Rate of raloxifene glucuronidation (pmol/min/mg) by human (A) and rat (B) microsomes. Raloxifene (8.7 μM) was added to the microsomes with cofactors as described under Materials and Methods. Incubation was carried out at 37°C for 2 h. Solid and shaded columns represent the rate of glucuronidation for M1 and M2, respectively. FHLM, FHJM, and FHIM represent pooled female human liver, jejunal, and ileal microsomes, respectively (A). FRLM, FRDM, FRJM, FRIM, and FRCM represent pooled female rat liver, duodenal, jejunal, ileal, and colonic microsomes, respectively (B). Each column represents the mean of three determinations, and the error bars are standard deviations of the mean.
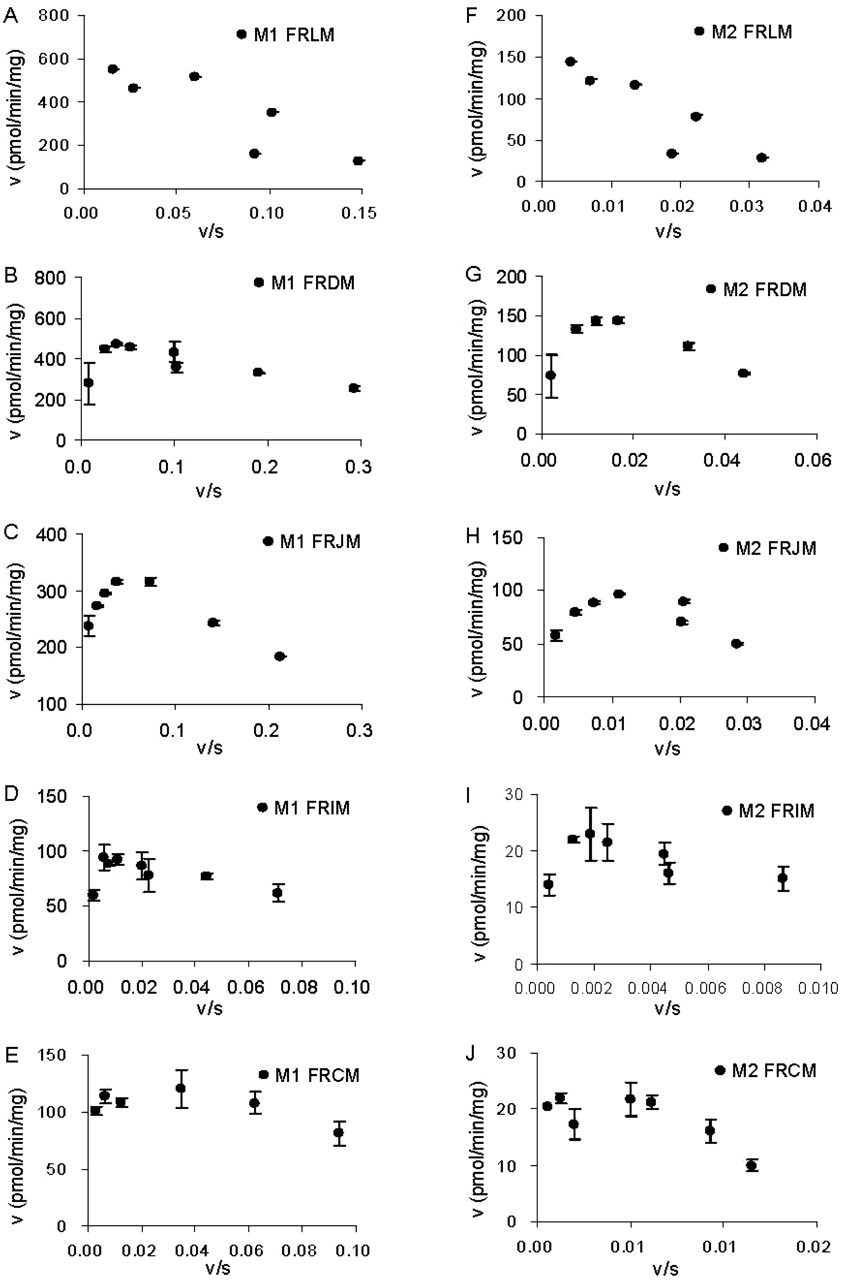
In rat intestinal microsomes, raloxifene was glucuronidated in all four sites with highest apparent CLint in duodenum and lowest apparent CLint in ileum for both M1 and M2 (Table 2). Eadie-Hofstee plots showed substrate inhibition at high substrate concentrations in all sites (Fig. 5), and therefore, eq. 3 was used to obtain kinetic parameters (Table 2). For M1 formation, duodenal and jejunal microsomes displayed lower apparent Km values (0.5∼0.8-fold) but higher apparent CLint, M1 value (1.5∼1.6-fold) than did liver (Table 2; Fig. 6). For M2 formation, duodenal and jejunal microsomes displayed higher apparent Km values (2.7-fold) than did liver but similar apparent CLint, M2 values (Table 2; and Fig. 6). On the other hand, ileal and colonal microsomes have lower apparent Km, CLint, M1, and CLint, M2 values (Table 2; Fig. 6).
When we compared raloxifene metabolism in human and rat microsomes, we found that the apparent CLint, M1 in humans was only 24∼41% of rat, but apparent CLint, M2 was substantially higher in humans (5.6-fold in liver, 33-fold in jejunum, 72-fold in ileum) than in rats (Table 2). Taken together, the total apparent CLint (sum of CLint, M1 and CLint, M2) of raloxifene in human intestinal microsomes was higher than the corresponding value in rat intestinal microsomes (3.3-fold for jejunum and 6.2-fold for ileum). On the other hand, the total apparent CLint in human liver microsomes was almost the same as that of the rat liver.
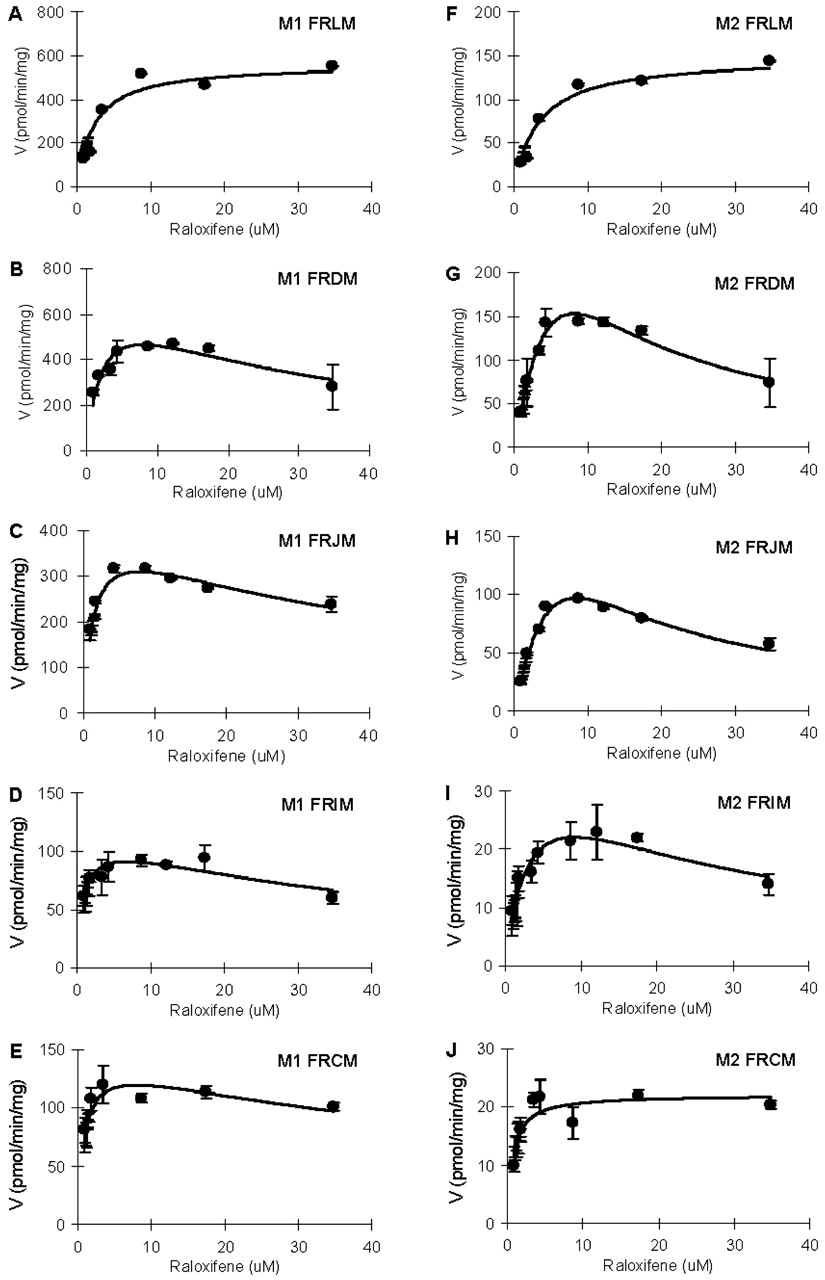
Effects of Flavonoids on Raloxifene Metabolism. The results indicated that metabolism of raloxifene in the rat microsomes, Caco-2 cell lysate, and human microsomes can be significantly and substantially decreased by the presence of genistein and apigenin (Fig. 7). Furthermore, the apparent IC50 values were in the range of 1 to 10 μM for Caco-2 cell lysate and rat microsomes (Table 3). In human intestinal microsomes, the apparent IC50 values were much higher (15–50 μM) (Table 3).
Inhibitory effect of genistein and apigenin on glucuronidation of raloxifene by human and rat jejunal microsomes and Caco-2 cell lysate Raloxifene (4.34 μM) and inhibitor (0∼100 μM genistein or apigenin) were added to the microsomes with cofactors as described under Materials and Methods. Apparent 50% inhibitory concentration (IC50) values were obtained by nonlinear regression of the data presented in Fig. 7 using SigmaPlot.

Effects of concentration on raloxifene glucuronidation by human liver and intestinal microsomes for M1 (A) and M2 (B). Raloxifene (0.87∼34.7 μM) was added to the microsomes with cofactors as described under Materials and Methods. Incubation was carried out at 37°C for 30 to 60 min, depending on the concentration of raloxifene. FHLM, FHJM, and FHIM represent pooled female human liver, jejunal, and ileal microsomes, respectively. Each data point represents the mean of three determinations, and the error bars are standard deviations of the mean. Each curve was obtained by nonlinear regression of the data points using SigmaPlot.
Discussion
Disposition of raloxifene is likely to have profound effects on its anticipated anticancer indication since consistent exposure levels are needed to sustain chemopreventive effects. Preclinical studies exploring raloxifene's anticancer effects have used rodents (e.g., rats) as their primary model (Merchenthaler et al., 1998; Kubatka et al., 2001; Cao et al., 2002; Ozgonul et al., 2003; Zheng et al., 2004). Because of the substantial difference in the metabolism of raloxifene (e.g., 39% bioavailability in rats and 2% in humans), the results derived from rodents must be weighed carefully before extrapolating to humans. This is because, in addition to typical species differences in pharmacological and pharmacodynamic responses, there are likely to be substantial differences in their pharmacokinetic responses as the result of disposition differences.
The first difference in disposition is the expression level of UGT1A10 in different species. UGT1A10 is the isoform responsible for M2 formation and rapid raloxifene clearance. Our result clearly demonstrated that UGT1A10 is a major isoform responsible for the selective formation of raloxifene 4′-β-glucuronide (M2) over M1, which is consistent with an earlier observation using human microsomes (Kemp et al., 2002). Its absence from the rats could largely, albeit not completely, explain why the extent of metabolism of raloxifene in rats is lower than in humans. In other words, the substantial difference in (intrinsic) clearance of M2 in the intestine of rats versus humans (with rat's clearance being 33∼72-fold less) clearly supercedes the fact that M1 clearance in rats is much faster (3∼4-fold) than that in humans (Table 2).
The second difference is where UGT1A10 is expressed. The presence of UGT1A10 in the human intestine and its absence from the human liver (Strassburg et al., 1999; Tukey and Strassburg, 2000) explains the higher formation rates of M2 and higher intrinsic glucuronidation clearance in humans than in rats. It also explains why rat intestine did not have much higher intrinsic clearance than rat liver. Similarly, in Caco-2 cells, which express UGT1A1, UGT1A3, UGT1A4, UGT1A6, and UGT1A8 (very low level) but not UGT1A10 (Fig. 2), much less M2 is formed than M1. The latter is somewhat unexpected since these cells were derived from human colon adenocarcinoma cells. Human colonic enterocytes were reported to express UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, and UGT1A10 mRNA (Strassburg et al., 1998, 2000; Tukey and Strassburg, 2000).

Effects of concentration on raloxifene glucuronidation by rat liver and intestinal microsomes for M1 (A–E) and M2 (F–J). Raloxifene (0.87∼34.7 μM) was added to the microsomes with cofactors as described under Materials and Methods. Incubation was carried out at 37°C for 30 to 60 min, depending on the concentration of raloxifene. FRLM, FRDM, FRJM, FRIM, and FHCM represent pooled female rat liver, duodenal, jejunal, ileal, and colonic microsomes, respectively. Each data point represents the mean of three determinations, and the error bars are standard deviations of the mean. Each curve was obtained by nonlinear regression of the data points using ADAPT II.
The third difference is where and how much UGT1A8 is expressed. Previously, UGT1A8 was reported to catalyze both M1 and M2 formation (Kemp et al., 2002). Since UGT1A8 is only expressed in human intestine, it helps to explain why there was more extensive metabolism of raloxifene in intestine than in liver. On the other hand, its ability to catalyze the formation of both M1 and M2 explains why M1 is present in the human intestinal microsome experiments. Formation of M1 may be aided by the presence of UGT1A1, which is known to be expressed by both liver and intestine. Taken together, UGT1A8 and, to a lesser extent, UGT1A1 in both human and rat (King et al., 2000) were responsible for M1 formation.
Eadie-Hofstee plot of concentration effect on raloxifene glucuronidation by rat liver and intestinal microsomes for M1 (A–E) and M2 (F–J). FRLM, FRDM, FRJM, FRIM, and FHCM represent pooled female rat liver, duodenal, jejunal, ileal and colonic microsomes, respectively. Each data point represents the mean of three determinations, and the error bars are standard deviations of the mean. Metabolite formation profiles by rat intestinal microsomes (except M2 formation by FRCM) showed substrate inhibition.
The fourth difference is probably the transporters that are responsible for the excretion of phase II conjugates and how they control the cellular excretion of phase II metabolites. This is becoming a more important issue, especially in our recent investigation of flavonoid and raloxifene disposition, where efflux transporters often serve as a “gatekeeper” or “the rate-limiting step” in cellular excretion of flavonoid and raloxifene conjugates (Chen et al., 2003, 2005; Hu et al., 2003; Jeong et al., 2004). In the present study, M1 was the main metabolite in Caco-2 cell lysate (60%). This finding is very different from our previous observation in intact Caco-2 cell monolayers, where M2 was the main glucuronide excreted by the Caco-2 cells (Jeong et al., 2004). It is probable that M1 is not well transported by the efflux transporter, or its efflux is competitively inhibited by raloxifene itself (a MRP2 substrate; Jeong et al., 2004). An additional possibility is that raloxifene sulfate, produced in Caco-2 cells at a much higher concentration (than glucuronide), inhibited the efflux of M1. Additional studies are necessary to delineate the mechanisms responsible for this unexpected difference.
Inhibitory effect of genistein (A) and apigenin (B) on glucuronidation of raloxifene by human jejunal microsomes, rat jejunal microsomes, and Caco-2 cell lysate. Raloxifene (4.34 μM) and inhibitor (0∼100 μM genistein or apigenin) were added to the microsomes or Caco-2 cell lysate with cofactors as described under Materials and Methods. Incubation was carried out at 37°C for 1 h (microsomes) or 4 h (Caco-2 cell lysate). Each data point represents the mean of three determinations, and the error bars are standard deviations of the mean. The mean control values (±S.D.) of rate of glucuronidation (in pmol/min/mg protein) were 64.2 ± 0.7 (M1, human, genistein), 530.9 ± 15.7 (M2, human, genistein), 116. 3 ± 9.1 (M1, rat, genistein), 30.1 ± 2.3 (M2, rat, genistein), 0.626 ± 0.075 (M1, Caco-2, genistein), 0.446 ± 0.044 (M2, Caco-2, genistein), 46.6 ± 17.3 (M1, human, apigenin), 500.4 ± 28.3 (M2, human, apigenin), 343.2 ± 13.3 (M1, rat, apigenin), 87.8 ± 2.4 (M2, rat, apigenin), 0.599 ± 0.072 (M1, Caco-2, apigenin), and 0.427 ± 0.042 (M2, Caco-2, apigenin), respectively. Each curve was obtained by nonlinear regression of the data points using SigmaPlot.
Taken together, these differences in disposition strongly support the role played by intestine in the first-pass metabolism of raloxifene. The results of the present study clearly showed the substantial preference (14∼16-fold higher CLint) for M2 over M1 in human intestine. This preference (or difference in CLint, M2) is a larger than 6-fold difference from that observed by Kemp et al. (2002) in human intestinal jejunal microsomes. Specifically, our results contained substantially smaller (>10 times lower) Km values, although the Vmax values were comparable (<3 times less). Multiple factors could contribute to this difference in the extent of preference, including the sources of human intestinal microsomes, concentrations of raloxifene used in the reaction (we could not reach 40 μM without using more than 1% organic phase), and methods of estimating Km, such as Kemp et al. (2002), who used truncated rates versus concentration data and the Michaelis-Menten equation to obtain Km. Despite the above differences, both sets of results are consistent with an earlier observation that M2 concentration in human systemic circulation was 8-fold higher than M1 (Jones, 1997). On the other hand, data from this study indicate that a lack of UGT1A10 expression in the rat intestine is a likely mechanism for increased raloxifene bioavailability in rats.
Finally, we investigated the potential metabolic interaction between raloxifene and flavonoids since they are likely to be taken together by postmenopausal women. The results showed that the potential for metabolic interaction between raloxifene and flavonoids (genistein and apigenin) in the intestine clearly exists. The apparent IC50 value of flavonoids in the tens of micromolar range (Fig. 7; Table 3) is achievable by the consumption of a dietary supplement (Setchell et al., 2001, 2003), especially in the intestinal lumen. Based on our previous experience, apigenin can easily achieve this apparent IC50 concentration in the lumen, whereas genistein can also achieve its IC50 concentration, although some formulation manipulation may be needed (Liu and Hu, 2002). Therefore, these isoflavones have the potential to increase the bioavailability of raloxifene when taken together. The higher inhibitory effect of apigenin over genistein may be explained by the fact that apigenin is a better substrate for various UGT isoforms (e.g., UGT1A1, UGT1A8, and UGT1A9) (King et al., 2000). The higher apparent IC50 values (8∼52 μM) exhibited by genistein against raloxifene glucuronidation suggest that the potential for metabolic interaction between raloxifene and genistein is low. Taken together, we did not expect to observe substantial interaction between raloxifene and flavonoids in humans since flavonoids did not appear to substantially inhibit raloxifene glucuronidation in UGT1A10-rich human intestinal microsomes at physiological concentrations.
In conclusion, the intestinal phase II metabolism catalyzed by the UGT1A family plays a more significant role in the extensive first-pass metabolism of raloxifene than does hepatic metabolism in both humans and rats. The contribution of intestinal metabolism to the low bioavailability of raloxifene is more prominent in humans than in rats because of the abundance of UGT1A10 isoform in the human intestine. Isoflavones and flavones have moderate potential to improve the bioavailability of raloxifene by inhibiting the intestinal conjugation of raloxifene.
Acknowledgments
We thank Dr. Vincent Tam and Yousif Rojeab for performing data fitting used in Fig. 5.
Footnotes
-
This study was supported by National Institutes of Health Grant CA 87779.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.001883.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; RT-PCR, reverse transcriptase-polymerase chain reaction; bp, base pair(s); CT, cycle threshold; CLint, intrinsic clearance.
-
↵1 Current address: Korea Institute of Toxicology, Daejeon, Republic of Korea.
-
↵2 Current address: Departments of Behavioral Neuroscience, Oregon Health and Science University, Portland, Oregon.
-
↵3 Current Address: Department of Pharmacological and Pharmaceutical Sciences, College of Pharmacy, University of Houston, Houston, Texas.
- Received August 23, 2004.
- Accepted March 10, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}