


Abstract
Raloxifene, a selective estrogen receptor modulator used for the treatment of osteoporosis, undergoes extensive conjugation to the 6-β- and 4′-β-glucuronides in vivo. This paper investigated raloxifene glucuronidation by human liver and intestinal microsomes and identified the responsible UDP-glucuronosyltransferases (UGTs). UGT1A1 and 1A8 were found to catalyze the formation of both the 6-β- and 4′-β-glucuronides, whereas UGT1A10 formed only the 4′-β-glucuronide. Expressed UGT1A8 catalyzed 6-β-glucuronidation with an apparent Km of 7.9 μM and aVmax of 0.61 nmol/min/mg of protein and 4′-β-glucuronidation with an apparent Kmof 59 μM and a Vmax of 2.0 nmol/min/mg. Kinetic parameters for raloxifene glucuronidation by expressed UGT1A1 could not be determined due to limited substrate solubility. Based on rates of raloxifene glucuronidation and known extrahepatic expression, UGT1A8 and 1A10 appear to be primary contributors to raloxifene glucuronidation in human jejunum microsomes. For human liver microsomes, the variability of 6-β- and 4′-β-glucuronide formation was 3- and 4-fold, respectively. Correlation analyses revealed that UGT1A1 was responsible for 6-β- but not 4′-β-glucuronidation in liver. Treatment of expressed UGTs with alamethicin resulted in minor increases in enzyme activity, whereas in human intestinal microsomes, maximal increases of 8-fold for the 6-glucuronide and 9-fold for the 4′-glucuronide were observed. Intrinsic clearance values in intestinal microsomes were 17 μl/min/mg for the 6-glucuronide and 95 μl/min/mg for the 4′-isomer. The corresponding values for liver microsomes were significantly lower, indicating that intestinal glucuronidation may be a significant contributor to the presystemic clearance of raloxifene in vivo.
Raloxifene (Evista) is an antiestrogen marketed for the treatment of osteoporosis. Clinical studies have shown that raloxifene blocks the unwanted effects of estrogen in breast and uterus while mimicking beneficial estrogen effects in other tissues such as bone (Jordan et al., 2001; O'Regan and Jordan, 2001). Raloxifene is rapidly absorbed after oral administration and is known to undergo extensive presystemic glucuronidation (Fig. 1), as evidenced by 2% bioavailability and apparent oral clearance of 44 l/kg/h (Hochner-Celnikier, 1999; Snyder et al., 2000). In addition, these clinical studies have shown that glucuronidation occurs primarily at the 4′-position of raloxifene, with lower levels of the 6-glucuronide found in vivo. Also, the plasma elimination half-life of approximately 28 h indicates systemic interconversion and entrohepatic cycling of the parent and glucuronides.
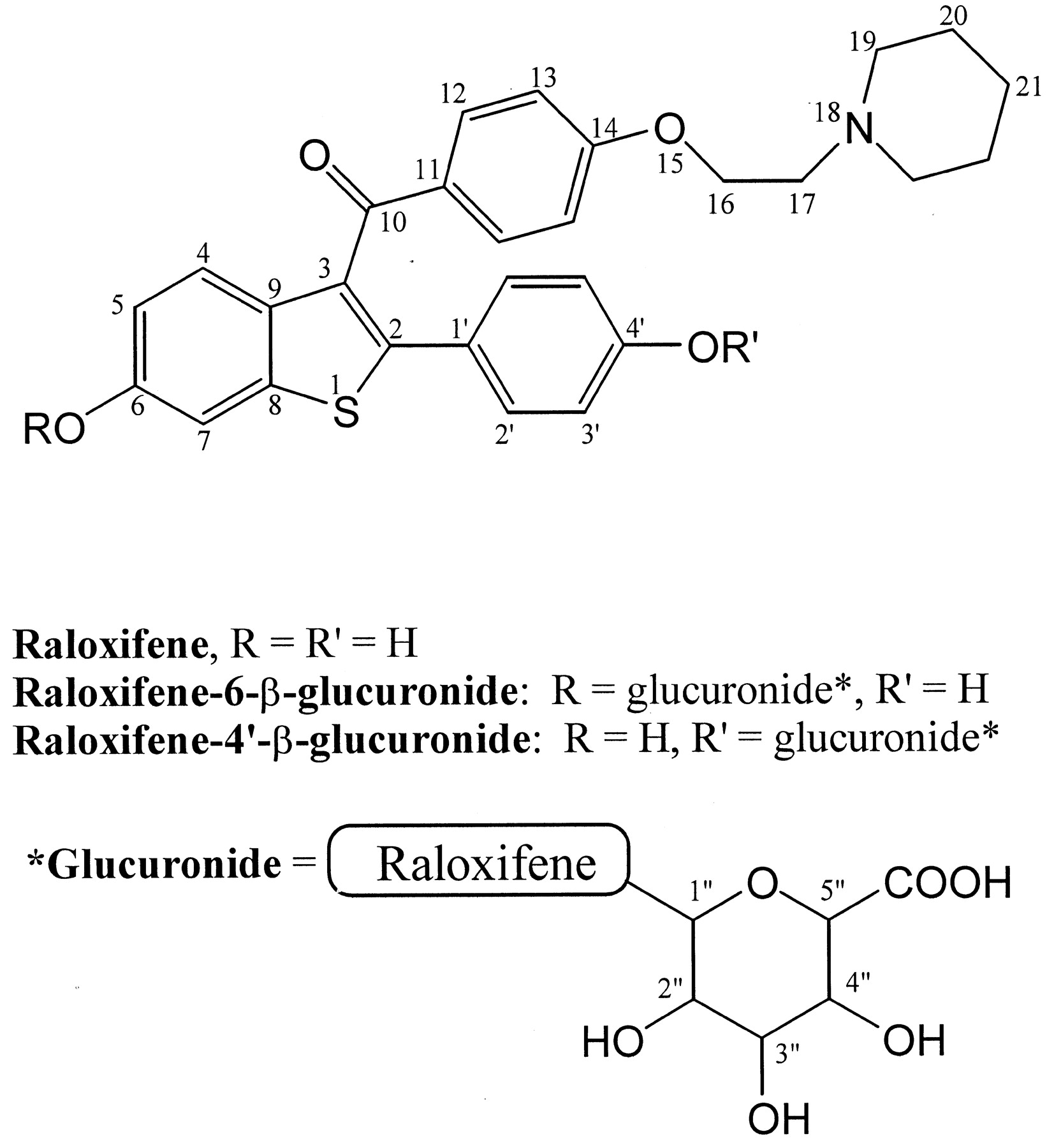
Structure of raloxifene and two major metabolites (R-6-G and R-4-G).
The numbers correspond to the proton assignments in Table 1.
The UDP-glucuronosyltransferases (UGTs1) catalyze the transfer of glucuronic acid to available substrates, resulting in conjugates with increased water solubility. Glucuronidation has become increasingly important in pharmaceutical drug development because biotransformation and elimination of drugs by this pathway may influence their potency, bioavailability, and pharmacokinetics. Several UGT isoforms have been cloned and categorized based on their protein sequence homology. In humans, tissue-dependent distribution of various UGT isoforms has been identified. For example, the expression of UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 has been detected in the liver, whereas UGT1A7, 1A8, and 1A10 are absent. In human intestine, UGT1A1, 1A4, 1A8, and 1A10 have been detected (Tukey and Strassburg, 2000).
Glucuronidation is one of several enzymatic processes that may contribute to presystemic or “first-pass” drug metabolism. Hepatic clearance is generally recognized as the primary contributor to presystemic metabolism, however, intestinal metabolism via the cytochrome P450 enzymes has also been shown to contribute (Watkins, 1992; Shen et al., 1997; Lown et al., 1998). For example, CYP3A4 is the predominant P450 form in the intestine, where it plays an important role in the metabolism of orally administered drugs such as cyclosporine, midazolam, nifedipine, saquinavir, and verapamil. In contrast, the contribution of glucuronidation and other phase II processes to first-pass metabolism has not been studied to the same extent. Examples to date include the sulfation of terbutaline and isoproterenol and the glucuronidation of morphine and labetalol (Shen et al., 1997).
Despite clinical data on the extensive glucuronidation of raloxifene, the role of specific hepatic and intestinal UGT isoform(s) has not been investigated. Therefore, the objectives of this study were to: 1) determine the major UGT isoform(s) responsible for the metabolism of raloxifene; 2) characterize the enzyme kinetics and interindividual variability of raloxifene glucuronidation in human liver microsomes; and 3) evaluate the contribution of liver and intestinal UGTs to the presystemic clearance of raloxifene.
Materials and Methods
Chemicals.
Chemicals were purchased from the following commercial sources. Alamethicin, ammonium acetate, magnesium chloride, saccharolactone, Tris-HCl, and UDPGA were purchased from Sigma-Aldrich (St. Louis, MO). HPLC-grade acetonitrile and methanol were from EM Science (Gibbstown, NJ). Raloxifene was extracted from Evista tablets (60 mg/tablet).
Biological Reagents.
Human liver samples were obtained through organ procurement agencies in accordance with proper ethical procedures for consent. Pooled human liver microsomes (mixed gender, n = 15) were obtained from Xenotech, LLC (Kansas City, KS). Pooled jejunum microsomes (mixed gender, n = 4) were purchased from Tissue Transformation Technologies (Edison, NJ). Individual liver microsomes from a panel of 13 donors were obtained from Gentest Corporation (Woburn, MA). Microsomes prepared from control or human lymphoblastoid cells transfected with cDNA from human UGT1A1, 1A4, 1A6, 1A8, 1A9, 1A10, 2B7, and 2B15 were purchased from Gentest. Microsomes prepared from control or SF9 insect cells infected with a baculovirus containing the cDNA for human UGT1A7 were purchased from Panvera (Madison, WI). Enzyme activities for these commercially available UGT forms were found to be comparable with product data provided by the manufacturers. β-Glucuronidase from Helix pomatia was purchased from Sigma-Aldrich.
Isolation and Characterization of R-4-G and R-6-G.
The glucuronides were synthesized, isolated, and characterized as follows. Expressed UGT1A8 (0.8 mg) in 50 mM Tris-HCl buffer (pH 7.5) and 5 mM MgCl2 was mixed with alamethicin in methanol (60 μg/mg of protein) on ice. The mixture was kept cold for 15 min before the addition of 200 μM raloxifene. The samples were preincubated for 3 min at 37°C in a shaking water bath, and the reaction was initiated by the addition of UDPGA (5 mM final concentration). The total volume of the incubation was 1 ml and the total organic solvent used did not exceed 1%. The incubation was allowed to continue overnight, and the reaction was terminated by the addition of 500 μl of cold acetonitrile. The protein was then precipitated by brief centrifugation. Finally, the supernatants of several incubations were combined and purified using a Luna 5μ C18(2) preparative column (250 × 21.2 mm; Phenomenex, Torrance, CA). The mobile phase consisted of 90% ammonium acetate buffer (pH 4.0) and 10% acetonitrile at a flow rate of 20 ml/min for 2 min, increased to 60% acetonitrile over the next 24 min, and then was held for 5 min before returning to initial conditions. UV detection was set at 255 nm. UDPGA-dependent peaks were collected, combined, and dried under vacuum overnight. The fractions were reconstituted in water and recovered using Oasis HLB extraction cartridges (Waters, Milford, MA). NMR (Table1) and mass spectrometry were used for the identification of the glucuronides. Raloxifene-6-β-glucuronide (R-6-G): positive ion electrospray LC/MS/MS: m/z 650, 474 (100%), rt 12 min. Raloxifene-4′-β-glucuronide (R-4-G): positive ion electrospray LC/MS/MS: m/z 650, 474 (100%), rt 14 min.
Proton NMR assignment of raloxifene and raloxifene glucuronides
Glucuronidation Assays.
In a typical kinetic experiment, assay mixtures contained 10 to 200 μM raloxifene, 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, alamethicin (60 μg/mg of protein), 2 mM UDPGA, and expressed UGT or human microsomes. For expressed UGT1A8, 25 μg of protein was used, and for UGT1A1, 1A9, and 1A10, the protein amount was increased to 50 μg. For human liver and jejunum microsomes, 25 μg of protein was used, and the incubation time was reduced to 5 min to keep total substrate consumption below 20%. Final incubation volumes were 250 μl and the maximal final methanol content was 1%. Samples were preincubated for 3 min at 37°C followed by the addition of UDPGA and incubation for 10 min. The reactions were stopped by the addition of 50 μl of cold acetonitrile, the samples were briefly centrifuged, and the supernatant was analyzed by HPLC using a Phenomenex Luna 5μ C18(2) analytical column (150 × 3.00 mm) and UV detection at 255 nm. The elution gradient was identical with the preparative HPLC method; however, the flow rate was adjusted to 0.7 ml/min.
Rates of enzymatic hydrolysis for R-4-G and R-6-G were measured using 0.25 units/ml β-glucuronidase from Helix pomatia. Incubations included either R-4-G or R-6-G (2.5 μM) in 100 mM ammonium acetate (pH 5.0) at 37°C. Aliquots were removed at time intervals ranging from 15 to 180 min and analyzed by HPLC for unhydrolyzed glucuronides. For confirmation of structure, an excess of glucuronidase (2000 units/ml) was incubated with either R-4-G or R-6-G (20 μM) at 37°C, and after 16 h, all glucuronides were hydrolyzed to the aglycone.
Data Analysis.
Apparent Km (Michaelis-Menten constant) andVmax (maximal metabolic velocity) were determined with Prism 3.0 (GraphPad, San Diego, CA) software using a one-site binding model (best-fit) and linear regression analysis of the Eadie-Hofstee plots. The latter method assessed the potential for atypical versus typical Michaelis-Menten kinetics (Obach et al., 2001).
Instrumentation.
Proton and COSY NMR spectra were obtained with an Avance DPX400 spectrometer (Bruker, Newark, DE). Preparative-HPLC and HPLC experiments were carried out on a Dynamax SD-1 dual pump system (Rainin Instruments, Woburn, MA) or a 200 Series LC system (PerkinElmer Life Sciences, Boston, MA), respectively. Mass spectrometry was performed on an LCQ instrument (Finnigan-MAT, San Jose, CA) tuned to unit mass resolution. This was directly coupled to the HPLC system (1050 Series pump; Hewlett Packard, Naperville, IL) through a Finnigan atmospheric pressure ionization source operated in the electrospray ionization mode. Chromatographic separations were performed using a Phenomenex Luna 5μ C18(2) analytical column (250 × 2 mm) with the previously described gradient, except that a flow rate of 0.25 ml/min was used. The mass spectrometer was operated in the positive ion mode, typically scanning from 150 to 1000 atomic mass units every 2 s. The capillary was operated at 230°C, the spray voltage was set to 5 kV, and nitrogen was employed as a drying gas at a sheath pressure of 70 psi and an auxiliary flow of 20 ml/min.
Results
Identification of Raloxifene Glucuronides.
Incubation of raloxifene and human liver microsomes in the presence of UDPGA resulted in the formation of two UDPGA-dependent peaks. Each peak gave an identical molecular ion at m/z 650 by LC/MS. Both peaks gave identical MS/MS spectra, further indicating that the analytes were structural isomers. Specifically, tandem MS analysis of the m/z 650 ion gave a daughter ion atm/z 474, which corresponded to the protonated aglycone formed from cleavage of the glycosidic bond (MH+−176). The structure of the raloxifene metabolites was established by comparison with the NMR data (Table 1) of unreacted raloxifene and that of synthetic raloxifene glucuronides previously characterized (Dodge et al., 1997). Specifically, the glucuronide at rt 12 min was identified to be R-6-G based on an exact match with the literature values for proton chemical shifts, proton integral ratios, and coupling patterns. An additional COSY experiment was performed to establish the proton chemical shift correlation of this glucuronide, and the proton assignments were made accordingly. In contrast, the1H NMR spectrum of the second UDPGA-dependent peak isolated (rt = 14 min) deviated from that of the synthetic standard reported in the literature. A selective NOE experiment performed on this peak demonstrated the close proximity ofH3′ on the phenolic ring to the anomeric proton (H1") of the glucuronide (Fig.2). Similarly, a corresponding nuclear Overhauser enhancement was observed for H5 and H7 on the hydroxybenzothienyl ring of R-6-G upon irradiation of its anomeric proton (H1", data not shown). On this basis, the 12 and 14 min metabolites were assigned as the R-6-G and R-4-G, respectively.

Characterization of the site of raloxifene glucuronidation by NMR analysis.
A, 1H NMR reference spectrum of R-4-G; B, the corresponding selective nuclear Overhauser enhancement using a gradient spectrum that illustrates the NOE effect upon irradiation of the anomeric proton (H1") at 4.77 ppm.
The β-anomeric configuration was assigned to both raloxifene glucuronides because the coupling constants of their anomeric protons (H1") were in the range of 7 to 8 Hz. Coupling constants in the range of 7 to 10 Hz have been documented to be characteristic of the β-anomers of various glucuronides, whereas the α-anomers have coupling constants in the range of 2 to 4 Hz (Green and Tephly, 1996).
Preliminary Assessment of in Vitro Conditions.
Several experiments were performed to fully characterize factors that could alter the rate of in vitro raloxifene glucuronidation and/or the stability of R-4-G or R-6-G. First, the formation of R-4-G and R-6-G by expressed UGT1A1, 1A8, 1A9, and 1A10, human liver, and intestinal microsomes was established to be linear as a function of incubation time up to 20 min using the protein concentrations described for each system under Materials and Methods. Also, buffer content and pH have been shown to effect rates of glucuronidation for expressed UGTs (Green and Tephly, 1996) and liver microsomes (Huskey et al., 1993), and, thus, these parameters were examined for raloxifene glucuronidation. Tris buffer was found to produce higher activity compared with phosphate buffer, and maximal activity was observed in the pH range of 7.5 to 8.0 regardless of the enzyme system or the metabolite measured (data not shown). Therefore, Tris buffer at a physiologically relevant pH of 7.5 was used for all experiments.
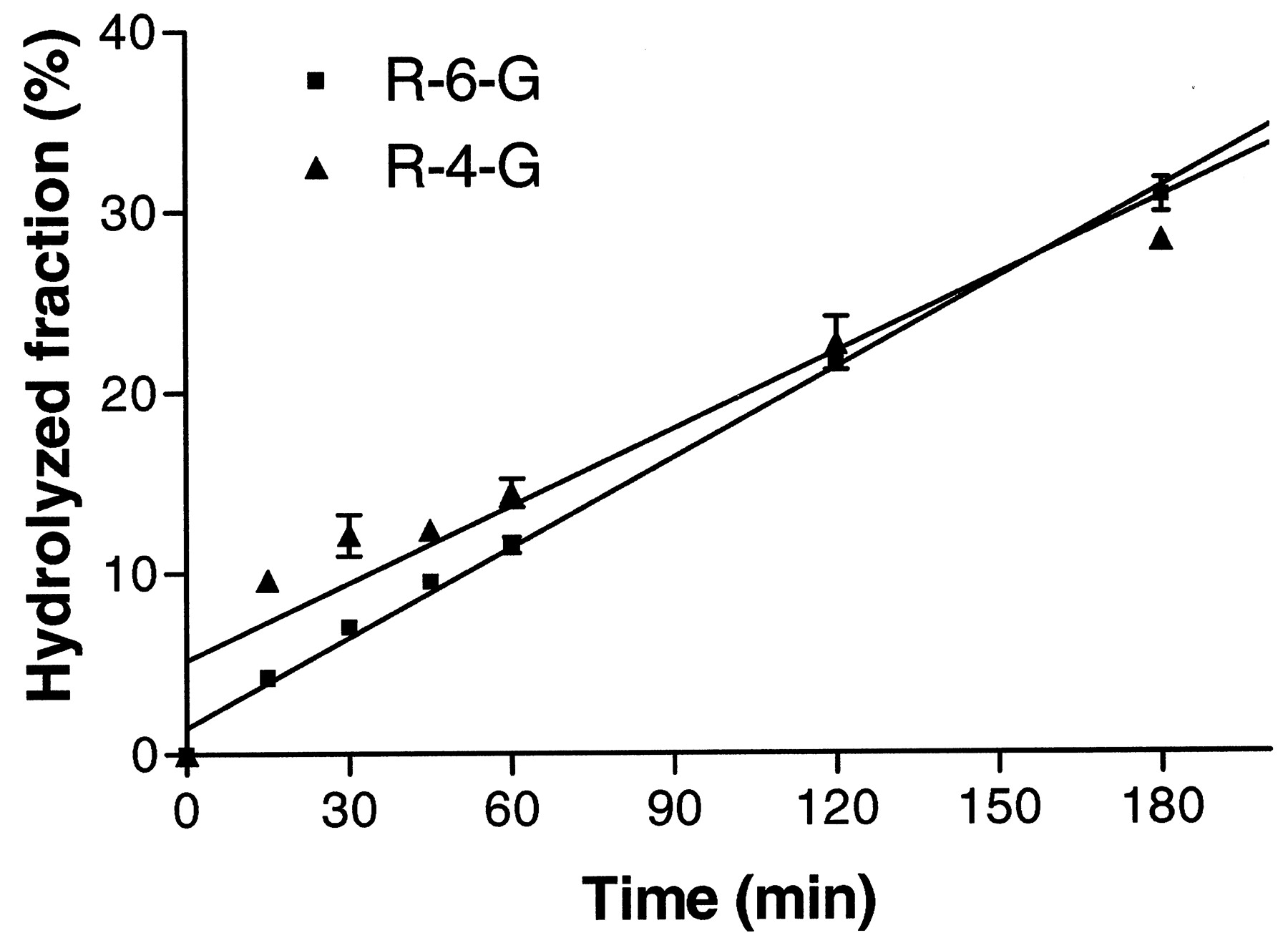
Saccharolactone has been shown to increase the apparent formation of glucuronides through the inhibition of β-glucuronidase (Ho et al., 1985). However, the addition of saccharolactone (5 mM final concentration) to human liver microsome incubations actually resulted in a decrease in the rate of raloxifene glucuronidation (data not shown). This component was, therefore, eliminated from future studies. Finally, clinical pharmacokinetic studies with raloxifene suggest substantial enterohepatic circulation (Hochner-Celnikier, 1999; Snyder et al., 2000). This would involve biliary excretion of glucuronides into the intestine, followed by enzymatic hydrolysis by β-glucuronidase and reabsorption of the aglycone. To simulate this process, the stability of R-4-G and R-6-G in the presence of β-glucuronidase was examined (Fig. 3). The results show that raloxifene glucuronides have similar rates of enzymatic hydrolysis.

Time course of the hydrolysis of the 6-and 4′-glucuronides with β-glucuronidase from Helix pomatia.
Raloxifene glucuronides (2.5 μM) were incubated separately with 0.25 unit/ml of the glucuronidase for varying amounts of time. Unhydrolyzed compounds were separated and quantitated by HPLC as described underMaterials and Methods. Determinations were performed in triplicate.
Raloxifene Glucuronidation by Expressed UGTs.
Various expressed UGT isoforms (1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B7, and 2B15) were tested for their ability to catalyze raloxifene glucuronidation. From the 10 isoforms tested, four (1A1, 1A8, 1A9, and 1A10) were able to catalyze the formation of either the 6- or 4′-glucuronides. After preliminary studies to determine the linearity of glucuronide formation with respect to the amount of microsomal protein and incubation time, the kinetic parameters of raloxifene glucuronidation were determined for the four different active UGT isoforms (Table 2). The results were analyzed by Eadie-Hofstee plots to distinguish the involvement of multiple enzymes. TheKm andVmax values for 6-glucuronidation ranged from 7.9 to 25 μM and from 0.32 to 0.61 nmol/min/mg, respectively. The Km andVmax values for 4′-glucuronidation ranged from 4.8 to 59 μM and from 0.25 to 2.0 nmol/min/mg, respectively. Whereas R-6-G and R-4-G formation by expressed 1A8 and 1A9 followed typical Michaelis-Menten kinetics for a one-enzyme system, 1A1 and 1A10 showed atypical kinetics.Km andVmax for the formation of R-6-G and R-4-G in the presence of UGT1A1 could not be estimated accurately from a one-enzyme system due to limited substrate solubility (200 μM). The lack of deviation from linearity between rate and substrate concentration up to 200 μM suggested a highKm value for raloxifene glucuronidation by UGT1A1. At a substrate concentration of 200 μM, the rates of R-4-G and R-6-G formation by 1A1 were 0.94 and 1.85 nmol/min/mg, respectively (Table 2).
Kinetic parameters for raloxifene glucuronidation by expressed UGTs, human liver, and intestinal microsomes
A dramatic decrease in the formation of R-4-G by UGT1A10 was observed with substrate concentrations of ≥25 μM, suggesting substrate inhibition may be occurring (Lin et al., 2001). Therefore,Km andVmax were calculated after truncation of the curve to exclude the inhibited rates and then overlaid onto the actual two-site binding curve fit for 1A10 (Fig.4). Interestingly, 1A10 displayed complete selectivity for R-4-G formation, because R-6-G was not detected in these kinetic experiments.

Michaelis-Menten kinetics of raloxifene 4′-glucuronidation by expressed UGT1A10.
Dotted line, a hyperbolic curve fitted after truncating the inhibited rates at high substrate concentrations; solid line, substrate inhibition curve fitted with a two-site binding model.
Raloxifene Glucuronidation by Human Liver and Intestinal Microsomes.
Kinetic parameters for raloxifene glucuronidation were also determined from human liver and intestinal microsomes (Table 2). TheKm andVmax values for 6-glucuronidation in liver microsomes could not be determined due to limited substrate solubility. The average intrinsic clearance (Vmax/Km) was highest for the 4′-glucuronide in jejunum microsomes, reaching 95 μl/min/mg, which was approximately 6-fold greater than that observed for R-6-G formation (17 μl/min/mg). In comparing liver and intestinal intrinsic clearance for R-4-glucuronidation, hepatic levels were found to be 3-fold lower. The rates of formation of R-6-G and R-4-G were then determined using a bank of 13 characterized human liver microsome samples. Based on previous experiments for the determination ofKm for raloxifene glucuronidation, a saturating substrate concentration (200 μM) was used. As shown in Fig. 5A, the rate of R-6-G formation ranged from 0.43 to 1.6 nmol/min/mg (∼4-fold) and for R-4-G, from 0.48 to 1.5 nmol/min/mg (3-fold). Furthermore, the rate of formation of R-6-G correlated strongly with UGT1A1-marker estradiol-3-glucuronidation (Fisher et al., 2000), exhibiting anr2 of 0.84 (p< 0.01) (Fig. 5B). In contrast, the rate of R-4-G showed no correlation with 1A1 activity (r2 = 0.13, p > 0.05). Finally, a weak correlation was observed between the rate of formation of R-6-G and R-4-G in human liver microsomes (r2 = 0.36,p = 0.03).

Rates of raloxifene glucuronidation for a panel of human liver microsomes (panel A) and correlation analysis of R-6-G formation with estradiol-3-glucuronidation activity (panel B).
Individual human liver microsomes (100 μg) were incubated with 200 μM raloxifene at 37°C for 10 min in the presence of UDPGA followed by reverse-phase HPLC/UV analysis and quantitation of R-6-G and R-4-G. Incubations were performed in triplicate. Individual human liver microsomes were purchased from Gentest as described underMaterials and Methods. The rates of estradiol-3-glucuronidation were obtained from the individual human liver microsomal activity data sheet published by Gentest.
Effect of Alamethicin on Expressed UGTs, Human Liver, and Intestinal Microsomal Activity.
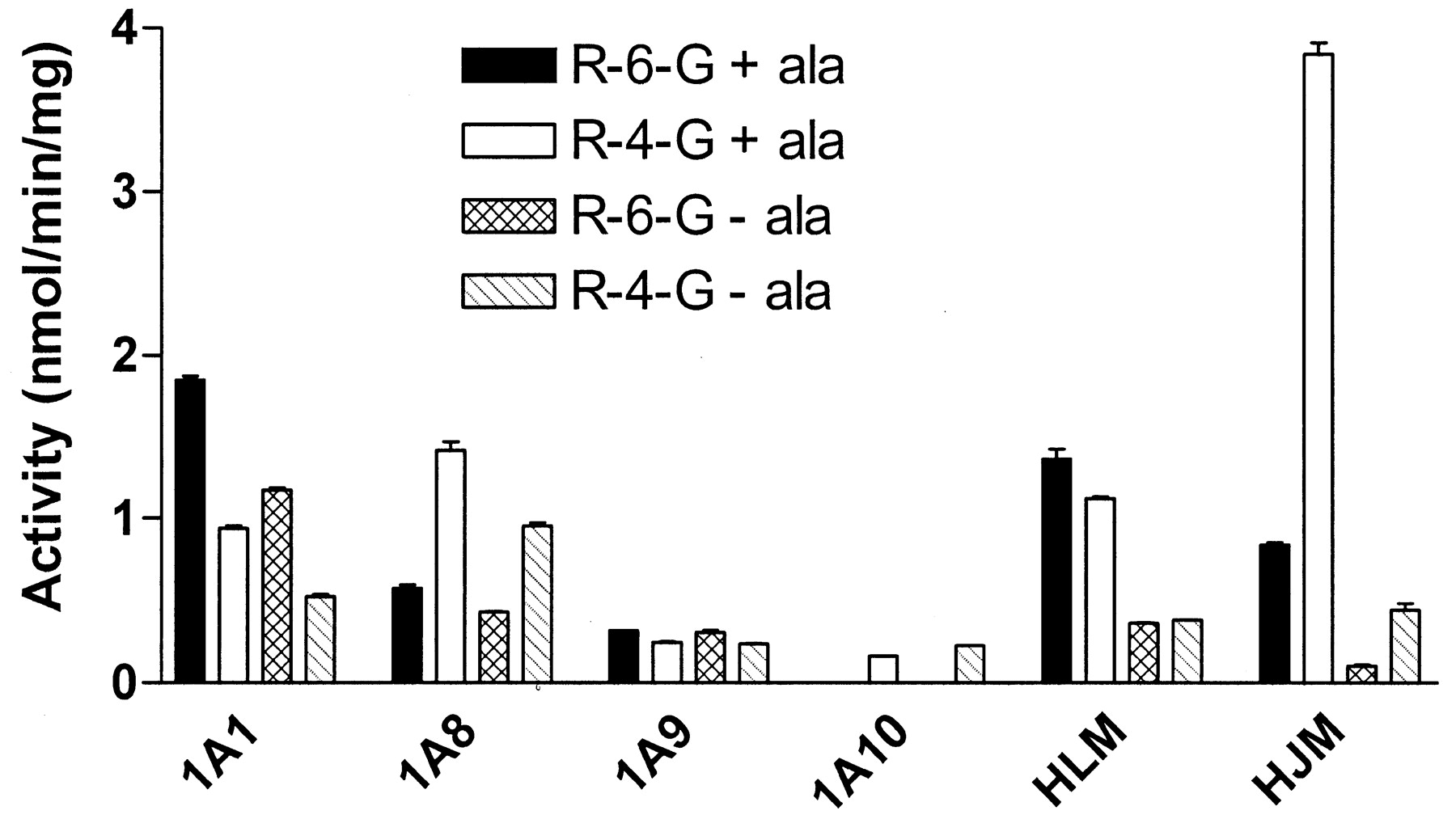
To examine the effect of alamethicin on UGT activity, raloxifene was incubated with different expressed UGT forms, human liver, or intestinal microsomes in the presence or absence of alamethicin (Fig.6). Regarding expressed UGT forms, the activity of expressed UGT1A9 and 1A10 was not increased significantly in the presence of alamethicin, whereas a more pronounced increase in activity was observed with UGT1A1 and 1A8. For example, the addition of alamethicin to UGT1A1 resulted in approximately a 40% increase in the amount R-6-G formed compared with control. In contrast, the addition of alamethicin to incubations of both pooled human liver and jejunum microsomes resulted in a significant increase in the rate of formation of both raloxifene glucuronides. An increase in activity from 0.11 to 0.85 nmol/min/mg (8-fold) was observed for the formation of R-6-G in jejunum microsomes. Similarly, an increase from 0.44 to 3.8 nmol/min/mg (9-fold) was observed for the formation of R-4-G in the same system. In liver microsomes, an increase from 0.36 to 1.4 nmol/min/mg (4-fold) and an increase from 0.38 to 1.1 nmol/min/mg (3-fold) were observed for the formation of R-6-G and R-4-G, respectively.

Effect of alamethicin on expressed UGTs, human liver (HLM), and jejunum microsomes (HJM).
Triplicate incubations included microsomes (25–50 μg), with (+ala) or without (−ala) alamethicin, 200 μM raloxifene, and 2 mM UDPGA. Incubations were carried out at 37°C for 5 to 10 min depending on the system (see Materials and Methods). Pooled human liver and jejunum microsomes were purchased from Xenotech and Tissue Transformation Technologies as described under Materials and Methods.
Discussion
The contribution of intestinal glucuronidation to the first-pass metabolism of orally ingested xenobiotics in humans has gained considerable interest in recent years (Shen et al., 1997; Czernik et al., 2000). Raloxifene represents a clear example of a drug that does not undergo significant P450-dependent oxidation (Hochner-Celnikier, 1999), but rather where glucuronidation is the major route of metabolism. The present studies used various in vitro systems to identify and characterize the UGT forms responsible for R-4-G and R-6-G formation and to differentiate the contribution of hepatic and intestinal tissues to raloxifene glucuronidation. Toward these objectives, systematic experimental approaches were taken which included activity measurements of raloxifene glucuronidation using expressed UGTs and microsomes from human liver and intestine in combination with correlation analyses. Consideration of literature reports of tissue-dependent expression of human UGT forms was also used to context results to existing data on the clinical pharmacokinetics of raloxifene.
After the isolation and characterization of the glucuronide isomers of raloxifene, the initial focus of these studies was to understand the role of hepatic UGTs in raloxifene conjugation. Several lines of evidence indicated hepatic UGT1A1 as a primary catalyst of raloxifene metabolic clearance via the 6-glucuronide pathway. For example, a strong correlation was observed between the interindividual variability in the rates of raloxifene glucuronidation by human liver microsomes and UGT1A1-marker estradiol-3-glucuronidation. This implies that the hydroxybenzothienyl group of raloxifene occupies the same binding space of UGT1A1 as the phenolic group of estradiol, which is consistent with the extensive structure-activity data used to design drugs such as raloxifene that mimic the receptor binding properties of estrogens. Although expressed UGT1A8 and 1A10 catalyzed raloxifene glucuronidation, these UGT forms are not expressed in human liver (Cheng et al., 1998; Tukey and Strassburg, 2000). UGT1A9 was shown to catalyze the formation of each isomer; however, the relative contribution of this form seems minor in relation to UGT1A1 and 1A8. Expressed UGT1A1 and pooled human liver microsomes were also found to generate slightly more R-6-G than R-4-G at a concentration of 200 μM raloxifene (Fig. 6). However, the relative levels of raloxifene glucuronide isomers produced from human liver microsome incubations are not in agreement with clinical studies showing the 4′-glucuronide to be the major metabolite in plasma (Hochner-Celnikier, 1999). There are several potential explanations for this inconsistency, including that an unknown higher affinity hepatic UGT form(s) could favor R-4-G formation at physiological concentrations. Our data showing similar rates of glucuronide isomer degradation by β–glucuronidase argue against R-4-G simply being more stable in vivo. Data presented in this report support an alternative hypothesis, that is, that intestinal UGT forms such as UGT1A10 and 1A8 catalyze a significant fraction of raloxifene metabolism in vivo. Among the UGT forms studied, UGT1A10 is unique in that it exclusively and efficiently (Clint = 115 μl/min/mg) catalyzed the formation of R-4-G. UGT1A8, which is 90% identical in primary amino acid sequence to UGT1A10 (Cheng et al., 1998), also seems to be involved in intestinal raloxifene glucuronidation. This hypothesis is consistent with published data on the metabolism of estrogens and related compounds by 1A8 (Cheng et al., 1998; Fisher et al., 2000). Because of the limited in vitro techniques presently available, enzyme activity relative to a marker substrate and relative amounts of individual intestinal UGT forms could not be determined.
Inhibition of enzyme activity at high substrate concentrations is known to occur with cytochrome P450 catalyzed oxidation (Lin et al., 2001). These authors have proposed that substrate inhibition may occur by the access of more than one substrate molecule to the active site, with a best fit to a two-site enzyme model. To the best of our knowledge, substrate inhibition has not been explored for UGTs. This mechanism is consistent for the inhibition of UGT1A10 by high concentrations of raloxifene. Hence, the substrate inhibition curve could not be fit to the standard Michaelis-Menten equation for one-site binding. Therefore, theoretical Km andVmax values were calculated for UGT1A10 by truncating the data before substrate inhibition occurs. In vivo, maximal plasma concentrations of raloxifene of approximately 1 μM (Hochner-Celnikier, 1999) suggest that substrate inhibition is not likely to occur at physiological conditions. Admittedly, the atypical kinetics of raloxifene glucuronidation by UGT1A10 is simply an observation, and a more complete understanding of the molecular basis will require better models of the UGT substrate binding and active sites.
The in vitro variability observed for raloxifene glucuronidation (3- to 4-fold for R-4-G and R-6-G, respectively) is comparable with the range of UGT activities reported for some phenolic compounds. For example,Fisher et al. (2000) observed modest (≤7-fold) in vitro variation in both acetaminophen-O- and morphine-3-glucuronidation. However, there are differences in variability. For example, these same authors reported a 30-fold variation in liver microsomal estradiol-3-glucuronidation, and Temellini et al. (1991) reported a 19-fold variation in the glucuronidation of ethinylestradiol. Given the overlapping substrate specificity of UGT1A1 for raloxifene and estradiol, such a difference in variability between our data and published observations could be due to different donors, sample suppliers, and/or sample preparation procedures.
UGTs are membrane-bound proteins found in the endoplasmic reticulum. Because of their location, a latency of activity has been suggested because the endoplasmic reticulum membrane provides a diffusional barrier for substrates (Meech and Mackenzie, 1997). Disruption of this barrier is required to remove the latency and observe optimal enzyme efficiency. Detergents are often used to accomplish this goal. However, high concentrations of detergents can hinder UGTs by disrupting their interaction with phospholipids that are necessary for catalytic activity (Parkinson, 1996). In addition, a study done by Little et al. (1997) showed that alamethicin, a pore forming peptide, is a more efficient activator of glucuronidation activity compared with detergent activation. With raloxifene as a substrate, alamethicin increased activity for UGT1A1 and 1A8 as compared with UGT1A9 and 1A10. Although the expression system was the same in each case, it is not possible to infer isozyme-specific effects due to potential confounding factors such as enzyme expression levels and differential flux of reaction components (i.e., UDPGA and substrate). The increase in UGT1A1 and 1A8 activities was still small when compared with pooled human intestinal microsomes, which showed 8-fold (R-6-G) and 9-fold (R-4-G) increases after treatment with alamethicin. This difference in activity could be due to the different lipid environments of the expressed UGTs and human microsomes or different UGT expression levels in these systems.
Three pharmacokinetic parameters from the human clinical data for raloxifene are suggestive of enterohepatic circulation: low bioavailability (2%), high oral clearance (44 l/kg/h), and an extended plasma elimination half-life (∼28 h) (Hochner-Celnikier, 1999). Therefore, the relationship of raloxifene-glucuronides to presystemic clearance and enterohepatic circulation was investigated using in vitro techniques. Our data show that these glucuronides are subject to hydrolysis by β-glucuronidase. This hydrolysis models the contribution of intestinal flora to enterohepatic cycling (Mey et al., 1999). Thus, competing processes of intestinal hydrolysis, conjugation, and absorption contribute to raloxifene pharmacokinetics.
In conclusion, we have demonstrated that raloxifene 4′-glucuronidation is catalyzed primarily by intestinal UGT1A8 and 1A10, whereas hepatic UGT1A1 preferentially forms R-6-G. Also, the high efficiency of human jejunum microsomes for raloxifene glucuronidation has been demonstrated with a 3-fold greater intrinsic clearance for R-4-G with jejunum compared with liver microsomes. The fact that intestinal microsomes produced more 4′-glucuronide than the 6-isomer is consistent with the in vivo data. Recent studies have demonstrated that, in contrast to liver, intestinal UGTs are subject to polymorphic regulation (Strassburg et al., 2000). However, the lack of availability of definitive characterization information for certain in vitro approaches (i.e., chemical and antibody inhibition, correlation analysis with specific enzyme activities, and expressed enzymes) hinders investigations to define the role of intestinal UGTs in drug conjugation. Certainly, tissue-specific expression and relative rates of raloxifene glucuronidation by UGT forms need to be contexted with in vivo factors that argue for either greater intestinal contribution (slow transit and high concentration of drug) or hepatic contribution (high 1A1 levels and blood flow). Quantitative in vitro/in vivo correlations for raloxifene glucuronidation taking into account both the hepatic and intestinal components of first pass metabolism will require further investigation.
Acknowledgments
We are grateful to Dr. Paul Fagerness for carrying out the selective NOE experiments and to Gregory Walker for assisting with LC/MS/MS operations.
Footnotes
- Abbreviations used are::
- UGT
- UDP-glucuronosyltransferase
- R-4-G
- raloxifene-4′-β-glucuronide
- R-6-G
- raloxifene-6-β-glucuronide
- ala
- alamethicin
- rt
- retention time
- MS/MS
- tandem mass spectrometry
- NMR
- nuclear magnetic resonance spectroscopy
- COSY
- proton-proton correlation spectroscopy
- NOE
- nuclear Overhauser enhancement
- LC/MS
- liquid chromatography mass spectrometry
- HPLC
- high-performance liquid chromatography
- UDPGA
- uridine 5′-diphosphoglucuronic acid
- Received December 13, 2001.
- Accepted March 5, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}