


Abstract
Cytochrome P450 3A4 (CYP3A4) is a major determinant of the metabolism of many drugs, including important anticancer drugs, with sometimes profound impact on therapeutic efficacy and toxic side effects. To study in vivo CYP3A(4) functions, we have generated and characterized transgenic mice with functional expression of human CYP3A4 cDNA in the liver. Two transgenic lines displayed substantial, physiologically relevant and stable CYP3A4 levels in liver and moderate levels in kidney, but not in small intestine. The mice did not display obvious physiological abnormalities. The CYP3A4 substrate drugs midazolam and cyclosporin A were used to test functional activity of CYP3A4 in liver. The area under the plasma concentration versus time curve (AUC) of intravenously administered midazolam (30 mg/kg) was 2.2-fold decreased in the transgenic mice compared with wild-type (5.45 ± 0.21 versus 11.7 ± 0.46 μg · hml–1; P < 0.01), and early formation of the primary metabolite 1-hydroxymidazolam was about 2-fold increased, demonstrating the functionality of CYP3A4 in the liver. Similarly, following intravenous administration of cyclosporin A (20 mg/kg), CYP3A4 transgenic mice displayed a reduced plasma AUC compared with wild-type (24.3 ± 0.66 versus 35.8 ± 0.53 μg · hml–1; P < 0.01). Thus, midazolam and cyclosporin A, compounds with markedly different clearance rates and half-lives, both demonstrated clearly accelerated kinetics in the CYP3A4 transgenic mice. We expect that this CYP3A4 transgenic model will provide a useful tool to study the impact of CYP3A4 on drug levels, especially when combined with other transgenic and knockout strains.
Cytochrome P450 3A (CYP3A) enzymes are heme-containing monooxygenases responsible for the oxidative metabolism of many endogenous and xenobiotic compounds. They are the most abundant P450s in human liver and small intestine and are involved in the metabolism of, among others, toxins, carcinogens, steroid hormones, and more than 50% of the drugs used in the clinic today (Guengerich, 1999). In humans, four functional CYP3As have been described (Lamba et al., 2002), of which CYP3A4 is generally the most abundant hepatic and intestinal form, accounting on average for 95% of the combined liver CYP3A mRNA pool in white people (Koch et al., 2002).
CYP3A expression varies as much as 40-fold in liver and small intestine donor tissues (Lamba et al., 2002). Through interaction with nuclear receptors, numerous drugs such as dexamethasone and rifampicin, endogenous CYP3A substrates such as bile acids (lithocholic acid), and food constituents such as vitamin D induce CYP3A (Pascussi et al., 2003). Additionally, CYP3A can be inhibited by food constituents (grapefruit juice) or drugs (HIV protease inhibitor ritonavir) (Dresser et al., 2000). These factors, together with genetic polymorphisms in CYP3A enzymes, contribute greatly to variation in oral availability and systemic clearance of CYP3A substrates. This variation is particularly undesirable for substrate drugs with narrow therapeutic indices, such as many cancer therapeutics and immunosuppressants. This is illustrated by St. John's wort, an herbal medicine that is frequently used for therapy of mild depression, which triggers adverse drug interactions with oral contraceptives, the HIV protease inhibitor indinavir, and the immunosuppressant cyclosporin A, as a consequence of activating CYP3A (and P-glycoprotein) drug-handling systems (Dresser et al., 2003; Izzo, 2004). The effects can be dramatic, including transplant rejection due to insufficient levels of cyclosporin A (Ruschitzka et al., 2000). It is thus important to systematically assess the in vivo impact of CYP3As on drug pharmacology, to minimize possible toxicity or inefficacy of drugs.
Midazolam is a frequently used probe drug for assessment of in vivo CYP3A(4) activity and widely used in clinical practice for preoperative sedation, induction, and maintenance of anesthesia, and sedation of patients in intensive care units. It is mainly metabolized by CYP3A4 and, in contrast to cyclosporin A, is not a substrate of P-glycoprotein.
To obtain useful model systems to study the in vivo impact of CYP3As on drug pharmacology, we have generated and characterized transgenic mice with substantial functional expression of human CYP3A4 cDNA in the liver. Using this transgenic model, we explored CYP3A4-dependent kinetics of midazolam and cyclosporin A in vivo, illustrating its potential for use in drug development and understanding of CYP3A metabolism.
Materials and Methods
Animals. Mice were housed and handled according to institutional guidelines complying with Dutch legislation. Mice (FVB/N) used in experiments were male and between 9 and 14 weeks of age. Animals were kept in a temperature-controlled environment with a 12-h light/dark cycle and received semisynthetic chow (Reference diet 20% casein, 4068.02; Hope Farms, Woerden, The Netherlands) and acidified water ad libitum.
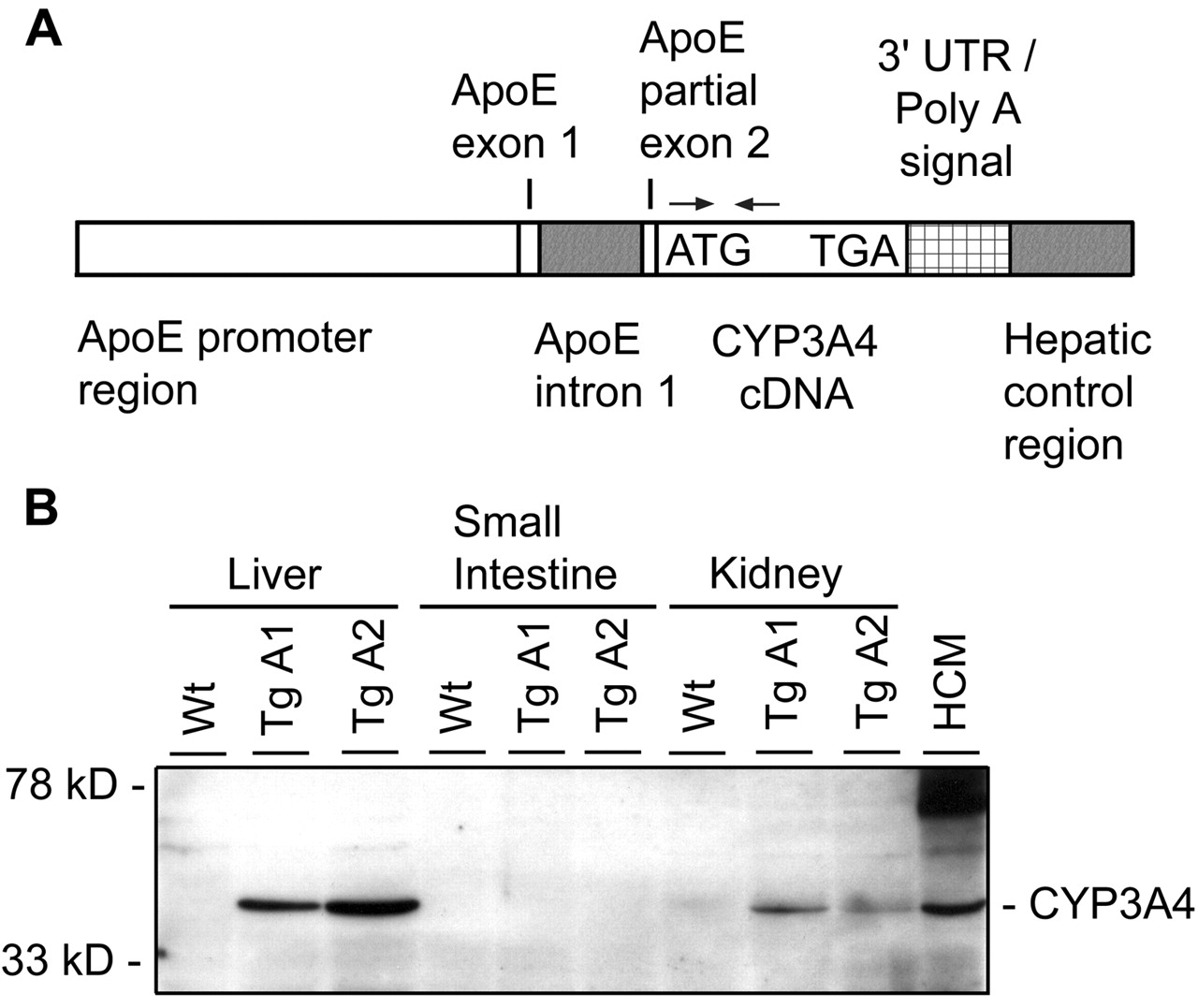
Transgene Construction. Liver-specific expression of human CYP3A4 was achieved by generating the following transgene construct (Fig. 1A). pLiv-LE.6 (kindly provided by J. Taylor, Gladstone Institute, University of California, San Francisco, San Francisco, CA) (Simonet et al., 1993) was digested with SalI and Asp718, releasing the 5′ segment of the human ApoE promoter region (including exon 1, intron 1, and part of exon 2 of ApoE) of ∼3.8 kb (ApoE), which was ligated in SalI- and Asp718-cut pSL1180 (Pfizer Inc., Cappelle a/d IJssel, The Netherlands). Wild-type human cytochrome P4503A4 cDNA was released as an ∼2.1-kb XbaI fragment from pUV1-CYP3A4 (GenBank accession number M18907; kindly provided by Dr. F. Gonzalez, National Institutes of Health, Bethesda, MD) and blunt ended. pSL1180-ApoE was digested with HpaI and blunt ended, and CYP3A4 was ligated into the HpaI site. pSL1180-ApoE-CYP3A4 clones were digested with SnaBI, and an ∼1-kb blunted 3′ polyadenylation signal sequence-hepatic control region 1 (HCR1), released from pLiv-LE6 by digestion with ClaI and SmaI, was inserted, yielding pSL1180-ApoE-CYP3A4-polyA-HCR1 (Fig. 1A). NotI fragments of this clone were used to inject pronuclei of FVB/N mice. Two-cell stage embryos were implanted into oviducts of pseudopregnant F1 fosters and carried to term.

A, structure of ApoE promoter-HCR1-driven expression cassette, containing human CYP3A4 cDNA. Functional elements are represented approximately to scale. Translational start and stop for CYP3A4 are indicated. Arrows indicate the primers used for PCR detection of the transgene. B, expression of human CYP3A4 in liver and kidney, but not in small intestine, of CYP3A4 transgenic mouse lines A1 and A2 (TgA1 and TgA2) as detected by Western blotting. Wt, wild-type; HCM, human crude liver membrane. Crude membrane protein (20 μg) was analyzed. Total protein staining (Ponceau S and India Ink) confirmed equal loading across the lanes (not shown).
PCR and Southern Analysis. Transgenic founder lines were detected by initial PCR screen with forward 5′-AGCAAAGAGCAACACAGAG-3′ and reverse 5′GACCATCATAAAAGCCCCAC-3′ primers located within the CYP3A4 cDNA to yield a 300-bp band (Fig. 1A). Southern analysis was used for definitive confirmation of transgenic founders. DNA was extracted from ear snips or tail tips of mice (Laird et al., 1991). An ∼2.1-kb XbaI CYP3A4 cDNA fragment was used as probe.
Western Analysis. Crude membrane fractions were prepared from mouse liver, kidney, and small intestine as described (Ogihara et al., 1996). Blots were probed with RDI-CYP3A4Mabr or RDI-CYP3A4abr (1:1000) (Research Diagnostics, Flanders, NJ), followed by horseradish peroxidase-labeled antibody (Santa Cruz Biotechnology, Santa Cruz, CA, or Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK). The microsomal protein was quantified by the Bio-Rad protein assay based on the method of Bradford (Bio-Rad, Veenendaal, The Netherlands). Crude membrane protein (20 μg) was analyzed. Equal loading across the lanes was confirmed with total protein staining (Ponceau S and India Ink). Quantification of transgenic CYP3A4 was assessed with dilution series, by comparison with a human CYP3A4 standard (cat. 456202, BD Biosciences, Alphen aan den Rijn, The Netherlands).
Clinical Chemical Analysis of Plasma. Standard clinical chemistry analyses on plasma were performed on a Hitachi 911 analyzer to determine levels of bilirubin, alkaline phosphatase, aspartate aminotransaminase, alanine aminotransaminase, γ-glutamyl transferase, lactate dehydrogenase, creatinine, urea, Na+, K+, Ca2+, phosphate, total protein, albumin, and cholesterol.
Pharmacokinetic Experiments. Midazolam (Dormicum; Roche Diagnostics, Almere, The Netherlands) (30 mg/kg) was dissolved in phosphate-buffered saline. Cyclosporin A (Sandimmun; Novartis, Arnheim, The Netherlands) (20 mg/kg) was dissolved in 5.2% (w/v) Cremophor EL in phosphate-buffered saline. These compounds were injected (bolus) into the tail vein of mice lightly anesthetized with methoxyflurane (Metofane; Medical Developments Australia Pty. Ltd., Springvale, VIC, Australia). Transgenic strains A1 and A2 were included in the pharmacokinetic studies. At indicated time points, blood samples were taken by cardiac puncture under anesthesia with methoxyflurane, after which mice were sacrificed by cervical dislocation (n = 3–5 for each time point). Midazolam, 1-hydroxymidazolam, and 4-hydroxymidazolam were determined in murine plasma by gas chromatography-mass spectrometry as modified from Thummel et al. (1994) and Eeckhoudt et al. (1998). To a 100-μl plasma sample, 800 μl of 0.02% (w/v) sodium hydroxide, 20 μl of internal standard solution (1.6 μg/ml diazepam for midazolam and 1 μg/ml temazepam for the hydroxy metabolites, in methanol) and 5 ml of a diethyl ethercyclohexane mixture (70/30 v/v) were added. After shaking for 10 min, the mixture was centrifuged 10 min at 4°C and 2000g. After storage for 1 h at –30°C, supernatant was evaporated under a stream of nitrogen at 40°C and additionally dried at 70°C for 5 min. Samples were reconstituted in 50 μl of 20% (v/v) t-butyl-dimethylsilyl-trifluoroacetamide in acetonitrile using vortex mixing and transferred to a 200-μl injection vial. Prior to injection of 3 μl into the gas chromatography-mass spectrometry apparatus, samples were heated for 2 h at 70°C. Three-microliter splitless injections (splitless time = 1 min) were made on a BPX5 column (44 mm × 0.25 mm, film thickness = 2.5 μm; SGE, Bester, The Netherlands). The constant helium flow was 1.1 ml/min at 140°C, and injector temperature was 260°C. The column temperature was raised 1 min after injection from the initial 85°C to 250°C at 30°C/min and further to 325°C at 15°C/min. Finally, the column was maintained at the final temperature for 4 min before full-speed cooling to 85°C. The mass spectrometry inlet probe was kept at 280°C, and the ions used for quantification were m/z 310 for midazolam, 398 for t-butyl-dimethylsilyl-1-hydroxymidazolam and t-butyldimethylsilyl-4-hydroxymidazolam, 256 for diazepam, and 357 for t-butyldimethylsilyl-temazepam. Calibration was performed in the 100 to 20,000 ng/ml range for midazolam and 1-hydroxymidazolam and in the 20 to 1000 ng/ml range for 4-hydroxymidazolam using peak area ratios of the analytes and their internal standard. Data were fitted by a least-squares power function because of the nonlinear response. Cyclosporin A was detected in whole blood samples by fluorescence polarization immunoassay, as previously described (Malingré et al., 2001).
Pharmacokinetic Calculations and Statistical Analysis. Concentrations are given as average ± standard deviation (n = 3–5, each time point). Averaged concentrations for each time point were subsequently used to calculate the area under the plasma (or blood concentration) versus time curve (AUC) from time = 0 to the last sampling point by the linear trapezoidal rule; standard errors (S.E.) were calculated by the law of propagation of errors (Bardelmeijer et al., 2000). The elimination constant (k) and the S.E.k were calculated by linear regression analysis of the natural log concentration versus time data points of the log-linear part of the concentration-time curve. The terminal half-life (t1/2) and S.E.t½ were calculated by the formulas: t1/2 = ln(1/2)/k and S.E.t½; = t1/2 · S.E.k/k. The plasma clearance (CL) and S.E.CL were calculated by the formulas: CL = dose/AUCi.v. and S.E.CL = CL · S.E.AUC/AUC. A two-tailed unpaired Student's t test was used to assess the significance of difference between two sets of data.
Results
Stable Liver-Specific Expression of Human CYP3A4 in Transgenic Mice. CYP3A4 transgenic mice were made using an ApoE promoter-HCR1 driven expression cassette, containing human CYP3A4 cDNA (Fig. 1A), aiming primarily for protein expression in the liver. The ATG translation start is located within the CYP3A4 cDNA. PCR and Southern analysis confirmed integration of the ApoE-CYP3A4-polyA-HCR1 transgenic construct into the genome (data not shown). Two independent founder lines were generated (A1 and A2) and each was inbred to obtain homozygous lines as determined by Southern analysis. Homozygous CYP3A4 transgenic mice were fertile, their life spans and body weights were not different from wild-type, and they were born at the expected Mendelian ratio. Clinical chemistry analysis of plasma did not reveal any abnormalities. Crude membrane fractions of liver, small intestine, and kidneys of the two strains were analyzed for expression of CYP3A4 by Western blotting. Transgenic CYP3A4 was expressed in high amounts in the liver of these two strains and had a mobility identical with that of CYP3A4 from a human crude liver membrane fraction (Fig. 1B). Assessed by a dilution series and compared with a human standard, the quantity of transgenic CYP3A4 in the crude membrane fraction was roughly 17 pmol/mg microsomal protein (not shown) and roughly 2-fold higher than in a human crude liver membrane fraction (Fig. 1B). Furthermore, as intended, we found no expression of the transgene in small intestine (Fig. 1B). The transgene was also moderately expressed in kidney, with somewhat higher expression in the A1 compared with the A2 line (Fig. 1B). An independent CYP3A4 antibody yielded similar results (not shown). Transgenic CYP3A4 expression was monitored over approximately eight mouse generations and was found to be stable (data not shown).

CYP3A4 expression in transgenic mice results in increased in vivo metabolism of midazolam and cyclosporin A. Plasma concentration versus time curves of midazolam (A), 1-hydroxymidazolam (B), and 4-hydroxymidazolam (C) after intravenous midazolam administration (30 mg/kg) are shown in wild-type and CYP3A4 transgenic mice. (Note the different axis scales for 4-hydroxymidazolam.) D, blood concentration versus time curve of cyclosporin A after intravenous administration (20 mg/kg) to wild-type and CYP3A4 transgenic mice. n = 3–5 for each time point. ★, P < 0.05; ★★, P < 0.01; ★★★, P < 0.001. Insets in A and D show semilog plots of the data.
CYP3A4 Expression in Transgenic Mice Results in Increased in Vivo Metabolism of Midazolam. We tested functionality of the transgenic CYP3A4 by measuring midazolam kinetics in CYP3A4 transgenic versus wild-type mice. Since midazolam is not a P-glycoprotein substrate and its clearance correlates with hepatic microsomal CYP3A content (Thummel et al., 1994), it is commonly used as an in vivo probe for CYP3A activity. It is metabolized by CYP3A to its primary metabolite 1-hydroxymidazolam and to a lesser extent to 4-hydroxymidazolam and 1,4-dihydroxymidazolam. At various time points after intravenous midazolam administration, blood samples were taken and plasma midazolam, 1-hydroxymidazolam, and 4-hydroxymidazolam concentrations were determined (Fig. 2). The AUC for midazolam in the CYP3A4 transgenic mice was 2.2-fold decreased compared with wild-type mice (5.45 ± 0.21 versus 11.7 ± 0.46 μg · hml–1; P < 0.01; Fig. 2A). Plasma midazolam levels decreased faster in transgenic mice compared with wild-type mice, as illustrated by a semilog plot (Fig. 2A). In addition, the AUC of the primary 1-hydroxy metabolite was higher in the CYP3A4 transgenic mice compared with the wild-type (14.2 ± 0.42 versus 10.6 ± 0.60 μg · hml–1; P < 0.05) (Fig. 2B), and metabolite formation was faster: within 15 min, plasma levels of 1-hydroxymidazolam surpassed the parent drug concentration. In wild-type mice this crossover point was only reached after 70 min (compare Fig. 2, A and B). Only a small amount of the minor 4-hydroxy metabolite was formed, and AUCs were not significantly different between CYP3A4 transgenic and wild-type mice (0.42 ± 0.06 versus 0.66 ± 0.03 μg · h ml–1; P = 0.07; Fig. 2C).
Human CYP3A4 Increases Cyclosporin A Clearance in Vivo. At several time points after 20 mg/kg intravenous cyclosporin A administration to CYP3A4 transgenic and wild-type mice, blood samples were taken and analyzed. There was a 1.5-fold lower blood AUC of cyclosporin A in CYP3A4 transgenic mice compared with wild-type mice (24.3 ± 0.66 versus 35.8 ± 0.53 μg · h ml–1; P < 0.01; Fig. 2D). Thus, midazolam and cyclosporin A, compounds with marked differences in clearance rates and half-lives, both demonstrated clearly accelerated kinetics in the CYP3A4 transgenic mice (Table 1). Note, that k values were calculated from the log-linear part of the concentration time curve, including the time points from 15 min after administration of midazolam and cyclosporin A.
Pharmacokinetic parameters estimated for midazolam and cyclosporin A in CYP3A4 transgenic and wild-type mice
Mice received midazolam (30 mg/kg) or cyclosporin A (20 mg/kg) by intravenous administration. Values represent the mean ± S.E.
Discussion
In this study, we describe the development and validation of a transgenic mouse model for studying in vivo hepatic metabolism by human CYP3A4. To avoid the complications of extensive regulatory mechanisms involved in modulation of CYP3A levels, we used the ApoE promoter, which gives stable expression of CYP3A4 in liver. We demonstrated stable DNA integration and protein expression of the CYP3A4 transgenic construct over a number of generations in the liver by Southern and Western analysis, respectively, as well as functional and substantial activity of CYP3A4 in liver. The metabolism and pharmacokinetics of two clinically widely used drugs and CYP3A4 substrates, midazolam and cyclosporin A, were clearly accelerated in the CYP3A4 transgenic mouse lines, demonstrating their potential to study CYP3A4-mediated metabolism in vivo. Especially for drugs with a narrow therapeutic window, such as cyclosporin A, the individual variation in CYP3A activity can cause significant adverse clinical differences in toxicity and efficacy. Recognition of the impact of CYP3A on the pharmacokinetics for drugs is therefore very important.
To demonstrate human CYP3A4 protein expression in the transgenic mice, we have used antibodies that recognize human CYP3A4. However, minor cross-staining with endogenous murine Cyp3as could be detected on prolonged exposures. A quantitative comparison of transgenic human CYP3A4 versus murine Cyp3a expression is not feasible due to different antibody affinities for the respective proteins. The marked additional metabolism of midazolam and cyclosporin A in the CYP3A4 transgenic mice compared with wild-type control mice (i.e., endogenous murine Cyp3a) demonstrates that transgenic CYP3A4 protein is sufficiently highly expressed to allow meaningful in vivo pharmacokinetic analyses.
Besides clear expression of CYP3A4 in the liver of our transgenic mice, we also observed (lower) CYP3A4 expression in the kidney, which is in accordance with Simonet et al. (1993), who demonstrated that the ApoE promoter-HCR1 expression cassette directs high liver gene expression, but also kidney expression. The contribution of this renal CYP3A4 to overall drug metabolism is probably modest compared with the hepatic CYP3A4; expression of CYP3A4 in kidney is clearly lower than in liver as normalized for protein content (especially in the A2 line), and the liver constitutes a much larger fraction of the total body weight of a mouse than kidney.
Both liver and intestine play substantial roles in CYP3A-mediated drug metabolism. This newly generated liver-specific transgenic model will form a useful element of a set of mouse models, in which in vivo CYP3A metabolism can be studied more systematically and in greater detail. Granvil et al. (2003) already described the generation of human CYP3A4 transgenic mice with CYP3A4 expression in the intestine, but not in the liver. Expression of human CYP3A4 in the intestine of these transgenic mice resulted in a 3-fold higher AUC of primary 1-hydroxymidazolam metabolite after oral administration of midazolam, indicating that the human CYP3A4 expressed in the intestine contributed to extrahepatic first-pass metabolism of orally dosed midazolam. Combinations of these (and related) liver- and intestine-specific models will allow systematic studies of the relative, separate, and combined impact of hepatic and intestinal CYP3A4 on the pharmacokinetics and metabolism of drugs.
Although test-tube assays are available that globally indicate to what extent a drug is broken down by CYP3A, this process does not always properly reflect the extent to which a drug is affected in an intact organism. In vivo factors such as blood flow, tissue distribution, and interactions with other drug-handling systems such as drug transporters can further complicate matters. We expect that this newly generated CYP3A4 transgenic mouse model will provide an appropriate tool to study the impact of CYP3A4 on drug levels in an in vivo situation, especially in combination with other transgenic and knockout strains.
Acknowledgments
We thank P. Krimpenfort and D. Meijer for oocyte injection of the ApoE-CYP3A4 cDNA construct and cyclosporin A measurements, respectively.
Footnotes
-
This investigation was supported in part by Grant NKI 2000-2143 of the Dutch Cancer Society.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.004721.
-
ABBREVIATIONS: ApoE, apolipoprotein E; kb, kilobase(s); HCR1, hepatic control region 1; PCR, polymerase chain reaction; AUC, area under the plasma (or blood concentration) versus time curve; CL, plasma clearance.
- Received March 15, 2005.
- Accepted April 20, 2005.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}