


Abstract
To determine the level of FMO1 protein present in human liver tissues, a monospecific antibody was prepared and a sensitive Western blotting procedure with enhanced chemiluminescence detection was developed. Human FMO1, purified from insect cells expressing the recombinant protein, was used as a protein standard for absolute quantification. The average concentrations of FMO1 in microsomes prepared from human liver, kidney, intestine, and fetal liver were found to be <1, 47 ± 9, 2.9 ± 1.9, and 14.4 ± 3.5 pmol/mg, respectively. Quantitation in intestinal microsomes was complicated by variable degrees of proteolytic degradation of FMO1, not seen in microsomes prepared from liver or kidney. Recombinant human FMO1 and detergent-solubilized human duodenal microsomes both metabolized p-tolyl methyl sulfide stereoselectively to the (R)-sulfoxide, indicating the expression of functional FMO1 in human intestine. The relatively high levels of immunoquantifiable FMO1 in human kidney and fetal liver complement our previous catalytic studies in these tissues, which also demonstrated preferential (R)-p-tolyl methyl sulfoxide formation. These data demonstrate a profound ontogenic change in expression of hepatic FMO1 in humans, such that in adult life FMO1 is exclusively an extrahepatic drug-metabolizing enzyme. The marked expression levels of FMO1 found in human kidney coupled to the high catalytic activity of this isoform toward a diverse array of sulfides and tertiary amines suggest the possibility that human renal FMO1 is a significant contributor to the metabolic clearance of drugs and other xenobiotics bearing these functionalities.
The flavin-containing monooxygenases (FMOs)2are a family of microsomal NADPH- and oxygen-dependent enzymes that are distributed ubiquitously in mammalian species. To date, five isoforms designated by an Arabic numeral prefix (FMO1–FMO5) have been characterized (Lawton et al., 1994; Lang et al., 1998). Mammalian FMOs readily N- and S-oxygenate various drugs and pesticides, as well as certain endogenous or dietary compounds such as trimethylamine (Dolphin et al., 1997). However, P450 monooxygenases are also capable of catalyzing many of the same reactions as FMO. Therefore, an assessment of the contribution that FMOs make to the metabolism of xenobiotics in humans requires knowledge of both the substrate specificity of the individual FMO isoforms and the concentration of each enzyme in metabolically important tissues, such as the liver, intestine, and kidney.
FMO3 is the primary form of the enzyme present in human liver, where it is expressed at concentrations up to 100 pmol/mg of microsomal protein (Overby et al., 1997; Haining et al., 1997). FMO5 is a lesser component of human liver microsomes and is present at about one-third the level of FMO3 (Overby et al., 1995). Preliminary data have shown that FMO5 protein is also present at very low levels in kidney (Code et al., 1998). However, FMO5 exhibits a severely restricted substrate specificity for most drugs and other xenobiotics examined to date (Fisher et al., 1995; Lang and Rettie, 1998a; Lang et al., 1998). Although FMO1 is the predominant hepatic enzyme in numerous animal species, Northern blot analysis indicates that the mRNA for FMO1 is present in extremely low abundance in adult human liver (Phillips et al., 1995). In contrast, FMO1 mRNA is readily detectable in human fetal liver and adult kidney (Dolphin et al., 1996). However, FMO1 protein concentrations have not been quantified in any human tissues to date.
Therefore, the first objective of the present study was to probe the apparent ontogenic change in human hepatic FMO1 content, indicated by the mRNA studies, at the protein level. Because FMO1 and FMO3 migrate with very similar mobilities on SDS-polyacrylamide gel electrophoresis (PAGE), this required the development of a high titer, monospecific antibody directed toward FMO1. A second objective was to quantitate FMO1 protein concentrations in human adult kidney and intestine microsomes to evaluate the possibility that FMO1 could contribute to the metabolic clearance of susceptible substrates at extrahepatic sites.
Materials and Methods
Chemicals.
DEAE-Sepharose (Fast Flow) and cyanogen bromide-activated Sepharose 4B were obtained from Amersham Pharmacia Biotech (Piscataway, NJ). Ceramic hydroxyapatite (Macro-Prep) was obtained from Bio-Rad Laboratories (Hercules, CA). Emulgen 911 was a gift from KAO Corp. (Japan). All other reagents were at least of analytical grade and obtained from Sigma Chemical Co. (St. Louis, MO) except for glycerol (anhydrous) obtained from Fisher Scientific (Fairlawn, NJ). The 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) phosphatase substrate was obtained from Kirkegaard and Perry Laboratories (Gaithersburg, MD). p-Tolyl methyl sulfide and its sulfoxide and sulfone metabolites were obtained or synthesized as previously described (Rettie et al., 1990).
Human Tissues.
Human adult liver (HL), kidney (HK), and intestinal tissues (HI) were obtained from the Human Tissue Bank at the University of Washington (Seattle, WA). Fetal liver tissue (FL) was obtained from the Central Laboratory for Human Embryology at the University of Washington. The age and gender of adult tissue donors were as follows: HL112: 24 years, male; HL122: 50 years, female; HL123: 15 years, male; HL131: 62 years, female; HK1: 76 years, female; HK2: 64 years, gender unknown; HK3: 73 years, gender unknown, HK112: 39 years, male; HIA: 38 years male; HIB: 10 years, male; HIC: 27 years, male; HID: 60 years, female; HI25: 27 years, male; HI30: 30 years, male; HI33: 46 years, male. The gestational ages of the fetal liver tissue donors was as follows: FL227: 98 days; FL306: 98 days; FL269: 110 days; FL343: 115 days; FL339: 120 days. Microsomes were prepared from tissue samples as described previously (Sadeque et al., 1992; Paine et al., 1997). All microsomal pellets were resuspended in 100 mM potassium phosphate, 20% glycerol, and 1 mM EDTA, pH 7.4, and stored at −80°C until use.
Expression and Purification of Human FMO1.
Human FMO1 was expressed in the baculovirus expression vector system, and insect cell membranes were prepared as described previously (Haining et al., 1997; Lang et al., 1998). FMO1 was purified for use as a standard for immunoquantitation by an adaptation of the protocol described by Sabourin et al. (1984). Briefly, insect cell membrane pellets containing approximately 200 nmol of FMO were solubilized for 1 h on ice with gentle stirring in buffer containing 10 mM phosphate buffer (pH 7.6), 20% glycerol, 0.1 mM EDTA, and 1.5% Emulgen 911 (buffer A). The insoluble material was pelleted by centrifugation at >100,000g for 40 min. The yellow supernatant was applied to a 70-ml DEAE-Sepharose column that had been equilibrated with buffer A. The column was washed with 3 column volumes of buffer A + 20 mM NaCl, and FMO1 was eluted with buffer A + 75 mM NaCl. The FMO1-containing fractions were pooled and dialyzed overnight at 4°C against 2 liters of 10 mM phosphate buffer (pH 7.6), 20% glycerol, 0.1 mM EDTA, 0.2% Emulgen 911 (buffer B). The dialyzed FMO1 preparation (total volume ∼60 ml) was loaded onto a small hydroxyapatite column that had been equilibrated with 30 ml of buffer B at a flow rate of 30 ml/h. The column was washed with 20 ml of buffer B, and FMO1 was eluted with buffer B + 250 mM phosphate buffer (pH 6.8). Highly purified human FMO1 (estimated to be greater than 90% pure by SDS-PAGE) was dialyzed against 2 liters of 100 mM phosphate buffer (pH 7.4), 20% glycerol, and 1 mM EDTA and stored in small aliquots at −80°C. The FAD content was determined by HPLC as previously described (Lang et al., 1998).
Purification of FMO1-Specific IgG.
Antisera raised previously against minipig liver FMO cross-reacted strongly with FMO1 and weakly with several other rabbit FMO isoforms (Rettie et al., 1994). Therefore, we attempted to improve the specificity of this antibody toward FMO1 by back-adsorption to 5 ml of CNBr-activated Sepharose 4B resin in 0.1 M Na2CO3, pH 8.5, to which 5 nmol each of rabbit FMO2 (Rettie et al., 1990), human FMO3, and human FMO5 (Lang et al., 1998) had been bound. Unreacted sites on the CNBr-activated Sepharose were capped with 0.1 M Na2CO3, 1 M glycine, pH 8.2, for 3 h, with the resin packed into a small column and equilibrated overnight in 20 mM phosphate buffer, 65 mM NaCl, pH 7.6. Nonspecific FMO1 IgG (25 mg, 10 mg/ml) was adjusted to 65 mM NaCl and run into the column, and the flow was stopped for 3 h to allow maximal binding to occur. The unbound IgG (anti-FMO1 IgG) was eluted with equilibration buffer and 0.5-ml fractions were collected. Absorbance of these fractions at 280 nm was determined, and the peak fractions were pooled. Recovery of monospecific IgG was ∼30%.
Electrophoresis and Immunoblotting.
SDS-PAGE (9% resolving gel) and Western blotting of microsomal proteins were performed as described previously (Thummel et al., 1988), except that the primary antibody was incubated with the nitrocellulose overnight at 4°C. Anti-minipig liver FMO IgG and anti-FMO1 IgG were used at a dilution of 1 μg/ml. In some experiments, an anti-FMO1 peptide antibody targeted against amino acids 408 through 419 of human FMO1 (Gentest Corp., Woburn, MA) was used at a dilution of 1:500. Immunoblots were visualized either with the BCIP/NBT colorimetric system or the Amersham Pharmacia Biotech enhanced chemiluminescence (ECL) kit with X-Omat Blue XB-1 scientific imaging film (Eastman Kodak, Rochester, NY) used according to the manufacturer's instructions. Secondary antibody dilutions were 1:1000 and 1:5000, respectively. The integrated optical density of resultant immunoreactive protein bands was determined on a Scanmaster 3+ densitometer (Howtek, Hudson, NH).
Metabolic Incubations.
Intestinal microsomes (4 mg; 1.3 mg/ml of protein) were preincubated, in triplicate, on ice with NADPH (0.5 mM) in glycine/pyrophosphate buffer, pH 8.5, supplemented with Lubrol PX (0.1% w/v) and glycerol (10% v/v) for 30 min in a final volume of 3 ml. This protocol has been shown previously to abrogate P450 activity in human liver microsomes while retaining FMO function (Sadeque et al., 1992; Haining et al., 1997). Vials were then transferred to a 37°C water bath and incubated for 3 min, and metabolic reactions were initiated by the addition ofp-tolyl methyl sulfide to a final substrate concentration of 1 mM. Control incubations contained no NADPH. After 30 min, reactions were terminated with 10 ml of cold dichloromethane, p-tolyl ethyl sulfoxide (5 μg) added as internal standard, and the samples were vortex-extracted for 60 s (Sadeque et al., 1992). Dichloromethane extracts were dried under nitrogen and subjected sequentially to silica and chiral-phase chromatography in hexane/isopropyl alcohol as detailed previously for analysis of reaction rates and sulfoxide stereochemistry (Sadeque et al., 1992;Rettie et al., 1995).
Results
Recombinant human FMO1 was expressed at a high level (∼200 nmol/l) with the baculovirus expression system and purified by anion exchange and hydroxyapatite chromatography to a final specific content of 13.7 nmol/mg. In contrast to several other FMO isoforms that we have isolated, recombinant human FMO1 solubilized from insect cells bound strongly to DEAE-Sepharose and could be eluted in a highly purified fashion with increasing salt concentrations. This distinctive chromatographic behavior is likely due to the increased acidity of human FMO1 (pI = 6.9) relative to the other members of this family where the pIs range from 8.3 to 9.1 (Phillips et al., 1995). Purified FMO1 was then used as a standard for immunoquantitation.
The specificity for human FMO1 of IgG isolated from antisera raised against minipig liver FMO was compared before and after back-adsorption of the IgG to a Sepharose column to which FMOs 2, 3, and 5 had been bound. Before back-adsorption, the antibody recognized human FMO3, FMO4, and FMO5 and protein bands of similar molecular weight to these recombinant proteins in human liver and kidney (Fig.1A). A similar lack of specificity toward recombinant rabbit FMO isoforms was noted earlier (Rettie et al., 1994). However, after back-adsorption, strong immunorecognition was limited to recombinant human FMO1 and human kidney microsomes (Fig.1B). This preparation was designated as anti-FMO1 and used for all subsequent immunoquantitation.

Western blots demonstrating selectivity of FMO1 antibodies.
A, anti-minipig liver IgG (1.0 μg/ml, 1 h); B, anti-FMO1 IgG (1.0 μg/ml, 1 h). Secondary antibodies were used at a dilution of 1:1000 and incubated for 1 h, and blots were developed with BCIP/NBT phosphatase substrate. Lane 1, 2.5 pmol of purified human FMO1; lane 2, 30 μg of human kidney microsomes; lane 3, 40 μg of human liver microsomes; lane 4, 5 pmol of human FMO5; lane 5, 6 pmol of human FMO4; lane 6, 5 pmol of human FMO3; lane 7, 2.5 pmol of human FMO1; lane 8, molecular weight standards. Lanes 4 through 6 contain FMO overexpressed in insect cell membranes.
Colorimetric overdevelopment of Western blots probed with anti-FMO1 revealed faint protein signals corresponding to the apparent molecular weight of purified FMO1 in human intestine microsomes (data not shown). Therefore, a method with ECL detection was developed to improve sensitivity. Signal response was linear over the concentration range of 0 to 3.5 pmol of FMO1, the limit of in-gel detection was 50 fmol of FMO1, and the lower limit of quantitation (50 μg of microsomal protein loaded per lane) was 1 pmol/mg.
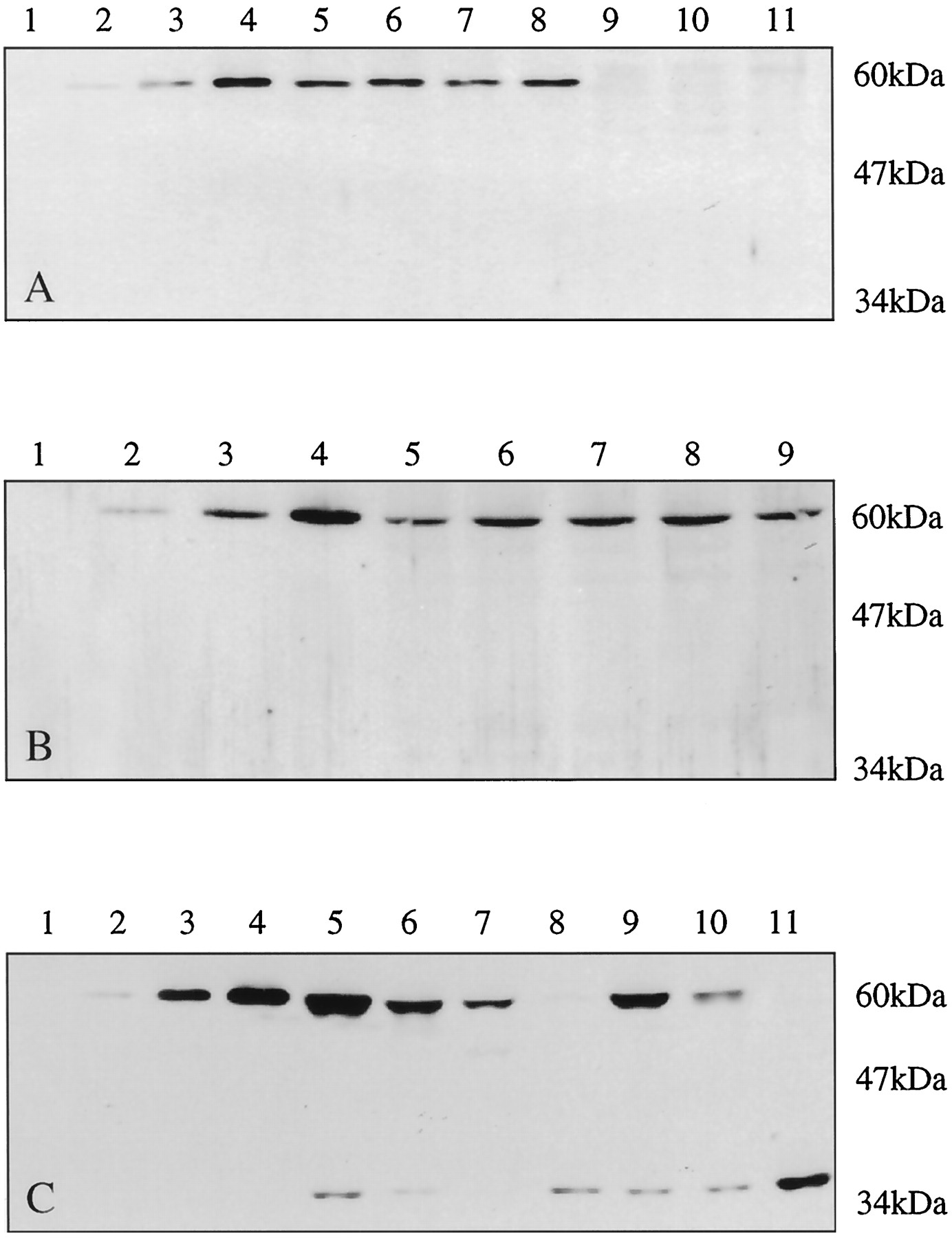
FMO1 levels in human kidney were the highest obtained of the four tissues examined with a range of values from 43.4 to 57.2 pmol/mg (mean ± S.D. = 47 ± 9 pmol/mg; n = 4) (Fig.2A; Table1). Mean adult liver and fetal liver levels were determined to be <1 pmol/mg of protein (n= 4), and 14.4 ± 3.5 pmol/mg (range, 9.7–18.2 pmol/mg;n = 4), respectively (Fig. 2, A and B; Table 1).

Western blots of human tissues probed with the FMO1-selective antibody.
Primary antibody (0.5 μg/ml × 1 h) followed by secondary antibody (1:5000, 1 h) and developed using chemiluminescence with 1 h film exposure. A: lane 1, blank; lane 2, 200 fmol of FMO1; lane 3, 400 fmol of FMO1; lane 4, 800 fmol of FMO1; lane 5, 10 μg of HK1; lane 6, 10 μg of HK2; lane 7, 10 μg of HK3; lane 8, 10 μg of HK112; lane 9, 30 μg of HL112; lane 10, 30 μg of HL122; lane 11, 30 μg of HL123. B: lane 1, blank; lane 2, 200 fmol of FMO1; lane 3, 400 fmol of FMO1; lane 4, 800 fmol of FMO1; lane 5, 20 μg of FL277; lane 6, 20 μg of FL306; lane 7, 20 μg of FL269; lane 8, 20 μg of FL343; lane 9, 20 μg of FL339. C: lane 1, blank; lane 2, 200 of fmol FMO1; lane 3, 400 fmol of FMO1; lane 4, 800 fmol of FMO1; lane 5, 100 μg of HI33; lane 6, 100 μg of HIA; lane 7, 100 μg of HIB; lane 8, 100 μg of HIC; lane 9, 100 μg of HID; lane 10, 100 μg of HI30; lane 11, 100 μg of HI25.
FMO1 content in human tissues
In contrast to the data obtained for human fetal and adult liver, and kidney tissue, Western blotting of intestinal microsomes provided two immunoreactive bands, one with an apparent molecular mass near 60 kDa, corresponding to full-length FMO1, and a second usually minor band that migrated with an apparent molecular weight near 35 kDa (Fig.2C; Table 1). In some intestinal preparations, only the lower molecular weight band was evident. Therefore, the densities of the two immunoreactive bands were summated to estimate the total FMO1 content of human intestinal microsomes. These values ranged from 1 to 6 pmol/mg of protein with a mean value ± standard deviation of 2.9 ± 1.9 pmol/mg (n = 7) (Table 1). If only the 60-kDa band was used to determine FMO1 content in human intestinal microsomes, the corresponding mean value ± standard deviation was 2.0 ± 2.0 pmol/mg (Table 1). The lower titer peptide antibody directed at the carboxyl terminus of FMO1 recognized the 60-kDa band but not the 35-kDa band (data not shown).
To provide further evidence that FMO1 is present in human intestinal microsomes, we determined the stereochemistry of arylalkyl sulfoxide formation by recombinant human FMO1 and by human intestinal microsomes treated with detergent to mask the contribution of P450 enzymes to overall stereochemistry (Rettie et al., 1995). As expected from previous stereochemical sulfoxidation studies conducted with rabbit FMO1 (Rettie et al., 1994) and rat FMO1 (Kashiyama et al., 1994), recombinant human FMO1 formed (R)-p-tolyl methyl sulfoxide in greater than 98% enantiomeric excess. Microsomalp-tolyl methyl sulfide metabolism was examined for human intestinal sample A, for which sufficient microsomal protein was available to conduct multiple incubations. p-Tolyl methyl sulfoxide was the only metabolite detected, and it was present at a level 11 times that of the reaction control (Fig.3, A and B). The rate of intestinal sulfoxide formation was 106 ± 4 pmol/mg/min, which is approximately 10% of the mean rates reported earlier for adult human liver microsomal FMO (Sadeque et al., 1992). As shown in Fig. 3C, the (R) enantiomer of p-tolyl methyl sulfoxide was generated stereoselectively by detergent-treated intestinal microsomes (90% R; 80% enantiomeric excess).

Products obtained from the incubation of detergent-solubilized human intestinal microsomes withp-tolyl methyl sulfide (1 mM).
A, intestinal microsomes incubated without NADPH and analyzed on a Partisil 10 column. The peak at 4.4 min is the p-tolyl ethyl sulfoxide internal standard. The peak at 8.4 min isp-tolyl methyl sulfoxide. B, intestinal microsomes incubated with NADPH. Peak identity is as above. C, chiral phase analysis of p-tolyl methyl sulfoxide formed enzymatically in B. The ratio of the S:Renantiomers is 10:90. Enantiomers were separated on a Chiralcel OB column.
Discussion
Current attempts to predict potential xenobiotic biotransformation pathways in humans have sparked interest in the FMO enzyme system. Most attention has been directed toward human FMO3, because it appears to be the major FMO isoform present in adult human liver. Recently, human FMO3 has been shown to readily N- or S-oxygenate trimethylamine (Lang et al., 1998), several tricyclic antidepressants (Adali et al., 1998), antipsychotics (Tugnait et al., 1997; Ring et al., 1999), H2 receptor antagonists (Overby et al., 1997), tamoxifen (Hodgson et al., 2000), amphetamine derivatives (Cashman et al., 1999), and the disulfiram metabolite,S-methyl-N,N-diethyldithiocarbamate (Pike et al., 1999). In contrast, the tissue distribution and catalytic properties of human FMO1 are much less well defined. Some indication of the tissue distribution of FMO1 can be gleaned from existing Northern blot analyses (Dolphin et al., 1996), although it is notable thatOverby et al., (1997) have reported a lack of concordance between mRNA levels and protein concentrations of human hepatic FMOs. Metabolic information for human FMO1 is even more limited, although preliminary data from our laboratory suggest that human FMO1 has probably the widest substrate specificity of the human isoforms and manifests substantial turnover numbers (20–40/min) at physiological pH (Lang and Rettie, 1998a,b).
The present studies show that FMO1 protein is not detectable (<1 pmol/mg) in adult human liver microsomes. However, FMO1 is clearly present in fetal liver. Previously we had reported that detergent-treated human fetal liver microsomes metabolizep-tolyl methyl sulfide to (R)-p-tolyl methyl sulfoxide in 86% enantiomeric excess (Sadeque et al., 1992), and the stereochemical composition we report here forp-tolyl methyl sulfoxide formed by recombinant human FMO1 (>98% enantiomeric excess, (R)-sulfoxide) are consistent with a dominant functional role for the FMO1 isoform in human fetal liver microsomes. In previous immunoblotting studies with anti-FMO3 (Sadeque et al., 1993) we were unable to detect this isoform in fetal liver microsomes, although a less sensitive visualization procedure was used. Nonetheless, when all the foregoing data are considered together, it is clear there is a profound ontogenic change in the FMO isoform complement in human liver. This is very reminiscent of the developmental profiles described for human liver cytochrome P450 (CYP)3A7 (fetal form) and CYP3A4 (adult form), a switch that occurs soon after birth (Lacroix et al., 1997). Further studies are required to determine exactly when the transition from FMO1 to FMO3 expression is triggered in human liver.
It is now well recognized that human jejunum and duodenum contain CYP3A4 in sufficient abundance to have a significant effect on first-pass metabolism and the bioavailability of certain drugs (Hall et al., 1999). As noted above, the catalytic activity of human FMO1 at physiological pH can be very high, and so it was of interest to determine the level of FMO1 protein in human small intestine microsomes. Immunoblots revealed a low and variable content of FMO1, which required visualization by ECL. However, quantitation was complicated by the presence of an additional band of ∼35 kDa on Western blots. On repeated freezing and thawing of intestinal preparations, additional lower molecular mass bands, immunoreactive toward both anti-FMO1 and the peptide antibody, could be identified (data not shown). Therefore, we assume that the initial 35-kDa protein is an FMO1 degradation product lacking the carboxyl terminus. Support for this interpretation came from additional immunoblotting studies with an anti-FMO1 peptide antibody directed against the carboxyl terminus of human FMO1 that recognized some, but not all, protein fragments detected by the nonpeptide antibody. Total FMO1 levels in human intestinal microsomes were between 1 and 6 pmol/mg, and stereochemical studies analogous to those described above for human fetal liver microsomes are consistent with the expression of functional FMO1 in human intestine. Interestingly, a rabbit FMO isoform, most likely FMO1 (Shehin-Johnson et al., 1995) is responsible for the majority of cimetidine sulfoxidation carried out by rabbit intestinal microsomes (Lu et al., 1998). However, as described for CYP2D6 (Madani et al., 1999), it seems unlikely that human FMO1 would be a significant contributor to intestinal first-pass metabolism of susceptible substrates unless the in vivo level of enzyme expression measured here is grossly underestimated due to proteolysis.
The highest levels of immunoreactive FMO1 were found in human kidney microsomes, where concentrations actually exceed some estimates of total P450 content in this organ (Jakobssen and Cinti, 1973). We reported previously that detergent-treated kidney microsomes also form (R)-p-tolyl methyl sulfoxide in high enantiomeric excess (Sadeque et al., 1992). Therefore, as for human fetal liver and small intestine, there is catalytic evidence for the expression of functional FMO1 in these tissues, although we cannot exclude the possibility that other, as yet unidentified, FMO isoforms with the appropriate metabolic stereoselectivity are expressed in these tissues. Nonetheless, FMO1 appears to be a major monooxygenase in human kidney. Indeed, it has been demonstrated that human kidney microsomal FMO plays an important role in the conversion of the cyclooxygenase inhibitor sulindac sulfide to its inactive sulfoxide metabolite (Eriksson and Bostrom, 1988). Moreover, it is possible that the “renal sparing effects” of sulindac could in part be due to facile FMO-mediated metabolism of sulindac sulfide within kidney tissue (S. D. Hall, personal communication). Therefore, the available immunochemical and catalytic data suggest that human kidney FMO1 could be a significant contributor to the extrahepatic clearance of drugs whereN-oxidation or sulfoxidation are important metabolic routes.
Acknowledgments
We acknowledge Jeannine Fisher and Dr. Mary Paine for their preliminary analysis of intestinal microsomes probed with the original nonspecific FMO1 antibody, and Charles L. Crespi and Erin L. Code from Gentest Corp. (Woburn, MA) for providing the anti-FMO1 peptide antibody .
Footnotes
-
Send reprint requests to: Allan E. Rettie, Department of Medicinal Chemistry, University of Washington, Box 357610, Seattle WA 98195. E-mail: rettie{at}u.washington.edu
-
↵1 Current address: Bayer AG, D-42096 Wuppertal, Germany.
-
This work was supported by National Institutes of Health Grant GM43511. C.K.Y. was supported by NIH Training Grant GM07750.
- Abbreviations used are::
- FMO
- flavin-containing monooxygenases
- PAGE
- polyacrylamide gel electrophoresis
- BCIP/NBT
- 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium
- HL
- human adult liver
- HK
- human adult kidney
- HI
- human adult intestine
- FL
- fetal liver
- CYP
- cytochrome P-450
- ECL
- enhanced chemiluminescence
- Received March 29, 2000.
- Accepted June 12, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}