


Abstract
The success of in vitro derived Ki values for predicting drug-drug interactions in vivo has been mixed. For example, the use of hepatic input concentration of inhibitor has resolved the negative and positive interactions on the qualitative level, eliminating false negative predictions. However, several examples of false positives and a high incidence of over-predictions of true positive interactions indicated a need for incorporation of additional factors. The aim of this study was to investigate the effect of parallel elimination pathways as a possible reason for false positives and over-predictions. Simulation studies indicated that the degree of interaction (assessed by area under the plasma concentration-time curve ratio in the presence and absence of inhibitor) depends largely on the fraction of substrate metabolized by the particular P450 enzyme (fmCYPi) that is inhibited. The current analysis focused on CYP2D6 interactions due to the well documented genetic polymorphism and the ability to estimate fmCYP2D6 readily from in vivo data obtained in extensive and poor metabolizers. Based on either a phenotype study or an alternative regression analysis approach, the fmCYP2D6 values of 0.37 to 0.94 and 0.25 to 0.89, respectively, were obtained for nine substrates. Prediction of 44 drug-drug interaction studies was improved by the combination of parallel pathways of elimination and their susceptibility to inhibition. The overall success of predicting positive and negative interactions was increased from 54% to 84%, and the number of over-predictions was substantially reduced. It is concluded that incorporating parallel pathways provides a valuable step forward in making quantitative predictions of drug-drug interactions from in vitro data.
Drug-drug interactions (DDIs) resulting from inhibition of P450-mediated drug metabolism by a coadministered drug continue to be a major cause of serious toxicities. The design of in vitro studies to predict the inhibition potential of a drug has benefited from substantial technological advances in recent years, yet the interpretation of the kinetic parameters generated from these studies remains problematic (Houston and Galetin, 2003), and hence there is a lack of confidence in in vivo predictions from in vitro data (Tucker et al., 2001; Bjornsson et al., 2003).
A statistically significant increase in the area under the plasma concentration-time curve (AUC) of the victim drug in the presence of inhibitor (administered by multiple oral dosing) is the commonly used metric for an in vivo drug-drug interaction. Assuming that hepatic metabolism of the victim drug by a single pathway is the only route of elimination, the AUC ratio for an orally administered substrate in the presence and absence of inhibitor reflects the ratio of the control and inhibited intrinsic clearances. It is also assumed that the conditions of the “well stirred” liver model apply and that there is no impact of inhibitor on either the fraction absorbed (Fa) or the fraction unbound in plasma (fu) for the substrate. Thus, in theory, the change in clearance, and hence AUC, may be predicted from the inhibitor concentration available to the hepatic enzymes in vivo [I] and the in vitro Ki (Ito et al., 1998, 2004; Yao and Levy 2002). 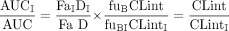
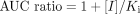 where subscript I indicates the presence of the inhibitor.
where subscript I indicates the presence of the inhibitor.
Although the use of eq. 2 to describe the degree of in vivo interaction between two drugs is widespread (von Moltke et al., 1994; Ito et al., 1998; Davit et al., 1999; Lin, 2000; Rodrigues, 2001; Tucker et al., 2001; Yao and Levy, 2002) and Ki values can be readily obtained from human liver microsomal studies, there is no general agreement over the selection of surrogate inhibitor concentration to reflect [I] (Tucker et al., 2001; Ito et al., 2002). In a recent analysis (Ito et al., 2004), the use of hepatic input concentration gave the best resolution between positive and negative interactions and, most importantly, eliminated false negatives. It was also noted that there was a high incidence of false positive predictions, and many true positives were substantially over-predicted. One possible reason for these limitations is the assumption that there is a single metabolic pathway responsible for the elimination of the victim drug. In practice, this is frequently not the case; renal clearance of unchanged drug, parallel pathways of metabolism, and involvement of multiple P450s for the same reaction may all contribute to elimination. Thus, incorporation of the fraction metabolized by the particular P450 enzyme subject to inhibition (fmCYPi) may be a critically important consideration. Although the kinetic consequences of parallel elimination pathways have been alluded to by various investigators (Rowland and Matin, 1973; Shaw and Houston, 1987; Ito et al., 1998; Yao and Levy, 2000; Rodrigues et al., 2001), there has been no comprehensive evaluation of its impact. In general, accurate values of fmCYPi in vivo are not known, but in the case of CYP2D6 substrates, substantial information is often available from genetic polymorphism studies. By comparison of the poor and extensive phenotypes, a direct measure of the importance of CYP2D6 (that is, fmCYP2D6) can be made. Previously, we (Ito et al., 2004) collated a number of in vivo studies involving CYP2D6 inhibition to assess the prediction of DDIs from the [I]/Ki ratio (see Fig. 1). These data are now used to explore the impact of parallel pathways on the prediction of DDIs. We also investigate the theoretical basis of the significance of parallel pathway and multiple P450 involvement and provide simulations of DDI based on a simple pharmacokinetic model. We demonstrate that prediction of DDIs from inhibitor characteristics (namely, [I] and Ki) is substantially improved by taking into account the fmCYPi for the victim drug.

Identification of the substrates in the 44 in vivo interaction studies involving CYP2D6 inhibition predicted from [I]/Ki ratios. Numbers in parentheses indicate the numbers of studies. Studies can be categorized into four zones: true positives (AUC ratio >2, [I]/Ki >1), true negatives (AUC ratio <2, [I]/Ki <1), false positives (AUC ratio <2, [I]/Ki >1), or false negatives (AUC ratio >2, [I]/Ki <1). The division between the four zones is shown by the dotted lines.
Materials and Methods
Impact of Parallel Metabolic/P450 Pathways of Drug Elimination on DDIs as Assessed by AUC Ratio. For the majority of drugs, elimination occurs via more than one pathway of metabolism involving more than one enzyme (frequently P450 enzymes) with a minor degree of renal clearance. Under such circumstances, hepatic intrinsic clearance, assuming a linear metabolism (substrate concentration much smaller than the Km), can be defined as the sum of the ratios of the Michaelis-Menten parameters (Vmax and Km) for the individual pathways/enzymes. In the simplest case of one P450 pathway and an undefined but distinct second pathway,  where 1 and 2 refer to a particular P450 pathway (e.g., CYP2D6) and other pathways, respectively.
where 1 and 2 refer to a particular P450 pathway (e.g., CYP2D6) and other pathways, respectively.
Case 1: Only One Pathway Subject to Inhibition. If the inhibitor does not affect both metabolic pathways, the intrinsic clearance in the presence of the inhibitor (CLintI) can be expressed as follows, again assuming linear metabolism, since the substrate concentration does not approach the Km and hence the distinction between competitive and noncompetitive inhibition mechanisms is not required:  where [I] is the inhibitor concentration, and Ki is the inhibition constant. Therefore, the ratio of the CLint in the presence and absence of the inhibitor is expressed by the following equation:
where [I] is the inhibitor concentration, and Ki is the inhibition constant. Therefore, the ratio of the CLint in the presence and absence of the inhibitor is expressed by the following equation: 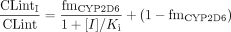
Combining the eqs. 1 and 5, the following equation describes the AUC increase by the inhibitor: 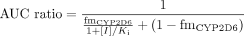
Case 2: Both Pathways Subject to Inhibition. If the inhibitor also inhibits the second pathway of substrate, then eq. 4 can be arranged as follows:  where Ki,1 and Ki,2 represent the Ki values for CYP2D6 and the second pathway, respectively. Therefore, the AUC increase by the inhibitor is expressed by the following equation, which is analogous to eq. 6:
where Ki,1 and Ki,2 represent the Ki values for CYP2D6 and the second pathway, respectively. Therefore, the AUC increase by the inhibitor is expressed by the following equation, which is analogous to eq. 6: 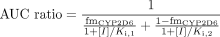
Simulations of AUC Ratio. First, the effect of a parallel elimination route was simulated according to eq. 6 for a range of values for fmCYP2D6 from 0.5 to 1. The effect of a parallel pathway subject to inhibition was simulated according to eq. 8, maintaining the fmCYP2D6 value at 0.8 and changing the relative affinity of the inhibitor for the two P450s, expressed as Ki,2/Ki, ratios of 1 to 10,000.
The CYP2D6 Paradigm: Estimation of fmCYP2D6 from Phenotype Studies and by Regression. CYP2D6 is known to show genetic polymorphism, and the clearance for CYP2D6 substrates in the extensive metabolizers (EMs) and poor metabolizers (PMs) can be expressed by eqs. 9 and 10, respectively, assuming that the CYP2D6 pathway is absent in the PMs: 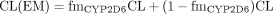
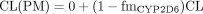 From these two equations, the following relationship between the fmCYP2D6 and the CL or AUC ratio in EMs and PMs follows:
From these two equations, the following relationship between the fmCYP2D6 and the CL or AUC ratio in EMs and PMs follows:  The values of fmCYP2D6 for nine CYP2D6 substrates were calculated by eq. 11 based on pharmacokinetic information in EM and PM subjects (see Table 1).
The values of fmCYP2D6 for nine CYP2D6 substrates were calculated by eq. 11 based on pharmacokinetic information in EM and PM subjects (see Table 1).
Details on the nine CYP2D6 substrates investigated in the drug-drug interaction studies
To obtain an alternative value of fmCYP2D6, the interaction data for each CYP2D6 substrate (see Fig. 1) were fitted to eq. 6 by nonlinear least-squares regression and the values obtained were compared to fmCYP2D6 from phenotype studies. For the interaction data involving desipramine as substrate, a regression analysis was also performed based on eq. 8 to obtain the best-fit for the Ki,2/Ki,1 ratio with the fixed value of fmCYP2D6 at 0.8. The total hepatic input concentration ([I]in) was used as the inhibitor concentration for all the predictions as this was found to be the most successful in qualitative zoning of DDIs (Ito et al., 2004).
Data Source. Our original database (Ito et al., 2004) contained 58 studies involving 14 substrates and 19 inhibitors. Overall, five substrates were excluded from the analysis due to the lack of phenotype studies or if they involved paroxetine, a mechanism-based inhibitor of CYP2D6 (Bertelsen et al., 2003). Thus, 44 studies were used in the current analysis involving nine substrates and 12 reversible inhibitors.
The in vitro Ki values were previously collected (Ito et al., 2004) and were all obtained against selective CYP2D6 probe reactions; in 25 cases the same probe was used in vitro and in vivo.
Results
Simulations of AUC Ratio for Drugs with Parallel Pathways of Elimination.Figure 2A shows the simulated effect of a parallel elimination route unaffected by the inhibitor on the AUC ratio according to eq. 6. A decrease in the value of fmCYP2D6 (from 1 to 0.5) gives a progressive reduction in the importance of the clearance pathway subject to inhibition up to 50%. The steepness of the curve changes depending on the fmCYP2D6 value, even with minor decreases in fmCYP2D6 (e.g., from 1 to 0.98). For the AUC ratio range covered in Fig. 2A, the maximum inhibitory effect is evident for values of fmCYP2D6 below 0.95. When fmCYP2D6 is reduced to 0.5, a maximum AUC ratio of 2 is predicted, which, for the criteria used in the present analysis, is considered not to constitute a significant DDI (see later). The maximum AUC ratio possible is solely dependent on fmCYP2D6 since eq. 6 will be reduced to eq. 12 when [I]>>Ki (i.e., potent CYP2D6 inhibition resulting in a large [I]/Ki ratio). 
When the second metabolic pathway is also affected by the same inhibitor, the AUC ratio depends also on the Ki ratio between the two pathways (Ki,2/Ki,1). In Fig. 2B, the effect of a parallel metabolic pathway subject to inhibition is simulated according to eq. 8. Fixing the fmCYP2D6 value at 0.8, the simulated line shifts progressively toward the right with the increase in the Ki ratio to finally give a line analogous to the Fig. 2A set as the Ki ratio exceeds 10,000.
The shifts in both the position and the shape of the AUC ratio relationship to [I]/Ki illustrated in Fig. 2, A and B, result in a movement of the borderline between negative and positive DDIs. For the case of fmCYP2D6 equals 1 (eq. 2), an [I]/Ki ratio <1 would represent a low risk (resulting in AUC ratio <2), whereas the [I]/Ki >1 case would be a high risk (AUC ratio >2). However, the critical [I]/KI value that separates negative from positive interactions increases from 1 as fmCYP2D6 is reduced, as can be seen when eq. 6 is solved for an AUC ratio of 2. 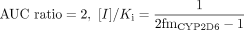
For fmCYP2D6 >0.75, the critical [1]/Ki value is relatively insensitive (between 1 and 2); however, as fmCYP2D6 decreases further, there is a marked increase in the borderline value as the limit of 0.5 is approached (see Fig. 2A, inset).
The CYP2D6 Paradigm. The effect of parallel pathways was first investigated in the interaction cases involving desipramine as the victim drug, since this was the substrate with the largest number of in vivo studies in our database (n = 16). Figure 3A includes studies showing an AUC increase in desipramine after both desipramine and imipramine dosing. The heavy broken line in Fig. 3A is the simulation line using eq. 6 with the fmCYP2D6 estimated from PM data. The light broken line is the regression line based on the same equation. The fmCYP2D6 values obtained from the PM data and regression analysis were comparable, with values of 0.88 and 0.8, respectively. A similar analysis using eq. 6 was performed for metoprolol (Fig. 4), the second most studied victim drug in our database. Although there were 10 studies, no major interactions were observed (AUC ratio ranged from 2.02 to 2.38). The fmCYP2D6 values obtained were 0.83 and 0.7 from PM data and regression approaches, respectively.

Effect of parallel pathways of drug elimination on predicted AUC ratio. A, the case of a secondary pathway unaffected by inhibitor, simulated from eq. 6. Inset shows the dependence of the borderline value between negative and positive interactions on the fraction metabolized by a particular P450. B, the case of a secondary pathway subject to inhibition, simulated from eq. 8 using the fmCYP2D6 value of 0.8.
In addition, the impact of the inhibition of the second pathway was investigated using the desipramine data. The Ki ratio for the inhibition of both pathways was estimated by regression using eq. 8 and fixing the fmCYP2D6 value at 0.8. The line agrees well with the observed data and corresponds to inhibition of the second pathway that is 360 times weaker than the major pathway (Fig. 3B).
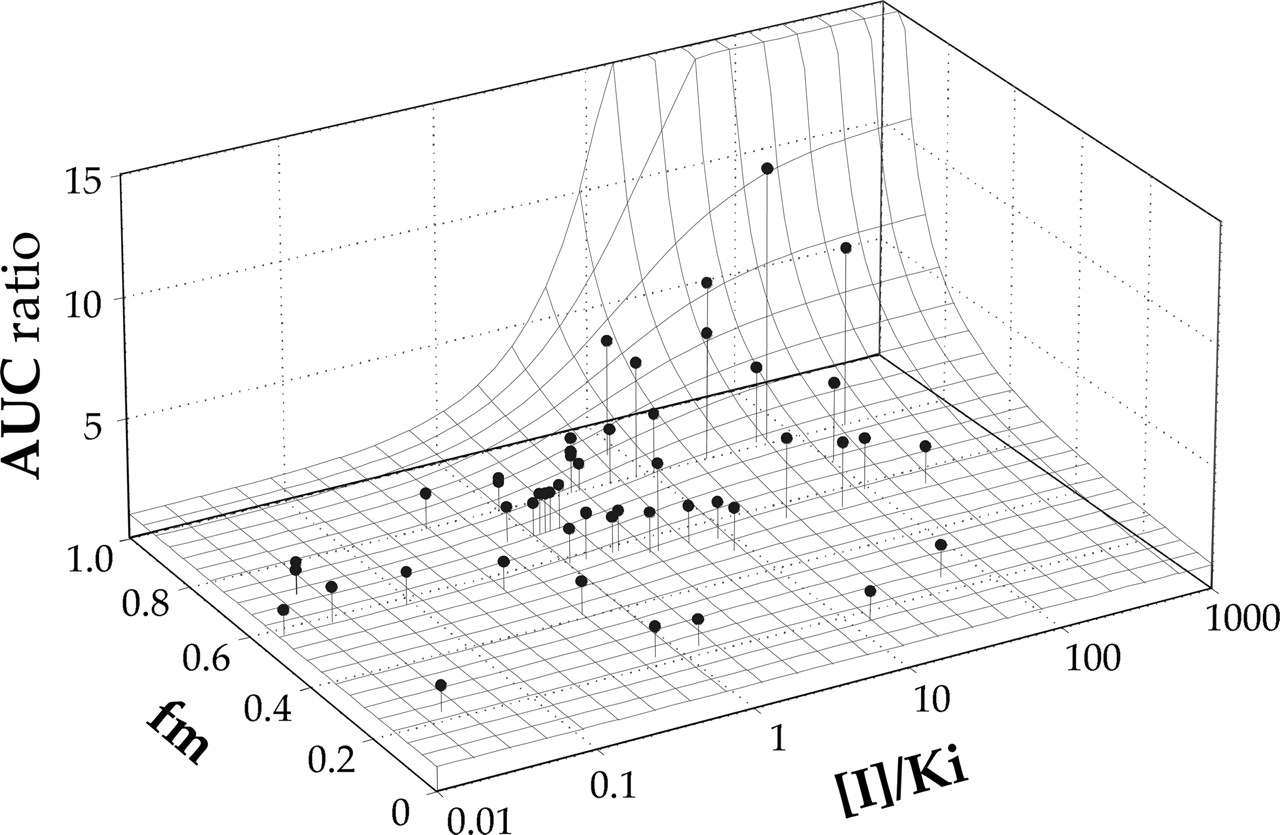
The effect of incorporating fmCYP2D6 in the prediction of AUC ratios for 44 DDI studies is shown in Fig. 5 as a three-dimensional surface. The fmCYP2D6 values for each substrate estimated by regression analysis were in good agreement with the PM-derived data (r = 0.748, p < 0.02; Table 1). For the nine substrates, a range of fmCYP2D6 values of 0.25 to 0.89 was found. The simulated surface is steepest as the fmCYP2D6 decreases from 1 to 0.8 and continues to decrease over the 0.8 to 0.5 range; below 0.5, all AUC ratios are less than 2. This indicates that many DDI over-predictions can be explained by taking parallel pathways into consideration and that false positives will occur when fmCYP2D6 is less than 0.5. This is clearly illustrated by the propranolol studies (fmCYP2D6 0.37).
Table 2 summarizes the improvement in positive and negative DDI zoning achieved by incorporating fmCYP2D6: a marked reduction in false positive prediction (with a corresponding increase in true negatives), with a minor increase in false negative predictions. Overall, the success of identifying true positive and negative DDI increased from 54 to 84%.
Zoning of 44 drug-drug interactions involving CYP2D6 into positive and negative events based on in vitro data
Discussion
The ability to predict a DDI from plasma concentrations of circulating inhibitor and Ki information from an in vitro study is a very attractive option. Based on eq. 2, interactions are regarded to be of low risk if the estimated [I]/Ki ratio < 1, whereas high risk is expected with an [I]/Ki ratio > 1, resulting in an AUC increase of at least 2-fold (Tucker et al., 2001). Hence, predictions can be categorized into four zones: true positives (AUC ratio >2, [I]/Ki >1), true negatives (AUC ratio <2, [I]/Ki <1), false positives (AUC ratio <2, [I]/Ki >1), or false negatives (AUC ratio >2, [I]/Ki <1), as illustrated in Fig. 1. However, this zoning is based on the assumption that there is complete metabolism of the drug by a single pathway and a single P450 enzyme. Figure 2A demonstrates the progressive increase in the critical [I]/Ki ratio (for an AUC ratio of 2) with fmCYPi, illustrating that this positive-negative interaction borderline value is substrate dependent. False negative and positive predictions will result if this factor is not appreciated.
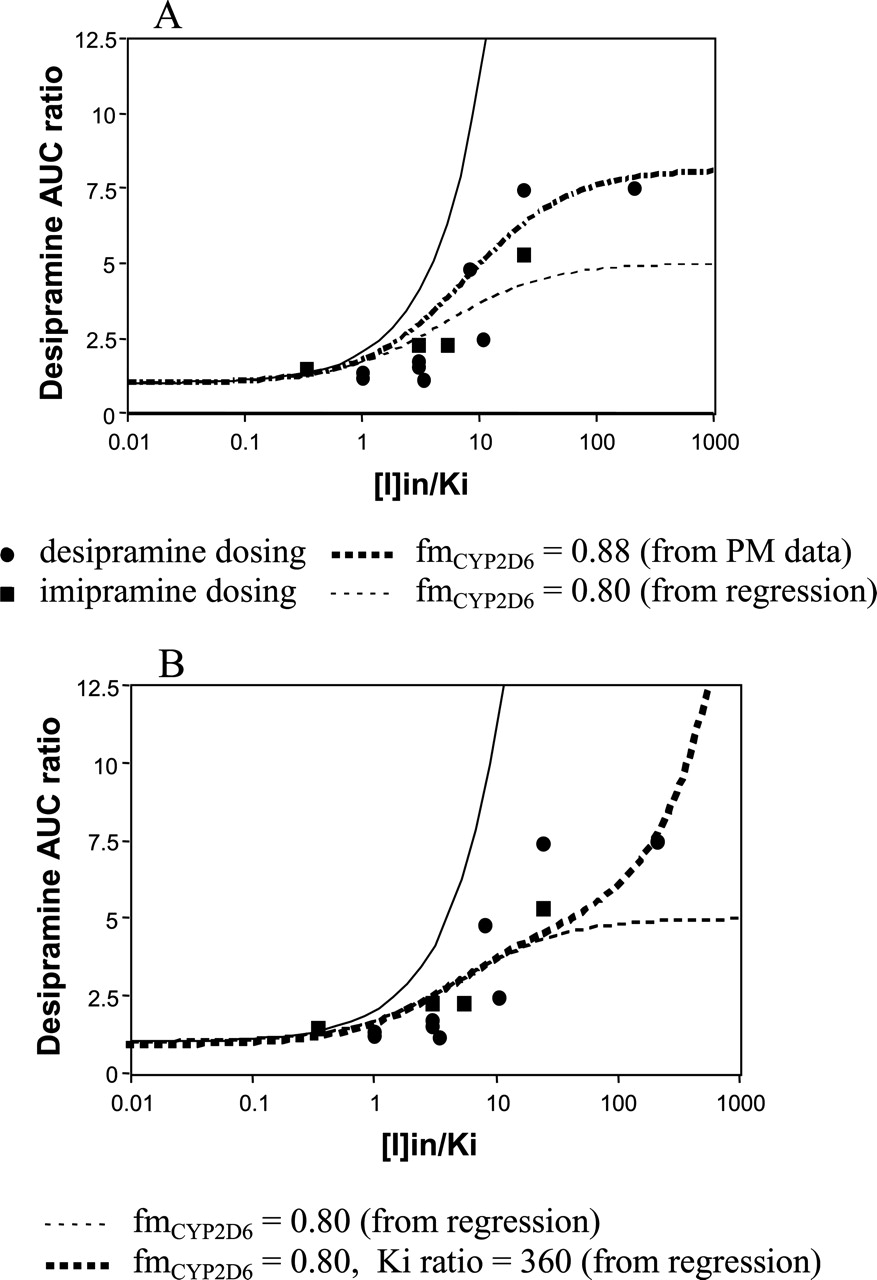
Analyses of the 16 desipramine drug-drug interaction studies taking into account the fraction metabolized by CYP2D6. A, no inhibition of the parallel metabolic pathway; B, parallel pathways subject to inhibition.
There have been numerous reports in the literature on the use of the [I]/Ki ratio approach (usually assuming a fmCYPi of 1), and they have been of mixed success. Recently, we (Ito et al., 2004) compiled a database of 193 DDIs for which in vitro data were available in addition to in vivo inhibitor concentrations. The study showed good agreement on a qualitative level between the prediction from in vitro data and clinical observations. This systematic approach highlighted the importance of the selection of inhibitor concentration, whether this is an average systemic circulating concentration, a maximum hepatic portal vein concentration during the absorption process, a total drug concentration, or an unbound concentration. In terms of qualitative zoning of positive and negative DDI, the most successful estimate of [I] was the total input concentration, i.e., the total concentration of an inhibitor in the hepatic portal vein (Kanamitsu et al., 2000). It was demonstrated that once mechanism-based inhibitors were excluded from the analysis, no false negative results were obtained. In terms of predicting positive interactions, more varied success was achieved, with a number of false positives and the majority of true positives being over-predicted. It was clear from this analysis that in order to predict DDIs quantitatively, additional considerations were necessary; hence the need for the present study that systematically explores the fraction metabolized by a particular cytochrome P450 enzyme (fmCYPi) as a substrate-dependent parameter.
The potential significance of parallel pathways of drug elimination in DDI prediction was pointed out as early as 1973 (Rowland and Matin, 1973). In more recent years, reviewers of the area have also cited parallel elimination pathways and multiple P450 involvement as an important consideration (Yao and Levy 2000; Rodrigues et al., 2001; Rostami-Hodjegan and Tucker, 2004). However, there has been little substantial analysis to date to support this claim. We have selected a number of CYP2D6 DDI studies (n = 44) as appropriate test cases in view of the validity of the fmCYPi parameter obtained for these substrates. By using genetic phenotyping studies and comparing PM and EM data, we were able to obtain unequivocal estimates of the importance of CYP2D6 to the elimination of the substrates investigated. For the nine substrates studied, the fmCYP2D6 from PM studies varied from 0.37 (for propranolol) to 0.94 (for tolterodine). A series of simulations illustrated the importance of this range of fmCYP2D6 on the predicted AUC ratio. This finding was confirmed using information from 44 clinical studies, and we were able to increase success from 54% to 84% for positive and negative predictions and to reduce the number of over-predictions (as summarized in Fig. 5).

Analyses of the 10 metoprolol drug-drug interaction studies taking into account the fraction metabolized by CYP2D6.

Three-dimensional surface for the relationship between the AUC ratio and the [I]/Ki and fmCYP2D6 for 44 drug-drug interaction studies.
The present study highlights the critical importance of fmCYPi values >0.5. If the particular enzyme under examination contributes less than 50% of the total clearance, then it may be argued that there is little reason for concern over DDIs or, indeed, genetic polymorphisms. The maximum consequence of either phenomenon will be a doubling of AUC. Equation 12 indicates that the maximum effect can be predicted simply from the fmCYPi term. Although this outcome will be true for the vast majority of individuals, it is of interest to speculate on the possible impact on extreme individuals in the population for DDIs involving drugs whose metabolic clearance results from both a polymorphic enzyme (e.g., CYP2D6) and a nonpolymorphic enzyme (e.g., CYP3A4). Although inhibition of the polymorphic enzyme may be of minimal consequence when fmCYPi is low (say 25%), inhibition of the nonpolymorphic enzyme may cause substantial effects and, more importantly, these may be very varied. The latter situation will result from the dependence of clearance on the activity of the polymorphic enzyme in the absence of the major route of metabolite clearance. Hence the importance of genetic polymorphism in DDIs may not be limited to direct inhibition effects.
Certain inhibitors in the CYP2D6 DDI database used (e.g., fluoxetine, sertraline; see Ito et al., 2004) have inhibitory metabolites; thus, the consequence of multiple inhibitors needs to be considered. As indicated (Rostami-Hodjegan and Tucker, 2004), it is important to distinguish whether the metabolite, or indeed any second inhibitor, will act on the same or different enzyme. If the former situation occurs, then the cumulative effect will be considerably less than in the latter situation. In our simulations, we were able to identify the consequences of a secondary inhibitory effect on the clearance of desipramine (see Fig. 3B) despite only minor potency on the second enzyme (indicated by a Ki ratio of 360).
Application beyond CYP2D6 to other polymorphic P450s (e.g., CYPC19, CYP2C9) should be readily achievable either by the method used here (comparison of PM and EM data) or by use of selective inhibitors in EM subjects to “phenocopy” the PM status. Extending this work to other P450s that do not show polymorphic metabolism is more problematic due to the need for an estimate of fmCYPi. The total fraction metabolized can be readily determined once the fraction of drug excreted unchanged via either renal clearance or other mechanisms is known. Considerable effort, including the use of radiolabeled drugs, is routinely expended to resolve issues of mass balance, oral absorption, and fecal/biliary excretion. More specific information from urinary recovery of drug metabolites may be obtained provided the complexity of the metabolic pathways allows this resolution. If sequential metabolism occurs from more than one primary metabolite to a common secondary metabolite, it may be impossible to quantitatively assess the importance of particular drug clearance routes. However, the key information needed is assignment of particular enzymes to those drug pathways; that is, fmCYPi rather than fraction metabolized by a particular pathway. The importance of this specific information is being increasingly realized (Bjornsson et al., 2003; Williams et al., 2003). The use of data from reaction phenotyping may be very useful in this area, but to date, this has tended to be more of a qualitative measure than a quantitative one (McGinnity et al., 2000; Lu et al., 2003). However, the success apparent with CYP2D6 substrates would indicate that the above are worthy of detailed consideration, since even approximate fmCYPi values may markedly improve a prediction. The use of fuzzy set theory (Nestorov et al., 2002) may provide a methodology to incorporate semiquantitative or qualitative estimates of fmCYPi.
The total hepatic input concentration was found to be the most useful estimate of [I] to predict the AUC ratio from the in vitro Ki value. This apparent need to ignore the consequences of plasma protein binding is surprising but similar to our previous experience with a larger database (Ito et al., 2004). Incorporation of fmCYP2D6 improves the identification of true positive and negative DDIs from 54% to 84%. Consideration of the false positives and negatives (Table 2) are particularly informative, the former reducing from 20 of 44 to 4 of 44. However, there are some false negatives (3 of 44) when fmCYP2D6 is included in the prediction and these particular studies deserve further comment. One study involves inhibition of propranolol by propafenone (observed AUC ratio of 2.13) and two studies inhibition of imipramine by fluoxetine and fluvoxamine (observed AUC ratio of 3.33 and 3.63, respectively). For both substrates, fmCYP2D6 is less than 0.5, and hence, AUC ratios would not have been predicted to be greater than 2. It is of interest that the eight other propranolol studies and the three other imipramine studies that are part of this 2D6 DDI compilation are true negative DDIs. Therefore, inaccurate fmCYP2D6 values appear not to be the explanation for these three anomalies. Inhibition of the other P450s responsible for the metabolism of both substrates (CYP1A2, CYP2C19, CYP2C9, and CYP3A4) is the likely explanation. For fluoxetine and fluvoxamine, this has been comprehensively documented (Olesen and Linnet, 2000; Hemeryck and Belpaire, 2002) but propafenone studies appear to have been limited to CYP1A2 (Kobayashi et al., 1998).
In conclusion, consideration of parallel elimination pathways and multiple P450 involvement provides extra information that improves prediction of CYP2D6 DDI. Incorporation of fmCYP2D6, readily available for CYP2D6 from PM and EM data, reduces the number of false positives and also the extent of quantitative over-predictions of true positives. This provides a valuable step forward in making quantitative predictions of DDIs, which, to date, have placed emphasis on delineating the inhibitor characteristics (namely [I] and Ki) and relatively little attention on the victim drug properties.
Footnotes
-
Financial support for this project was provided by the following Centre for Applied Pharmacokinetic Research (CAPkR) Consortium members: Bristol Myers-Squibb, GlaxoSmithKline, Novartis, Pfizer, Roche, and Servier. Part of this study was presented at the 8th European ISSX Meeting, Dijon, April 27–May 1, 2003 and was published in abstract form in Drug Metab Rev35 (Suppl 1):49.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.003715.
-
ABBREVIATIONS: DDI, drug-drug interaction; AUC, area under the plasma concentration-time curve; CLint, intrinsic clearance; EM, extensive metabolizer; fmCYPi, fraction metabolized by a particular P450 enzyme i; [I], concentration of inhibitor available to the enzyme; PM, poor metabolizer.
- Received January 14, 2005.
- Accepted March 9, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}