


Abstract
Flavin-containing monooxygenases (FMOs) have a significant role in the metabolism of small molecule pharmaceuticals. Among the five human FMOs, FMO1, FMO3, and FMO5 are the most relevant to hepatic drug metabolism. Although age-dependent hepatic protein expression, based on immunoquantification, has been reported previously for FMO1 and FMO3, there is very little information on hepatic FMO5 protein expression. To overcome the limitations of immunoquantification, an ultra-performance liquid chromatography (UPLC)-multiple reaction monitoring (MRM)-based targeted quantitative proteomic method was developed and optimized for the quantification of FMO1, FMO3, and FMO5 in human liver microsomes (HLM). A post-in silico product ion screening process was incorporated to verify LC-MRM detection of potential signature peptides before their synthesis. The developed method was validated by correlating marker substrate activity and protein expression in a panel of adult individual donor HLM (age 39–67 years). The mean (range) protein expression of FMO3 and FMO5 was 46 (26–65) pmol/mg HLM protein and 27 (11.5–49) pmol/mg HLM protein, respectively. To demonstrate quantification of FMO1, a panel of fetal individual donor HLM (gestational age 14–20 weeks) was analyzed. The mean (range) FMO1 protein expression was 7.0 (4.9–9.7) pmol/mg HLM protein. Furthermore, the ontogenetic protein expression of FMO5 was evaluated in fetal, pediatric, and adult HLM. The quantification of FMO proteins also was compared using two different calibration standards, recombinant proteins versus synthetic signature peptides, to assess the ratio between holoprotein versus total protein. In conclusion, a UPLC-MRM-based targeted quantitative proteomic method has been developed for the quantification of FMO enzymes in HLM.
Introduction
Flavin-containing monooxygenases (FMOs; EC 1.14.13.8) are FAD- and NADPH-dependent microsomal enzymes that have a significant role in the metabolism and detoxification of pharmaceutical, endogenous substances, and environmental compounds. FMOs catalyze the oxygenation of soft nucleophilic heteroatom-containing (e.g., N, S, and P) organic substances, converting them to more readily excreted polar metabolites. Five functional human FMO isozymes have been discovered; among these, FMOs 1, 3, and 5 are relevant to hepatic drug metabolism (Krueger and Williams, 2005; Cashman and Zhang, 2006; Mitchell, 2008).
FMO1 and FMO3 are differentially expressed in the liver during development (i.e., undergo a developmental transition). FMO1 expression, the major fetal isozyme, peaks early in gestation (first and second trimesters) and gradually decreases to undetectable at birth (Koukouritaki et al., 2002). In contrast, FMO3 expression, the major adult isozyme, turns on after birth and increases over time, reaching an adult level in the early teenage years (Koukouritaki et al., 2002). This differential enzyme expression has garnered much attention, specifically in terms of adjusting the dosage of FMO substrate drugs for infants and children (Yokoi, 2009; Yanni et al., 2010). FMO5 mRNA expression in adult liver exceeds that of FMO3 (Cashman and Zhang, 2006); however, earlier reports have suggested the opposite (Cashman, 1995, 2000). In addition, FMO5 mRNA expression in fetal livers is approximately one-sixth of that in adult livers (Cashman and Zhang, 2006). However, the ontogeny of hepatic FMO5 protein expression has not yet been characterized.
Traditionally, FMO enzyme quantification has relied on isozyme-specific antibody-based immunoquantification via Western blots. For absolute quantification, FMO content has been determined based on FAD content, the tightly bound prosthetic group required for the catalytic activity of FMO holoproteins (Lang et al., 1998). Recombinant FMOs (e.g., heterologously expressed in baculovirus-infected insect cells or Supersomes) have served as calibration standards (Yeung et al., 2000; Koukouritaki et al., 2002). Thus, previous studies reported the quantification of FMO holoproteins, rather than total FMO proteins (i.e., holoprotein + apoprotein).
To overcome the common limitations of immunoquantification (i.e., cross-reactivity, dynamic range, reproducibility, and multiplexity), liquid chromatography-tandem mass spectrometry (LC-MS/MS)- and multiple reaction monitoring (MRM)-based targeted quantitative proteomic methods have been developed for the absolute quantification of cytochrome P450s (CYPs), UDP-glucuronosyltransferases, and membrane drug transporters (Fallon et al., 2008; Kamiie et al., 2008; Li et al., 2008; Wang et al., 2008). However, targeted quantitative proteomic methods for FMOs have yet to be reported. The term “absolute” quantification in these publications and the report herein refers to a type of proteomic quantification that produces protein concentration or amount, rather than “relative” protein expression profiles. A targeted quantitative proteomic method for absolute protein quantification relies on the use of either synthetic signature peptides of known concentration or signature peptides derived from the tryptic digest of target proteins of known concentration as calibration standards. The selection of appropriate signature peptides involves the in silico tryptic digestion of target proteins, followed by evaluation of the resulting candidate peptides based on several selection criteria to ensure specificity, stability, and digestion efficiency (Wang et al., 2008; Michaels and Wang, 2014; Peng et al., 2015). Candidate signature peptides (usually at least two for each protein) then can be synthesized and used to tune the MS (typically, a triple-quadrupole MS) for optimal MRM detection. However, some candidate signature peptides may not perform optimally because of poor digestion efficiency, chromatography, or ionization during MS analysis, therefore rendering expensive signature peptides useless. Hence, it is desirable to incorporate an additional process(es) to verify candidate signature peptides after in silico prediction but before their synthesis.
The primary objective of the current study was to develop an ultra-performance liquid chromatography (UPLC)-MRM-based targeted quantitative proteomic method for the absolute quantification of FMO1, FMO3, and FMO5 in human liver microsomes (HLM). The secondary objective was to evaluate post-in silico product ion screening of the target protein tryptic digest as a way to verify candidate signature peptides prior to their synthesis.
Materials and Methods
Chemicals, Enzymes, and Liver Tissues.
Optima-grade acetonitrile, water, formic acid, and acetic acid were obtained from Fisher Scientific (Pittsburgh, PA). Ammonium bicarbonate, dimethylsulfoxide, dithiothreitol, iodoacetamide, and cimetidine were purchased from Sigma-Aldrich (St. Louis, MO). Cimetidine sulfoxide was acquired from Abcam Biochemicals (Cambridge, UK). Famotidine sulfoxide was acquired from Toronto Research Chemicals (Toronto, Canada). Recombinant human FMO1, FMO3, and FMO5 Supersomes, prepared from baculovirus-infected insect cells expressing human FMO enzymes, were purchased from Corning Gentest (Woburn, MA). The FMO concentration (pmol/ml and pmol/mg protein) of each Supersome, based on the FAD content determined by an high-performance liquid chromatography-fluorescence method (Lang et al., 1998), was provided by the supplier. Control Supersomes (Corning Gentest) contained microsomes from insect cells infected with wild-type baculovirus. Synthetic unlabeled AQUA Ultimate-grade signature peptides (5 pmol/μl ± 5% by amino acid analysis) were ordered from Thermo Scientific (Ulm, Germany). Peptide purity (>97%), determined by reverse-phase high-performance liquid chromatography-ultraviolet (detection wavelength of 215 nm) and matrix-assisted laser desorption/ionization-time of flight MS, was provided by the manufacturer. Synthetic 13C and 15N stable isotope-labeled crude signature peptides also were acquired from Thermo Scientific. All synthetic peptide sequences were confirmed by MS/MS fragmentation analysis using a Waters Xevo TQ-S triple-quadrupole MS (Milford, MA). Sequencing-grade modified trypsin was purchased from Promega (Madison, WI). Pooled HLM (XTreme 200) and nine individual adult donor HLM (Supplemental Table 1) were purchased from XenoTech, LLC (Lenexa, KS). Liver tissues from 7 fetal (14–20 weeks gestation) donors and 16 pediatric (aged 5 months–10 years) donors were obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders (Contract #HHSN275200900011C, Ref. No. NO1-HD-9-0011; Baltimore, MD) under an approved University of North Carolina-Chapel Hill Institutional Review Board and were used to prepare fetal and pediatric HLM (Supplemental Table 1).
In Silico Selection of FMO Signature Peptides.
Candidate tryptic signature peptides for FMO quantification were selected in silico using criteria described previously (Wang et al., 2008; Michaels and Wang, 2014; Peng et al., 2015). The selected candidate peptides for each FMO protein are listed in (Supplemental Table 2).
Trypsin Digestion.
The tryptic digestion of FMO Supersomes and HLM was performed as described previously with minor modifications (Wang et al., 2008; Michaels and Wang, 2014). Briefly, protein samples (30 μg) were reduced in ammonium bicarbonate buffer (pH 8.0, 50 mM final concentration) containing dithiothreitol (4 mM final concentration) and heated at 60°C for 60 minutes to denature the proteins. After cooling to room temperature, the samples (90 μl total volume) were alkylated with iodoacetamide (10 mM final concentration) for 20 minutes in the dark before digestion with 1 µg trypsin at 37°C for 4 hours unless stated otherwise. All reactions were carried out in Eppendorf Protein LoBind microcentrifuge tubes (Hamburg, Germany) to minimize protein and peptide loss due to binding. Solvent evaporation during the incubations was minimized by sealing the capped tubes with parafilm and applying pressure with an aluminum block. To optimize the trypsin digestion protocol, different digestion times (0.5, 1, 2, 4, 6, 8, 12, and 24 hours) and protein-to-trypsin ratios (10, 20, 30, 40, 50, 60, 80, and 100:1) were examined. Reactions were cold-quenched with storage at −80°C. A mixture of stable isotope-labeled signature peptides (1 µl; internal standards) was spiked into the thawed samples before loading into a 6°C autosampler.
Signature Peptide Verification by Post-In Silico Product Ion Screening.
After vortexing and centrifugation (16,000 g for 10 minutes at 4°C), the supernatants (10 µl) of the quenched digestion mixtures underwent UPLC-MS/MS analysis. The UPLC-MS/MS instrument, consisting of a Waters Acquity UPLC I-class binary solvent manager coupled with a Waters Xevo TQ-S triple-quadrupole MS, was operated under positive electrospray ion mode. Chromatographic separation of the peptides was carried out on a reversed-phase column (Waters UPLC BEH-C18, 1.7 μm, 2.1 × 100 mm), fitted with an in-line column filter and a VanGuard guard-column (Waters). The mobile phases consisted of (A) water containing 0.1% (v/v) formic acid and (B) acetonitrile containing 0.1% (v/v) formic acid. A 13.5 minute gradient (0.4 ml/min) began with 2% B held for 1 minute, followed by an increase to 15% B over 2 minute, and to 30% B over the next 7 minute. The column was washed with 95% B for 1.5 minute and then re-equilibrated with 2% B for 2 minutes before the next injection.
To detect the in silico-selected candidate signature peptides, product ion scans were set up using selected precursor ions corresponding to the doubly protonated ions of the candidate peptides in the Q1 quadrupole, fragmenting these precursor ions with a collision energy ramp (15–40 V) in the Q2 quadrupole, and mass analysis of the product ions in the Q3 quadrupole mass analyzer under a scan rate of 5000 amu/s. Extracted product ion (EPI) chromatograms of all the possible y ions of each candidate peptide were generated using Masslynx (Version 4.1; Waters) to allow visual inspection for product ion screening. A salient peak shared by most or all y ion EPI chromatograms verified the detection of the corresponding signature peptide. Upon detection verification, the signature peptide sequences were sent for synthesis (Thermo Scientific).
UPLC-MRM Analysis.
Lyophilized stable isotope-labeled signature peptides were dissolved in 1 mL of 1:1 (v/v) acetonitrile:water solution. The solution was diluted further to approximately 2–4 μg/ml and then infused into the Xevo TQ-S MS at 5 μl/min with an LC flow of 50% B at 0.4 ml/min. MRM parameters were optimized using IntelliStart (Waters) under positive electrospray ion mode: capillary voltage, 1.5 kV; cone voltage, 40 V; source offset, 40 V; dissolvation temperature, 500°C; dissolvation gas, 1000 l/h; nebulizer gas, 7 bar. The optimum collision energy and precursor/production masses for the signature peptides are summarized in Table 1. UPLC-MRM quantification was performed using the peak area ratios of signature peptides to corresponding stable isotope-labeled signature peptides (internal standards).
Signature peptides for human FMO1, FMO3, and FMO5
Preparation of Calibration Standards.
Two types of calibration standards were prepared for the absolute quantification of FMOs in HLM. First, recombinant FMO1, FMO3, and FMO5 Supersomes of known concentrations (based on FAD content) were used to build calibration standards (0.005 to 20 pmol/digestion). Quality controls (QCs), consisting of FMO Supersomes at 0.2, 1, and 10 pmol/digestion, were prepared in triplicate. All recombinant protein standards and QCs were denatured, alkylated, and trypsin-digested as described above before UPLC-MRM analysis. Because of the varying amount of total proteins in the standards, additional trypsin (2 µg total) was used to keep the protein:trypsin ratio ≤30:1 in the high concentration standards. Second, synthetic signature peptides of known concentrations (based on amino acid analysis) were used to build calibration standards (0.02 to 20 pmol/digestion). To normalize total protein loading, control Supersomes (30 µg) were spiked into the peptide standards. The spiked peptide standards also were denatured, alkylated, and trypsin-digested before UPLC-MRM analysis. The lower limit of quantification was defined as the lowest standard concentration with signal-to-noise ratio >5 and acceptable precision and accuracy (within 20%).
FMO Marker Substrate Activity Assay.
Cimetidine sulfoxidation was used to measure FMO functional activity as described previously (Cashman et al., 1993; Overby et al., 1997). Cimetidine (1 mM; reported Km values are 4 mM for FMO3 and >10 mM for FMO5) was preincubated with HLM (0.1 mg/mL) in a phosphate buffer (pH 7.4, 100 mM) containing 3.3 mM MgCl2 for 5 minutes at 33°C. Although these conditions were different from what was used by Zane et al. (submitted manuscript), i.e., substrate concentration and incubation temperature, they served the purpose of validating protein quantification by correlating marker substrate activity and protein expression. Reactions (200 µl final volume) were initiated by the addition of NADPH (1 mM final concentration). Aliquots (10 µl each) were removed from each reaction at 1 and 5 minutes and transferred to tubes containing ice-cold acetonitrile (300 µl) and famotidine sulfoxide (10 nM; internal standard). Quenched reaction mixtures were centrifuged (2250 g for 20 minutes at 4°C), and the resulting supernatants (100 µl) were dried under nitrogen at 50°C. The dried samples were reconstituted in water (150 µL) before UPLC-MS/MS quantification of cimetidine sulfoxide using the Xevo TQ-S triple-quadrupole MS operated under positive electrospray ion mode. Analytes were separated on a reversed-phase analytical column (Thermo Scientific Aquasil C18, 2.1 × 50 mm, 3 µm; Bellefonte, PA). The gradient (0.4 ml/min) began at 0% B for 0.5 minutes, then quickly increased to 5% B and was held there for 3 minutes. The column was washed with 100% B for 1 minute and re-equilibrated at 0% B for 0.5 minutes before the next injection. UPLC-MS/MS quantification was performed using the peak area ratios of cimetidine sulfoxide to famotidine sulfoxide. Cimetidine sulfoxide calibration standards ranged from 0.1 to 100 µM. Cimetidine sulfoxidation rates were determined from the amount of metabolite generated between the 1- and 5-minute reaction times. Cimetidine sulfoxide formation was linear for a minimum of 30 minutes under the described conditions (data not shown). Because FMO enzymes are heat labile in the absence of NADPH, their stability was examined during the preincubation (5 minutes at 33°C) with substrate only. Results showed no significant difference in cimetidine sulfoxidation activities of recombinant FMO1, FMO3, and the pooled HLM between preincubation with substrate cimetidine only and preincubation with NADPH (data not shown), indicating stability of FMO enzymes during the preincubation with substrate only.
Data Analysis.
The final FMO protein concentration was the average value determined using two signature peptides for each FMO protein. All average values were calculated as the mean. For correlation analysis, measured cimetidine sulfoxide formation rates in HLM were plotted versus FMO protein concentration in the same sample, and the Pearson r and P values were reported because all relevant data passed normality test (Supplemental Table 3). The slope and Y-intercept values were determined by least-square linear regression analysis. Student’s t tests (two-tailed, unpaired) were used to compare the pairs of signature peptides. One-way analysis of variance followed by post hoc test using Tukey’s adjustment was used to compare FMO5 expression in the fetal, pediatric, and adult HLM. P < 0.05 was considered significant. All data analyses were performed using GraphPad Prism (v. 5.0; San Diego, CA).
Results
Verification of Signature Peptides by Post-In Silico Product Ion Screening.
After the initial in silico selection of human FMO3 signature peptides, eight candidate peptides (Supplemental Table 2) satisfied every selection criteria described previously (Wang et al., 2008; Peng et al., 2015). To select the final signature peptides (two for each protein) from the candidate peptides, recombinant FMO3 was reduced, alkylated, and trypsinized, and the resulting digest was separated on a UPLC analytical column. Analysis was completed through product ion screening of the doubly charged ions of the candidate peptides. Representative EPI chromatograms of predicted y ions for the two final FMO3 signature peptides selected for use in this study (FMO3_pep1_L and FMO3_pep4_L; Table 1) are shown in Fig. 1, A and B, respectively. The signature peptides produced salient peaks in each EPI chromatogram (2.5-minute peak for FMO3_pep1_L and 5.5-minute peak for FMO3_pep4_L) and the product ion mass spectra integrated across the peaks matched each peptide sequence (Fig. 1, C and D). In addition, EPI chromatograms of predicted y ions for the remaining six FMO3 candidate signature peptides are shown in (Supplemental Fig. 1). Likewise, EPI chromatograms of predicted y ions for the final FMO1 and FMO5 signature peptides (Table 1) also were examined and verified for optimal UPLC-MRM detection (data not shown).

Post-in silico product ion screening of FMO3 signature peptides (FMO3_pep1_L and FMO3_pep4_L). Extracted product ion chromatograms of predicted y ions (A and B) and MS/MS spectra (C and D) of the detected FMO3 signature peptides are shown after product ion screening analysis of a recombinant FMO3 Supersomes tryptic digest (40.5 pmol FMO or 50 μg total protein). FMO3_pep1_L and FMO3_pep4_L eluted at 2.5 and 5.5 minutes, respectively.
After identification and verification of the predicted signature peptides, unlabeled signature peptides and corresponding 13C and 15N stable isotope-labeled signature peptides (Table 1) were synthesized and used for the development and optimization of a UPLC-MRM method. This method allows for the multiplexed detection and quantification of FMO1, FMO3, and FMO5 in HLM. Representative UPLC-MRM chromatograms of signature peptides in tryptic digests of adult HLM and fetal HLM are shown in Fig. 2.
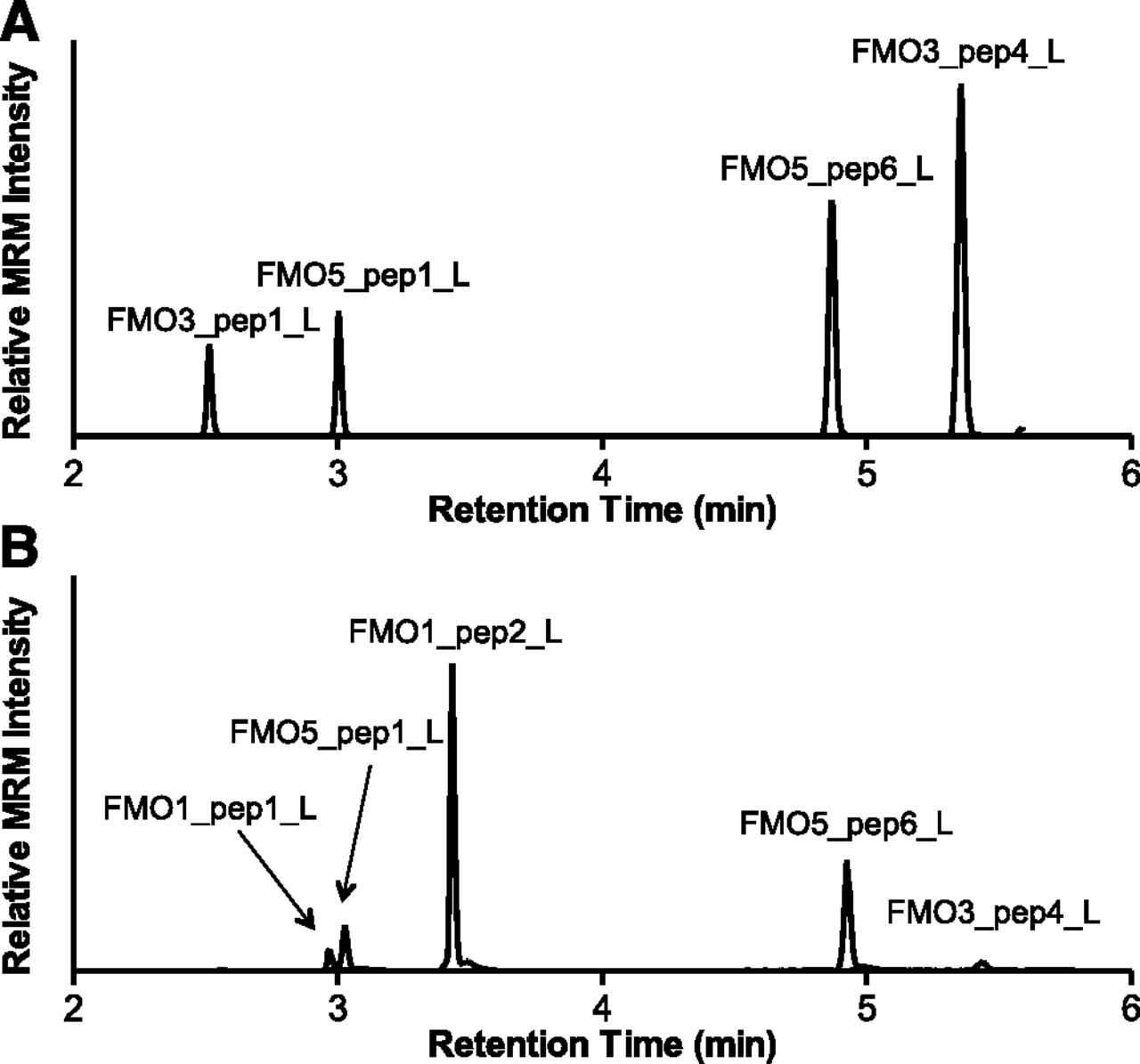
Representative MRM chromatograms of FMO signature peptides in tryptic digests of adult (A) and fetal (B) HLM. Digestion mixtures, containing 30 μg HLM and 1 μg trypsin, were incubated for 4 hours at 37°C before UPLC-MRM analysis.
Effects of Trypsin Digestion Time and Protein:Trypsin Ratio.
To optimize trypsin digestion conditions and determine the dynamic range, the effects of digestion time and protein:trypsin ratio on the absolute quantification of FMOs in pooled HLM were evaluated using the developed UPLC-MRM method. The relative UPLC-MRM signals of the signature peptides reached a maximum after 4 hours of digestion and plateaued (or decreased slightly in some cases) thereafter (Fig. 3, A and B). Because of the low expression of FMO1 in pooled HLM, only one of the two FMO1 signature peptides was detected and evaluated (Fig. 3B). As a result, tryptic digestion was carried out for 4 hours for the remainder of the study. In addition, the relative UPLC-MRM signals of the signature peptides increased linearly with respect to HLM protein loading between 10 to 100 μg when 1 μg trypsin was used (Fig. 3, C and D); however, a slight downward deviation was noticed above 50 μg of HLM protein. Thus, optimized trypsin digestion conditions, 4 hours digestion and 30:1 protein:trypsin ratio, were selected and used for the absolute quantification of FMO1, FMO3, and FMO5 in HLMs.

Effects of trypsin digestion time and HLM protein loading on the UPLC-MRM signals of FMO signature peptides derived from pooled HLM. The UPLC-MRM peak areas of FMO signature peptides were normalized by those of corresponding stable isotope-labeled signature peptides spiked in as internal standard. For the digestion time study (A and B), each reaction contained 30 μg of pooled HLM and 1 μg of trypsin. For the protein loading study (C and D), each reaction contained 1 μg of trypsin and varying amounts of HLM proteins. Symbols and error bars represent the mean and standard deviation of triplicate determinations. In many cases, error bars are too small to be seen. Dashed lines (C and D) represent the best-fit lines of least-square linear regression analysis.
Absolute Quantification of FMO3 and FMO5 in Adult HLM and Correlation to Marker Substrate Activity.
Similar to the immunoquantification and targeted proteomic quantification of CYPs (Wang et al., 2008; Michaels and Wang, 2014), recombinant FMO Supersomes of known concentrations were used initially to create calibration standards. The concentrations of the recombinant FMO Supersomes, based on FAD content, were provided by the vendor. The calibration curves for each recombinant FMO Supersome (0.01 to 4 pmol/ digestion; 10–12 concentrations) demonstrated good linearity (r2 > 0.99). By using 30 μg of HLM, the observed lower limit of quantification for the three FMOs was 0.33 pmol/mg HLM protein. The intraday accuracy (percent deviation) and precision (CV) of the analytical method, based on QC samples, were within 15%.
Method coherence was evaluated by comparing protein quantification results from two different signature peptides of the same protein (i.e., FMO3_pep1_L versus FMO3_pep4_L and FMO5_pep1_L versus FMO5_pep6_L). In each case, a strong correlation, near-unity slope and near-zero Y-intercept were observed (Fig. 4, B and C), indicating consistent protein quantification results between the different signature peptides. In addition, good coherence was observed for two FMO1 signature peptides when fetal HLM were analyzed (Fig. 4A; described below). As a result, final protein concentrations were calculated as the average of the quantification results from the two signature peptides.

Coherence analysis of FMO protein quantification by UPLC-MRM-based targeted proteomic approach using different signature peptides and recombinant FMO Supersomes-generated calibration standards. Quantification of FMO1 (A) was performed using the fetal HLM panel, whereas quantification FMO3 (B) and FMO5 (C) was performed using the adult HLM panel. Symbols and error bars represent the mean and standard deviation of triplicate determinations for an individual donor HLM. In many cases, error bars are too small to be seen. Dotted lines represent the best-fit lines of least-square linear regression analysis.
By using a panel of adult HLM (n = 9 individual donors and 1 pooled), the protein concentrations of the three FMOs were determined using the developed targeted quantitative proteomic method. The FMO1 concentration in adult HLM was below the lower limit of quantification (<0.33 pmol/mg HLM protein). The final FMO3 and FMO5 average protein concentrations (range and 95% confidence interval [CI]) were 46 (26–65 and 36–56) and 27 (11.5–49 and 18.5–36) pmol/mg HLM protein, respectively. Furthermore, cimetidine sulfoxidation activities were measured in the HLM panel and compared with FMO protein concentrations. A strong correlation was observed between cimetidine sulfoxidation activity and FMO3 protein concentration (r2 = 0.86, P = 0.0001; Fig. 5A) but not FMO5 protein concentration (r2 = 0.30, P = 0.103; Fig. 5B).

Correlation analysis of FMO3 (A) and FMO5 (B) protein content and measured marker activity in the adult individual donor HLM panel. Symbols and error bars represent the mean and standard deviation of triplicate determinations for an individual donor HLM. In many cases, error bars for protein concentration are too small to be seen. Dotted lines represent the best-fit lines of least-square linear regression analysis.
Absolute Quantification of FMO1 and FMO5 in Fetal HLM and Correlation to Marker Substrate Activity.
To evaluate the method for absolute quantification of FMO1, a panel of fetal HLM (n = 7 individual donors) was analyzed; the adult HLM panel lacked FMO1 expression. The final FMO1 average protein concentration (range and 95% CI) in the fetal HLM panel was 7.0 (4.9–9.7 and 5.2–8.7) pmol/mg HLM protein. In addition, there were appreciable amounts of FMO5, which averaged 21 (14–32 and 14–29) pmol/mg HLM protein (Fig. 6A). In contrast to adult HLM, FMO3 was barely above lower limit of quantification (0.33 pmol/mg HLM protein) in fetal HLM, averaging 0.7 pmol/mg HLM protein with a highest concentration of 2.2 pmol/mg HLM protein. In addition, cimetidine sulfoxidation activity also was measured in the fetal HLM panel and compared with FMO protein concentrations. Neither FMO1 (r2 = 0.41, P = 0.12) nor FMO5 (r2 = 0.01, P = 0.83) protein concentration correlated with the marker substrate activity (data not shown). Because FMO5 was reported to lack appreciable cimetidine sulfoxidation activity (Overby et al., 1997; Hai et al., 2009), correlation using a relative activity factor-adjusted FMO expression was not attempted.
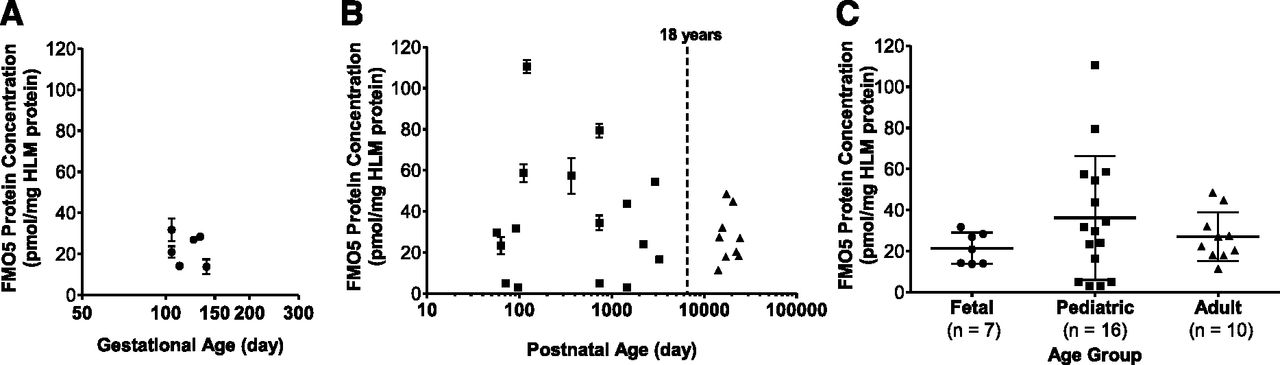
Comparison of FMO5 protein expression in the fetal, pediatric, and adult HLM. FMO5 protein concentration was determined by UPLC-MRM-based targeted proteomic approach using recombinant FMO5 Supersomes-generated calibration standards. Donor age (gestational [A] and postnatal [B] age) was plotted in logarithm scale. Symbols in the scatterplots represent the mean of triplicate determinations of an individual HLM sample. Lines and error bars represent the mean and standard deviation of all HLM samples in an age group. One-way analysis of variance was used to compare all three age groups (C; P = 0.317).
FMO5 Expression in Fetal, Pediatric, and Adult HLM.
In addition to fetal and adult HLM described above, a panel of pediatric HLM (n = 16 individual donors; Supplemental Table 1) was analyzed for FMO1, FMO3, and FMO5 expression. The FMO1 and FMO3 expression in the pediatric HLM has been reported (Zane et al., submitted manuscript). The final FMO5 average protein concentration (range and 95% CI) in the pediatric HLM panel was 36.2 (2.9–110 and 20.1–52.3) pmol/mg HLM protein (Fig. 6B). There was no statistically significant difference among the three age groups (P = 0.317; Fig. 6C).
Comparison of Recombinant Proteins versus Synthetic Peptides as Calibration Standards for Absolute Quantification.
Previously, our laboratory and others reported signature peptide-dependent absolute quantification of CYPs and drug transporters using synthetic peptides as calibration standards (Wang et al., 2008; Balogh et al., 2013; Michaels and Wang, 2014; Prasad et al., 2014; Peng et al., 2015). To assess such a scenario for the absolute quantification of FMOs, two signature peptides were selected for each FMO isozyme (Table 1), and quantification coherence between the two peptides was evaluated. When recombinant FMO Supersomes of known concentration were used to generate signature peptide standards, good coherence was observed, as described above (Fig. 4). However, when synthetic peptides of known concentrations were used to generate signature peptide standards, good coherence was observed for FMO1 but not for FMO3 or FMO5 (Fig. 7).

Comparison of FMO protein quantification by UPLC-MRM-based targeted proteomic approach using different signature peptides and synthetic signature peptide-generated calibration standards. Symbols represent the mean of triplicate determinations for an individual donor HLM. Lines and error bars represent the mean and standard deviation for a panel of HLM. Student’s t tests (two-tailed, unpaired) were used to compare the pairs of signature peptides for FMO1 (A), FMO3 (B), and FMO5 (C).
Absolute FMO concentrations measured using synthetic peptide standards were substantially greater than those determined using recombinant protein standards (i.e., Supersomes) (Fig. 7 versus Fig. 4). For example, the average FMO1 concentration in fetal HLM was 7.0 pmol/mg HLM protein with recombinant protein standards. In contrast, it was 29 or 32 pmol/mg HLM protein (4- to 5-fold higher) with synthetic FMO1_pep1_L or FMO1_pep2_L standards, respectively. Similarly, the average FMO3 and FMO5 concentrations in adult HLMs were 46 and 27 pmol/mg HLM protein, respectively, with recombinant protein standards. In contrast, they were 259 or 412 pmol/mg HLM protein (5.6- to 9-fold higher) for FMO3 with synthetic FMO3_pep1_L or FMO3_pep4_L standards and 21 or 32 pmol/mg HLM protein (0.8- to 1.2-fold higher) for FMO5 with synthetic FMO5_pep1_L or FMO5_pep6_L standards.
Absolute Quantification of FMOs in Recombinant FMO Supersomes using Synthetic Peptide-Generated Calibration Standards.
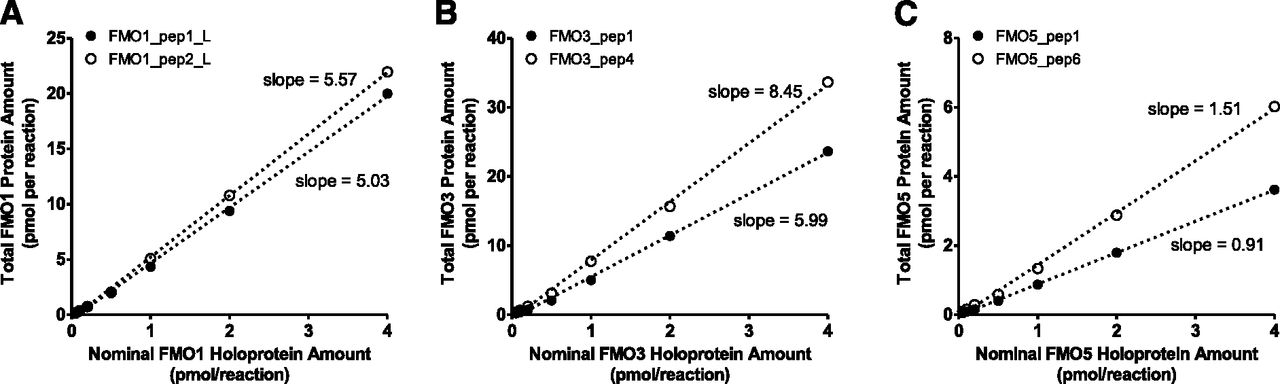
To further investigate discrepancies in the absolute quantification of FMOs when recombinant proteins versus synthetic peptides were used as standards and determine the ratios of holoprotein versus total protein, total FMO protein was quantified in recombinant FMO Supersomes of different concentrations using synthetic peptides as calibration standards. The measured total FMO protein amount was plotted against the nominal FMO protein amount based on FAD content, which represents the FMO holoprotein (Fig. 8). Similar to the previously described signature peptide-dependent quantification, the ratio of total protein versus holoprotein (slopes in Fig. 8) for each recombinant FMO Supersomes also was dependent upon the signature peptide used. The ratio ranged from 5.0 to 5.6 for FMO1, 6.0 to 8.4 for FMO3, and 0.9 to 1.5 for FMO5.

Comparison between total (holoprotein + apoprotein) FMO protein concentration and nominal holoprotein concentration in FMO1 (A), FMO3 (B), and FMO5 (C) Supersomes. The total FMO protein concentration was determined using synthetic signature peptides as calibration standards, whereas the nominal holoprotein concentration was determined based on FAD content (provided by the vendor). Dotted lines represent the best-fit lines of least-square linear regression analysis.
Discussion
In this study, a UPLC-MRM-based targeted quantitative proteomic method has been developed for the multiplexed absolute quantification of FMOs 1, 3, and 5 in HLM. This method has a lower limit of quantification of 0.33 pmol/mg HLM protein for each FMO when 30 µg of HLM is used. By using the developed quantitative proteomic method, protein concentrations of the three FMOs were determined in three panels of HLM, one derived from adult livers (n = 10), one from pediatric livers (n = 16), and one from fetal livers (n = 7). In the adult HLM, FMO3 was more abundant than FMO5 (46 versus 27 pmol/mg HLM protein), which supports earlier reports (Cashman, 1995, 2000) but not the most recent report (Cashman and Zhang, 2006), both of which were based on mRNA expression. FMO1 was below the lower limit of quantification (0.33 pmol/mg HLM protein). In the fetal HLM (14–20 weeks gestation), FMO1 was expressed at relatively high levels (7.0 pmol/mg HLM protein), similar to what was reported previously (7.8 pmol/mg HLM protein; 8–15 weeks gestation) (Koukouritaki et al., 2002). FMO3 was barely above the lower limit of quantification, averaging 0.7 pmol/mg HLM protein. Interestingly, FMO5 was the predominant FMO isozyme in fetal HLM, averaging threefold greater protein expression than FMO1 (21 versus 7.0 pmol/mg HLM protein). In the pediatric HLM, FMO5 also appeared to be the predominant FMO isozyme (36.2 versus 20.0 pmol/mg HLM protein for FMO3), whereas FMO1 was barely detected (Zane et al., submitted manuscript). Although FMO5 expression was not significantly different among the three age groups, larger interindividual variability was observed in the pediatric HLM (38-fold versus 4.3- and 2.3-fold in adult and fetal HLM, respectively) (Fig. 6). These targeted quantitative proteomic results confirm previous reports that FMO1 and FMO3 expression undergo a developmental transition and also discovered that FMO5 has relatively stable expression throughout development. However, these results should be interpreted with caution, because our study only included a small number of HLM from each age group, fetal samples only represented the second trimester, and neonatal samples (birth to first month) were absent (Supplemental Table 1). As such, future studies employing larger panels of HLM are warranted.
LC-MRM-based targeted quantitative proteomic methods for the absolute quantification of CYPs, UDP-glucuronosyltransferases, and drug transporters were first reported in the late 2000s (Fallon et al., 2008; Kamiie et al., 2008; Li et al., 2008; Wang et al., 2008). These methods rely on the identification and detection of signature peptides for each target protein. The selection and verification of suitable signature peptides can be time-consuming and costly, mainly because of peptide synthesis after in silico selection. The ability to verify LC-MRM detection of the selected signature peptides in a protein digest before committing to peptide synthesis is therefore desirable. As such, we implemented a post-in silico product ion screening step to verify the detection of selected signature peptides (Fig. 1) before their synthesis to reduce unnecessary peptide synthesis and costs. For example, only two FMO3 signature peptides (FMO3_pep1_L and FMO3_pep4_L) were synthesized in this study, rather than all eight candidate signature peptides (Supplemental Table 2).
To achieve absolute quantification using an LC-MRM-based targeted proteomic approach, two types of standards are typically employed, recombinant proteins of known concentration or synthetic signature peptides of known concentration. Because of the poor coherence (i.e., signature peptide-dependent quantification) when synthetic peptides were employed as standards (Wang et al., 2008; Balogh et al., 2013; Michaels and Wang, 2014; Prasad et al., 2014; Peng et al., 2015), we prefer to use recombinant proteins when available (e.g., CYPs) to generate standards and employ at least two signature peptides for each protein to ensure quantification coherence. In the current study, poor quantification coherence was observed for FMO3 and FMO5 when synthetic peptides were used as standards (Fig. 7, B and C). Presumably, the presence of multiple acidic amino acid residues (D or E) close to the tryptic cleavage sites (e.g., FMO1_pep1, FMO3_pep1 and FMO5_pep1; Table 1) could cause missed cleavage (Yen et al., 2006), resulting in lower recovery of signature peptides and underestimation of protein concentration. In contrast, quantification was coherent between signature peptides for all three FMOs when recombinant FMO Supersomes were used to generate standards (Fig. 4). Therefore, we recommend the use of recombinant proteins, when available, to generate standards for LC-MRM-based targeted protein quantification. Moreover, we call for a coordinated effort to produce reference protein standards, especially in the case of drug transporters, for use as calibration standards for targeted quantitative proteomics. It is not completely understood yet what may cause the lack of coherence in signature peptide-dependent quantification when synthetic peptides are used as standards. We have proposed that different digestion efficiencies (e.g., missed cleavage) and/or unexpected posttranslational modifications of signature peptides were the underlying causes (Peng et al., 2015) and warrant future investigation.
FMOs, specifically the holoprotein, require an FAD prosthetic group for catalytic activity. Recombinant FMO Supersomes can be quantified based on their FAD content to give a holoprotein concentration. In contrast, the use of synthetic peptides as standards for targeted proteomic quantification provides a total protein concentration (i.e., holoprotein + apoprotein) for a sample. Such a distinction was seen (Fig. 8), because the total FMO protein amount exceeded its nominal holoprotein amount 5- to 6.6-fold for FMO1 and 6- to 8.5-fold for FMO3, whereas only a small difference (0.9- to 1.5-fold) was seen for FMO5. These results suggest that a large portion of FMO1 and FMO3 proteins in Supersomes is present as apoprotein without the FAD prosthetic group, whereas most FMO5 proteins are holoproteins. This is consistent with a much greater FAD content in FMO5 Supersomes (2700 pmol/mg protein; lot#3154943) relative to those in FMO1 and FMO3 Supersomes (500 and 810 pmol/mg protein, respectively; lot#3098891 and lot#3130681, respectively) reported by the vendor, although differential expression efficiency also could contribute to FAD content differences in FMO Supersomes.
For both conventional immunoquantification and the targeted proteomic quantification described here, an assumption was made that the holoprotein:apoprotein ratio remains the same between a recombinant system (e.g., Supersomes) and HLM. However, this assumption remains to be examined. Deviation from this assumption could result in either underestimation or overestimation of enzymatic activity in HLM, depending on how the ratio in HLM deviates relative to that in the recombinant system. For example, if the ratio deviates upward in HLM (i.e., higher proportion of holoproteins), this will result in an underestimation of HLM holoprotein concentration, and the measured HLM activity will exceed the predicted activity calculated as the product of recombinant enzyme activity and HLM protein expression. To test this, one could first determine the rate of a probe substrate reaction, which needs to be catalyzed exclusively by the enzyme of interest, in HLM and then compare the measured HLM activity with the predicted activity based on the measured activity of the recombinant enzyme and measured expression level of the enzyme in HLM. By using FMO3 and cimetidine sulfoxidation as an example, the average measured cimetidine sulfoxidation activity in the adult HLM panel was 1.25 nmol/min/mg HLM (Fig. 5A), the measured cimetidine sulfoxidation activity of recombinant FMO3 was 6.0 nmol/min/nmol FMO3 (unpublished data), and the measured FMO3 expression in HLM was 0.046 nmol/mg HLM (Fig. 4B). The predicted activity is 0.28 nmol/min/mg HLM, substantially less than the measured activity of 1.25 nmol/min/mg HLM. Thus, an upward deviation of the holoprotein:apoprotein ratio in HLMs could have contributed to the underprediction, in addition to other possibilities proposed in the companion paper (Zane et al., submitted manuscript). The questionable assumption regarding the holoprotein:apoprotein ratio for FMOs, as well as for CYPs, is underappreciated and requires further investigation using newly available analytical tools (e.g., targeted quantitative proteomics).
In summary, a UPLC-MRM-based targeted proteomic assay has been developed for the absolute protein quantification of FMOs 1, 3, and 5 in HLM. Our results corroborated the developmental transition in FMO1 and FMO3 expression and revealed relatively stable FMO5 expression throughout development. The developed FMO assay and other previously developed targeted quantitative proteomic assays are expected to assist in addressing previously unanswered questions in quantitative pharmacology.
Authorship Contributions
Participated in research design: Chen, Zane, Thakker, and Wang.
Conducted experiments: Chen and Zane.
Performed data analysis: Chen, Zane, Thakker, and Wang.
Wrote or contributed to the writing of the manuscript: Chen, Zane, Thakker, and Wang.
Footnotes
- Received October 1, 2015.
- Accepted January 19, 2016.
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant R01GM089994] and by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) of the National Institutes of Health [Award number 5 T32 GM086330-04]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CI
- confidence interval
- CYP
- cytochrome P450
- EPI
- extracted product ion
- FMO
- flavin-containing monoxygenase
- HLM
- human liver microsomes
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MRM
- multiple reaction monitoring
- QC
- quality control
- UPLC
- ultra-performance liquid chromatography
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}